

Advances in Research on Type 2 Diabetes Mellitus Targets and Therapeutic Agents

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Incretin-Based Targets and Therapeutic Medicines
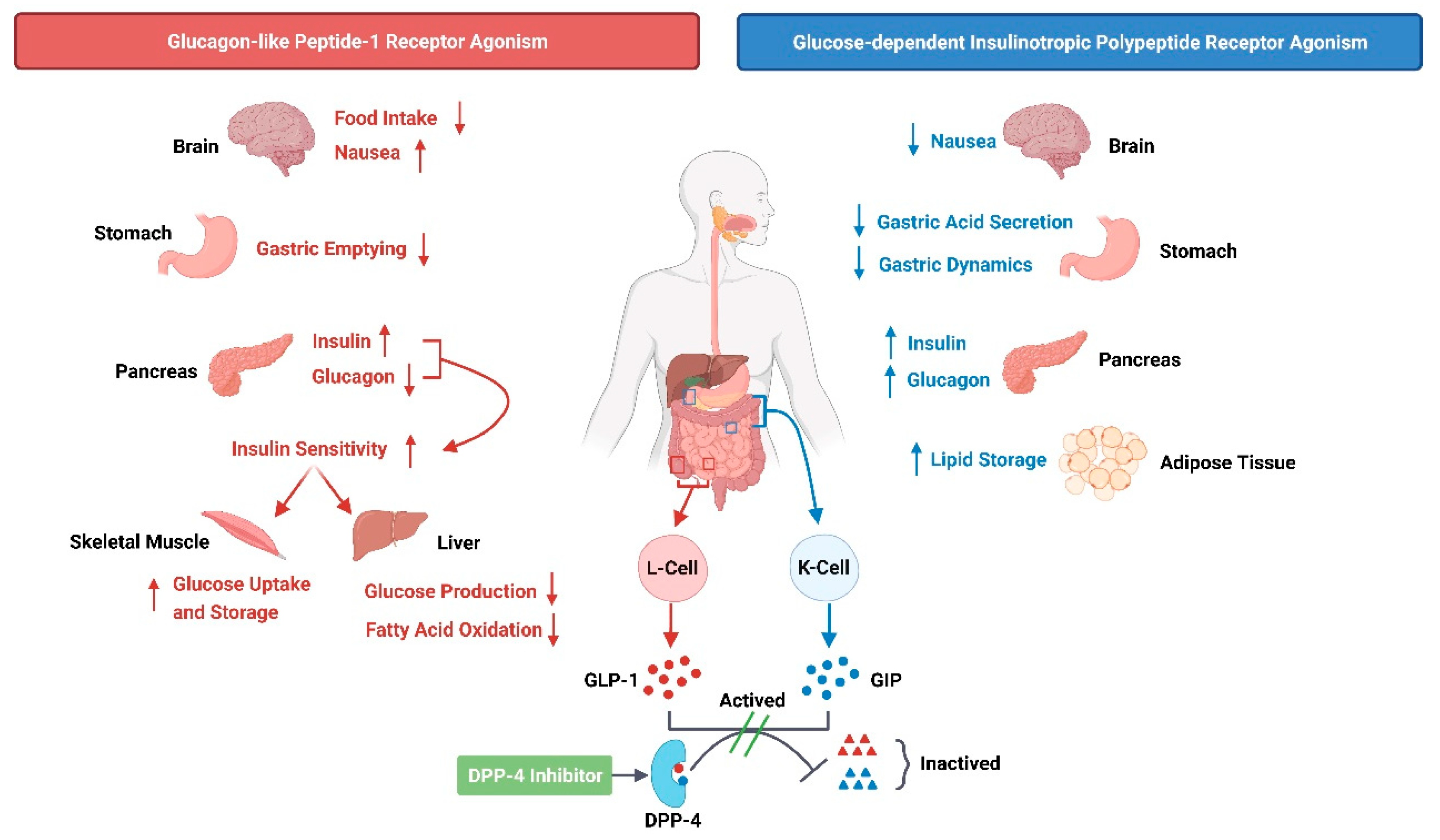
3.1. Glucagon-like Peptide-1 (GLP-1) and Glucose-Dependent Insulinotropic Polypeptide (GIP)
3.2. Dipeptidyl Peptidase-4 (DPP-4)
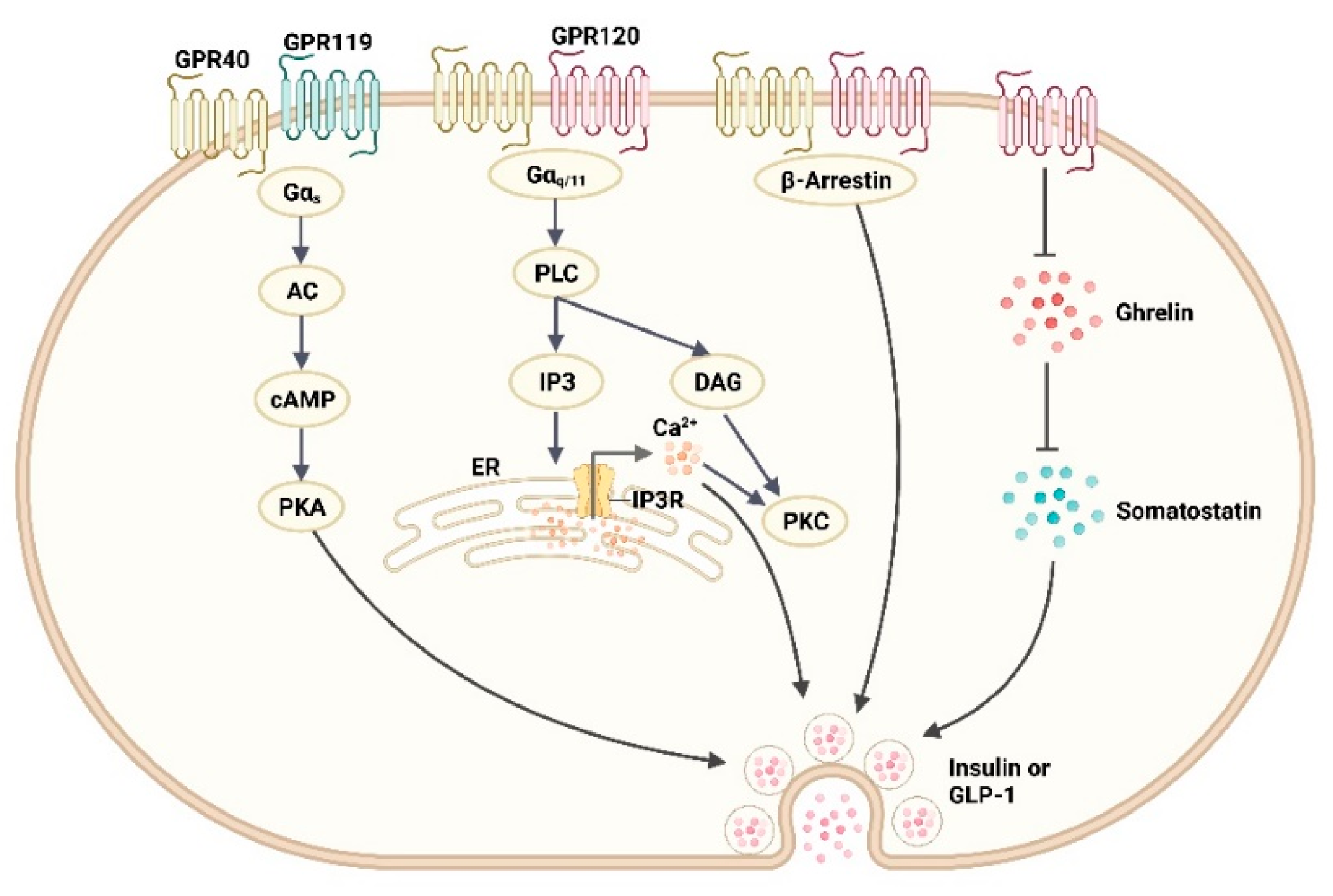
3.3. G protein-Coupled Receptors (GPCRs)
4. Glucose Metabolism Pathway-Based Targets and Therapeutic Medicines
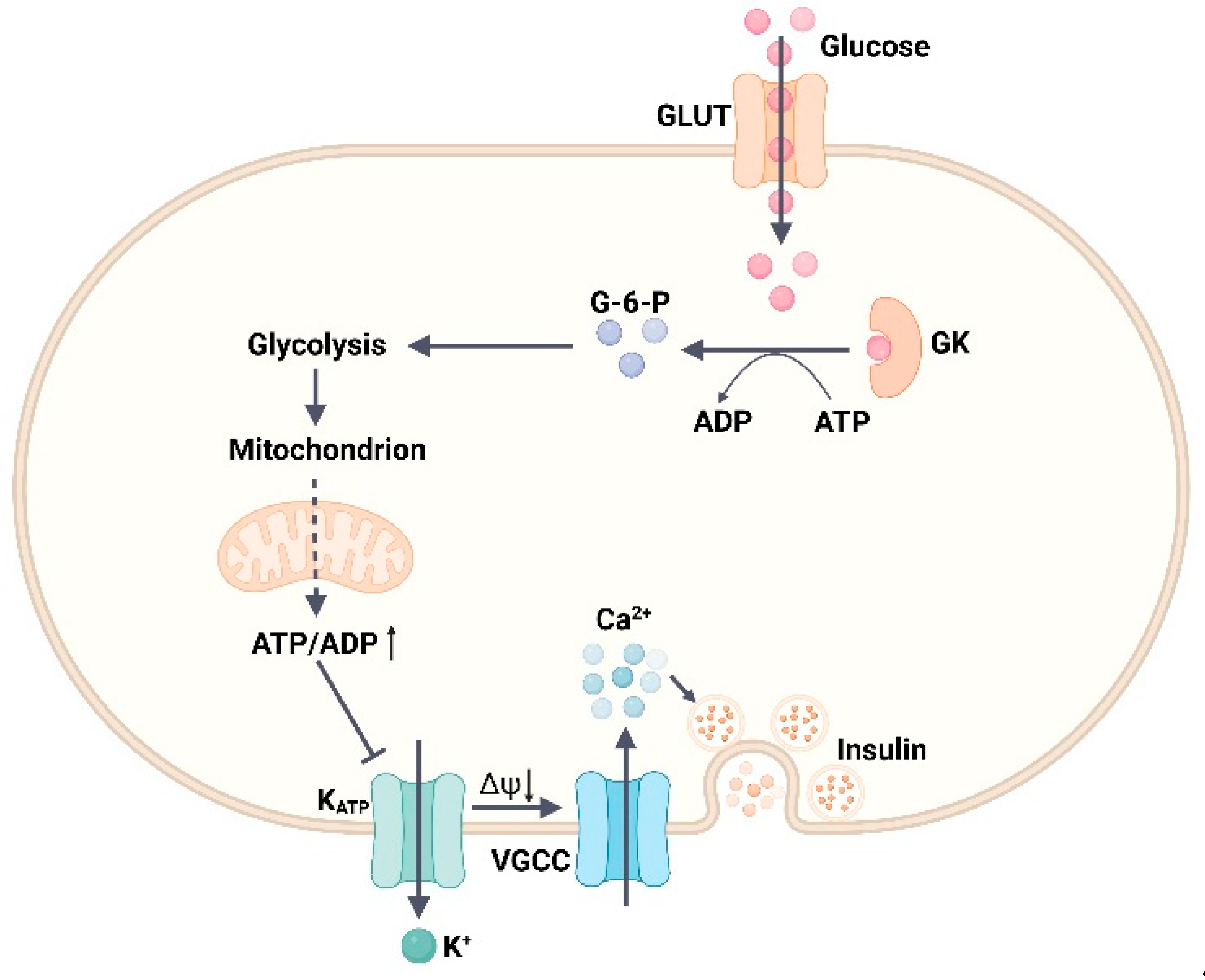
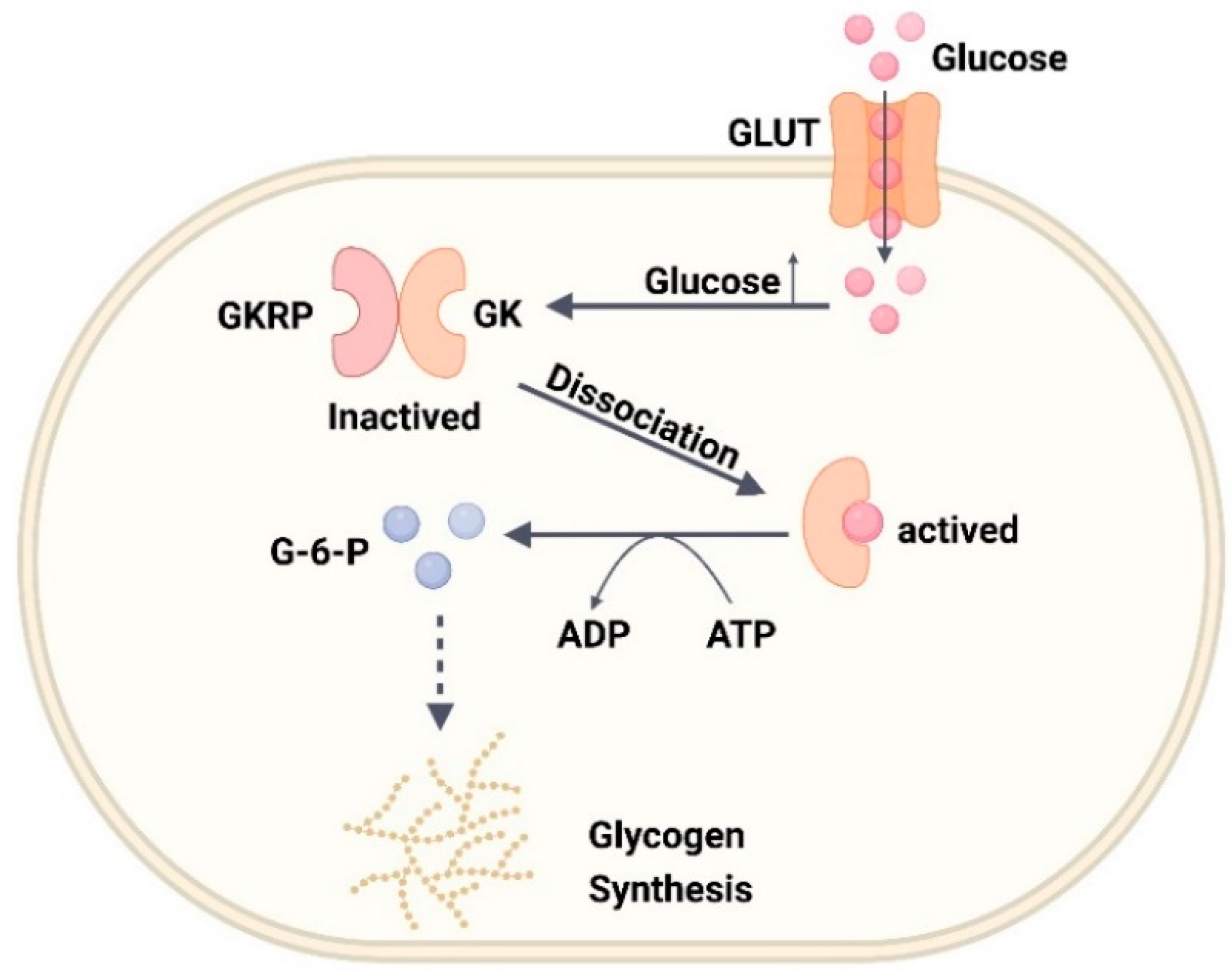
4.1. Glucose Kinase (GK)
4.2. Protein Kinase B (AKT/PKB)
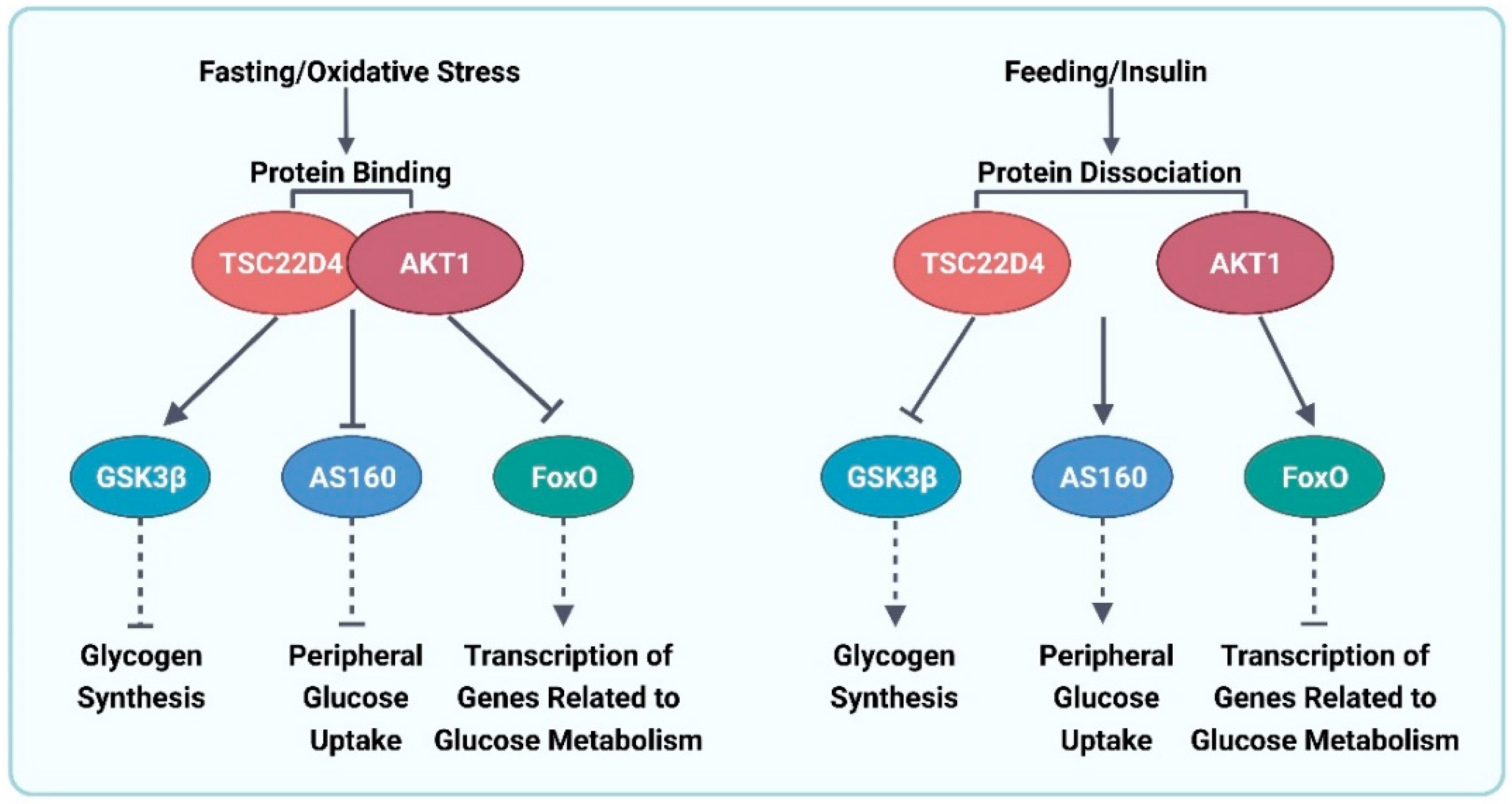
4.3. Transforming Growth Factor-β1 Stimulated Clone 22 D4 (TSC22D4)
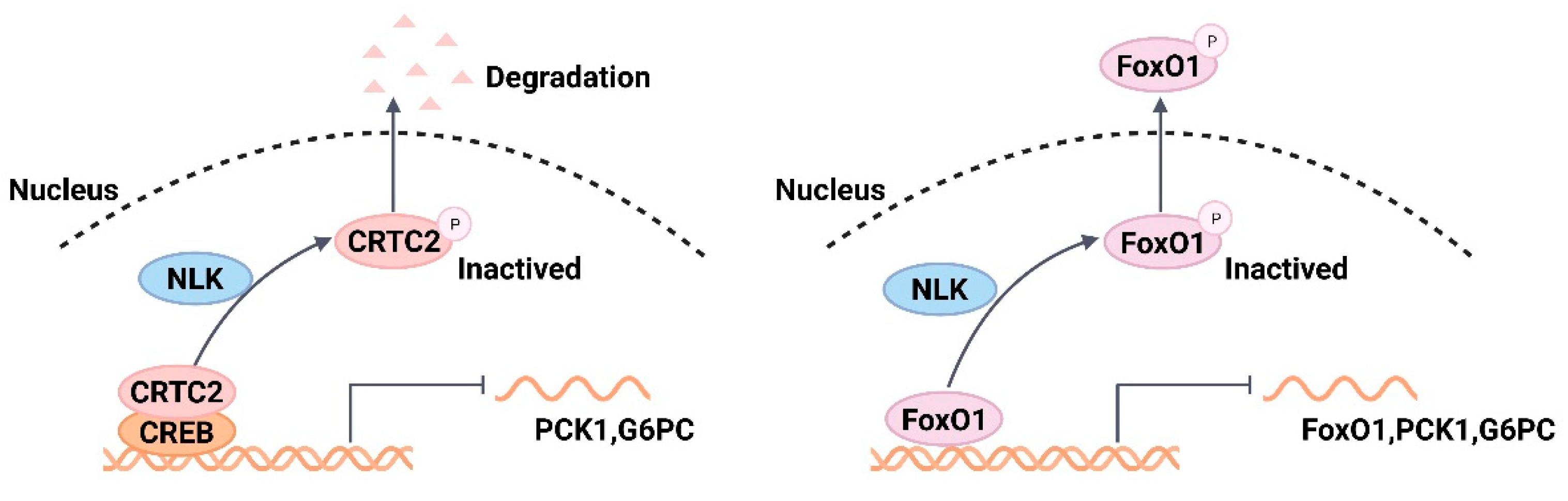
4.4. Nemo-like Kinase (NLK)
5. Insulin-Based Targets and Therapeutic Medicines
5.1. Fibroblast Growth Factor 21 (FGF21)
5.2. Protein Tyrosine Phosphatase 1B (PTP1B)
6. Other Targets and Therapeutic Medicines
6.1. Sodium-Glucose Cotransporter Protein-2 (SGLT-2)
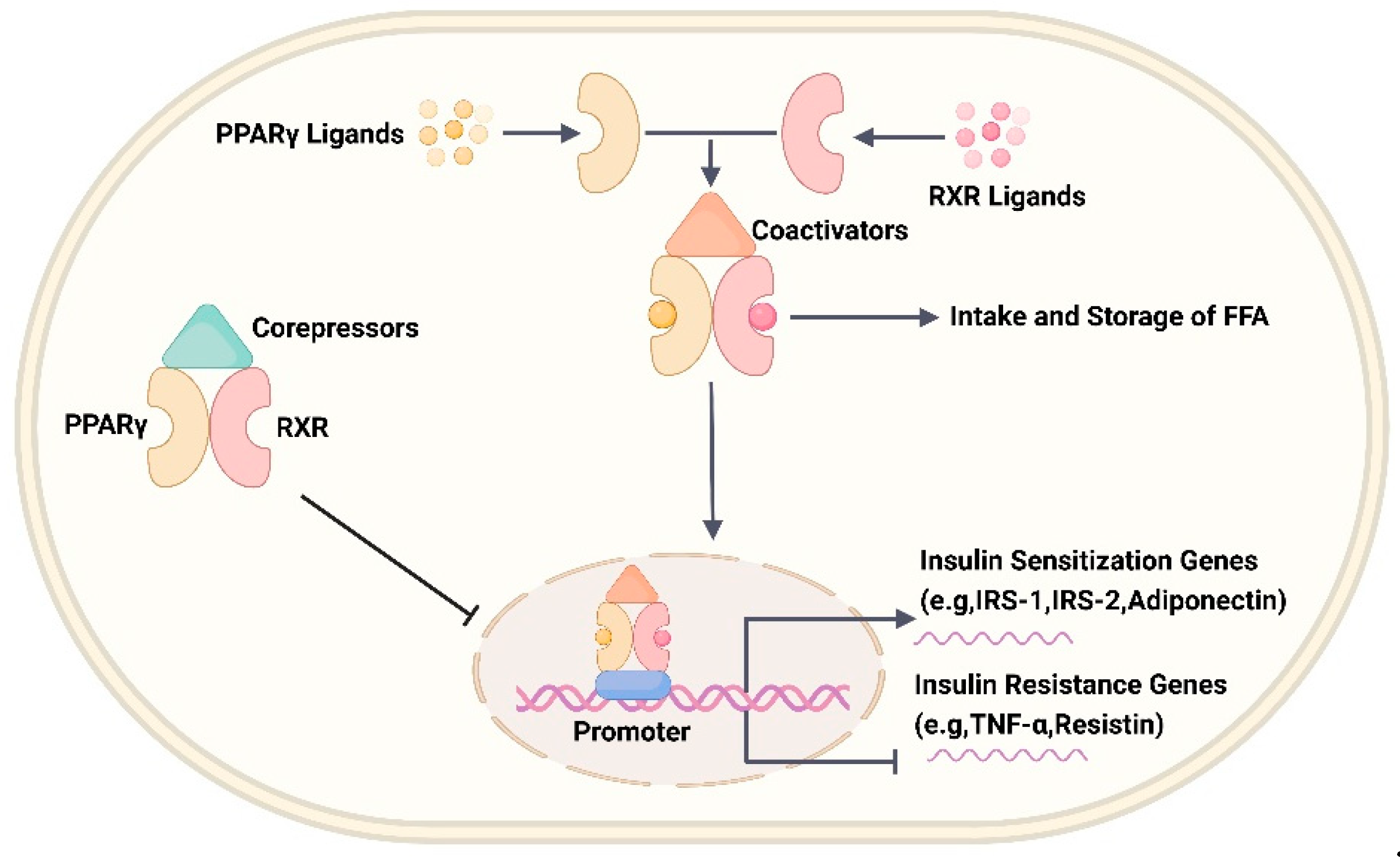
6.2. Peroxisome Proliferator-Activated Receptor (PPAR)
6.3. Gut Microbiota
6.4. microRNA (miRNA)
6.5. Glucose-Sensitive Neurons (GSNs)
6.6. Carbohydrate Response Element-Binding Protein (ChREBP)
6.7. Islet Microexons (IsletMICs)
7. Diet- and Exercise-Based Therapy
8. Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Diabetes Association Professional Practice Committee. 6. Glycemic Targets: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S83–S96. [Google Scholar] [CrossRef]
- Janssen, J.A.M.J.L. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef]
- Tomic, D.; Shaw, J.E.; Magliano, D.J. The burden and risks of emerging complications of diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 525–539. [Google Scholar] [CrossRef]
- Lim, S.; Bae, J.H.; Kwon, H.-S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021, 17, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Jagat, J.M.; Kalyan, K.G.; Subir, R. Use of pioglitazone in people with type 2 diabetes mellitus with coronavirus disease 2019 (COVID-19): Boon or Bane? Diabetes Metab. Syndr. 2020, 14, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.; Avgerinos, I.; Liakos, A.; Del Prato, S.; Matthews, D.R.; Tsapas, A.; Bekiari, E. Management of type 2 diabetes with the dual GIP/GLP-1 receptor agonist tirzepatide: A systematic review and meta-analysis. Diabetologia 2022, 65, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Samms, R.J.; Coghlan, M.P.; Sloop, K.W. How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol. Metab. 2020, 31, 410–421. [Google Scholar] [CrossRef]
- Nauck, M.A.; Müller, T.D. Incretin Hormones and Type 2 Diabetes. Diabetologia 2023. [Google Scholar] [CrossRef]
- Drucker, D.J. GLP-1 physiology informs the pharmacotherapy of obesity. Mol. Metab. 2021, 57, 101351. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Metabolic Messengers: Glucagon-like peptide 1. Nat. Metab. 2021, 3, 142–148. [Google Scholar] [CrossRef]
- Hayes, M.R.; Borner, T.; De Jonghe, B.C. The Role of GIP in the Regulation of GLP-1 Satiety and Nausea. Diabetes 2021, 70, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Rosenkilde, M.M. GIP as a Therapeutic Target in Diabetes and Obesity: Insight From Incretin Co-Agonists. J. Clin. Endocrinol. Metab. 2020, 105, e2710–e2716. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Wysham, C.; Frías, J.P.; Kaneko, S.; Lee, C.J.; Fernández Landó, L.; Mao, H.; Cui, X.; Karanikas, C.A.; Thieu, V.T. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): A double-blind, randomised, phase 3 trial. Lancet 2021, 398, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Wefers, J.; Meier, J.J. Treatment of type 2 diabetes: Challenges, hopes, and anticipated successes. Lancet Diabetes Endocrinol. 2021, 9, 525–544. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef]
- Borner, T.; Geisler, C.E.; Fortin, S.M.; Cosgrove, R.; Alsina-Fernandez, J.; Dogra, M.; Doebley, S.; Sanchez-Navarro, M.J.; Leon, R.M.; Gaisinsky, J.; et al. GIP Receptor Agonism Attenuates GLP-1 Receptor Agonist–Induced Nausea and Emesis in Preclinical Models. Diabetes 2021, 70, 2545–2553. [Google Scholar] [CrossRef]
- Sattar, N.; McGuire, D.K.; Pavo, I.; Weerakkody, G.J.; Nishiyama, H.; Wiese, R.J.; Zoungas, S. Tirzepatide cardiovascular event risk assessment: A pre-specified meta-analysis. Nat. Med. 2022, 28, 591–598. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Thomas, M.K.; Nikooienejad, A.; Bray, R.; Cui, X.; Wilson, J.; Duffin, K.; Milicevic, Z.; Haupt, A.; Robins, D.A. Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-Cell Function and Insulin Sensitivity in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, 388–396. [Google Scholar] [CrossRef]
- Huang, J.; Liu, X.; Wei, Y.; Li, X.; Gao, S.; Dong, L.; Rao, X.; Zhong, J. Emerging Role of Dipeptidyl Peptidase-4 in Autoimmune Disease. Front. Immunol. 2022, 13, 830863. [Google Scholar] [CrossRef]
- Yin, R.; Xu, Y.; Wang, X.; Yang, L.; Zhao, D. Role of Dipeptidyl Peptidase 4 Inhibitors in Antidiabetic Treatment. Molecules 2022, 27, 3055. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.D.; Solomon, A. Inhibitors of dipeptidyl peptidase-4 as therapeutic agents for individuals with type 2 diabetes: A 25-year journey. Diabet. Med. 2020, 37, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Razavi, M.; Wei, Y.-Y.; Rao, X.-Q.; Zhong, J.-X. DPP-4 inhibitors and GLP-1RAs: Cardiovascular safety and benefits. Mil. Med. Res. 2022, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Im, K.; Murphy, S.A.; Kuder, J.F.; Rodriguez, D.A.; Lopes, R.D.; Green, J.B.; Ruff, C.T.; Sabatine, M.S. Re-adjudication of the Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS) with study-level meta-analysis of hospitalization for heart failure from cardiovascular outcomes trials with dipeptidyl peptidase-4 (DPP-4) inhibitors. Clin. Cardiol. 2022, 45, 794–801. [Google Scholar] [CrossRef]
- Lyu, Y.S.; Oh, S.; Kim, J.H.; Kim, S.Y.; Jeong, M.H. Comparison of SGLT2 inhibitors with DPP-4 inhibitors combined with metformin in patients with acute myocardial infarction and diabetes mellitus. Cardiovasc. Diabetol. 2023, 22, 185. [Google Scholar] [CrossRef]
- Xie, Y.; Bowe, B.; Xian, H.; Loux, T.; McGill, J.B.; Al-Aly, Z. Comparative effectiveness of SGLT2 inhibitors, GLP-1 receptor agonists, DPP-4 inhibitors, and sulfonylureas on risk of major adverse cardiovascular events: Emulation of a randomised target trial using electronic health records. Lancet Diabetes Endocrinol. 2023, S2213-8587(23)00171-7. [Google Scholar] [CrossRef]
- Jedlowski, P.M.; Jedlowski, M.F.; Fazel, M.T. DPP-4 Inhibitors and Increased Reporting Odds of Bullous Pemphigoid: A Pharmacovigilance Study of the FDA Adverse Event Reporting System (FAERS) from 2006 to 2020. Am. J. Clin. Dermatol. 2021, 22, 891–900. [Google Scholar] [CrossRef]
- Velazhahan, V.; McCann, B.L.; Bignell, E.; Tate, C.G. Developing novel antifungals: Lessons from G protein-coupled receptors. Trends Pharmacol. Sci. 2023, 44, 162–174. [Google Scholar] [CrossRef]
- Slosky, L.M.; Caron, M.G.; Barak, L.S. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol. Sci. 2021, 42, 283–299. [Google Scholar] [CrossRef]
- Ghislain, J.; Poitout, V. Targeting lipid GPCRs to treat type 2 diabetes mellitus—Progress and challenges. Nat. Rev. Endocrinol. 2021, 17, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G Protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Governa, P.; Caroleo, M.C.; Carullo, G.; Aiello, F.; Cione, E.; Manetti, F. FFAR1/GPR40: One target, different binding sites, many agonists, no drugs, but a continuous and unprofitable tug-of-war between ligand lipophilicity, activity, and toxicity. Bioorg. Med. Chem. Lett. 2021, 41, 127969. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.-P.; Xiong, Y. Learn from failures and stay hopeful to GPR40, a GPCR target with robust efficacy, for therapy of metabolic disorders. Front. Pharmacol. 2022, 13, 1043828. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Zhang, L. Current status of GPR40/FFAR1 modulators in medicinal chemistry (2016–2019): A patent review. Expert Opin. Ther. Pat. 2020, 30, 27–38. [Google Scholar] [CrossRef]
- Nishizaki, H.; Matsuoka, O.; Kagawa, T.; Kobayashi, A.; Watanabe, M.; Moritoh, Y. SCO-267, a GPR40 Full Agonist, Stimulates Islet and Gut Hormone Secretion and Improves Glycemic Control in Humans. Diabetes 2021, 70, 2364–2376. [Google Scholar] [CrossRef]
- Li, H.; Fang, Y.; Guo, S.; Yang, Z. GPR119 agonists for the treatment of type 2 diabetes: An updated patent review (2014-present). Expert Opin. Ther. Pat. 2021, 31, 795–808. [Google Scholar] [CrossRef]
- Im, D.-S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. Int. J. Mol. Sci. 2021, 22, 1034. [Google Scholar] [CrossRef]
- Qian, Y.; Wang, J.; Yang, L.; Liu, Y.; Wang, L.; Liu, W.; Lin, Y.; Yang, H.; Ma, L.; Ye, S.; et al. Activation and signaling mechanism revealed by GPR119-Gs complex structures. Nat. Commun. 2022, 13, 7033. [Google Scholar] [CrossRef]
- Nema, P.; Asati, V.; Kendya, P.; Gupta, T.; Agarwal, S.; Kori, S.; Kashaw, V.; Iyer, A.K.; Kashaw, S.K. Structural Insight on GPR119 Agonist as Potential Therapy for Type II Diabetes: A Comprehensive Review. Mini Rev. Med. Chem. 2023. [Google Scholar] [CrossRef]
- Li, G.; Meng, B.; Yuan, B.; Huan, Y.; Zhou, T.; Jiang, Q.; Lei, L.; Sheng, L.; Wang, W.; Gong, N.; et al. The optimization of xanthine derivatives leading to HBK001 hydrochloride as a potent dual ligand targeting DPP-IV and GPR119. Eur. J. Med. Chem. 2020, 188, 112017. [Google Scholar] [CrossRef] [PubMed]
- Al Mahri, S.; Malik, S.S.; Al Ibrahim, M.; Haji, E.; Dairi, G.; Mohammad, S. Free Fatty Acid Receptors (FFARs) in Adipose: Physiological Role and Therapeutic Outlook. Cells 2022, 11, 750. [Google Scholar] [CrossRef]
- Carullo, G.; Mazzotta, S.; Vega-Holm, M.; Iglesias-Guerra, F.; Vega-Pérez, J.M.; Aiello, F.; Brizzi, A. GPR120/FFAR4 Pharmacology: Focus on Agonists in Type 2 Diabetes Mellitus Drug Discovery. J. Med. Chem. 2021, 64, 4312–4332. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Lloyd, M.; Haythorne, E.A. Glucokinase activity in diabetes: Too much of good thing? Trends Endocrinol. Metab. 2023, 34, 119–130. [Google Scholar] [CrossRef]
- Toulis, K.A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.H.; Tahrani, A.A. Glucokinase Activators for Type 2 Diabetes: Challenges and Future Developments. Drugs 2020, 80, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, L.; Wan, L.; Huang, Y.; Cao, S. Glucokinase as an emerging anti-diabetes target and recent progress in the development of its agonists. J. Enzyme Inhib. Med. Chem. 2022, 37, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Li, X.; Ma, J.; Zeng, J.; Gan, S.; Dong, X.; Yang, J.; Lin, X.; Cai, H.; Song, W.; et al. Dorzagliatin in drug-naïve patients with type 2 diabetes: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2022, 28, 965–973. [Google Scholar] [CrossRef]
- Omori, K.; Nakamura, A.; Miyoshi, H.; Yamauchi, Y.; Kawata, S.; Takahashi, K.; Kitao, N.; Nomoto, H.; Kameda, H.; Cho, K.Y.; et al. Glucokinase Inactivation Paradoxically Ameliorates Glucose Intolerance by Increasing β-Cell Mass in db/db Mice. Diabetes 2021, 70, 917–931. [Google Scholar] [CrossRef]
- Nakamura, A.; Omori, K.; Terauchi, Y. Glucokinase activation or inactivation: Which will lead to the treatment of type 2 diabetes? Diabetes Obes. Metab. 2021, 23, 2199–2206. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, S.; Sharma, N.; Singla, D.; Guarve, K.; Grewal, A.S. Targeting human glucokinase for the treatment of type 2 diabetes: An overview of allosteric Glucokinase activators. J. Diabetes Metab. Disord. 2022, 21, 1129–1137. [Google Scholar] [CrossRef]
- Syed, Y.Y. Dorzagliatin: First Approval. Drugs 2022, 82, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.; Wang, K.; Lim, C.K.P.; Tsoi, S.T.F.; Fan, B.; Poon, E.; Luk, A.O.Y.; Ma, R.C.W.; Ferrannini, E.; Mari, A.; et al. Dorzagliatin, a Dual-Acting Glucokinase Activator, Increases Insulin Secretion and Glucose Sensitivity in Glucokinase Maturity-Onset Diabetes of the Young and Recent-Onset Type 2 Diabetes. Diabetes 2023, 72, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhu, D.; Gan, S.; Dong, X.; Su, J.; Li, W.; Jiang, H.; Zhao, W.; Yao, M.; Song, W.; et al. Dorzagliatin add-on therapy to metformin in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2022, 28, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Guerau-de-Arellano, M.; Piedra-Quintero, Z.L.; Tsichlis, P.N. Akt isoforms in the immune system. Front. Immunol. 2022, 13, 990874. [Google Scholar] [CrossRef]
- Tsai, P.-J.; Lai, Y.-H.; Manne, R.K.; Tsai, Y.-S.; Sarbassov, D.; Lin, H.-K. Akt: A key transducer in cancer. J. Biomed. Sci. 2022, 29, 76. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Chen, J.; Wang, J.; Liu, J.; Jiang, Y. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Fang, X.; Wei, J.; Wu, H.; Wang, X.; Tian, J. Akt: A Potential Drug Target for Metabolic Syndrome. Front. Physiol. 2022, 13, 822333. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/MTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 2021, 64, 994–1006. [Google Scholar] [CrossRef]
- Herman, R.; Kravos, N.A.; Jensterle, M.; Janež, A.; Dolžan, V. Metformin and Insulin Resistance: A Review of the Underlying Mechanisms behind Changes in GLUT4-Mediated Glucose Transport. Int. J. Mol. Sci. 2022, 23, 1264. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Di, L.-J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Ali, I.; Yang, M.; Yang, C.; Li, Y.; Li, L. Non-Esterified Fatty Acid-Induced Apoptosis in Bovine Granulosa Cells via ROS-Activated PI3K/AKT/FoxO1 Pathway. Antioxidants 2023, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, L.; Wang, K.; Ke, J.; Li, S.; Meng, T.; Yuan, H.; Zhang, Q.; Hu, F. SC79 promotes efficient entry of GDNF liposomes into brain parenchyma to repair dopamine neurons through reversible regulation of tight junction proteins. Nano Res. 2023, 16, 2695–2705. [Google Scholar] [CrossRef]
- Wang, T.; Zheng, L.; Wang, S.; Zhao, M.; Liu, X. Anti-diabetic and anti-hyperlipidemic effects of sea cucumber (Cucumaria Frondosa) gonad hydrolysates in type II diabetic rats. Food Sci. Hum. Wellness 2022, 11, 1614–1622. [Google Scholar] [CrossRef]
- Liu, Z.; Meng, L.; Wang, M.; Wang, L.; Liu, Y.; Hou, G.; Li, S.; Kang, W. New iridoids from Patrinia Scabiosaefolia and their hypoglycemic effects by activating PI3K/Akt signaling pathway. Fitoterapia 2023, 165, 105423. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Wolff, G.; Wieder, A.; Maida, A.; Bühler, L.; Brune, M.; Hautzinger, O.; Feuchtinger, A.; Poth, T.; Szendroedi, J.; et al. TSC22D4 interacts with Akt1 to regulate glucose metabolism. Sci. Adv. 2022, 8, eabo5555. [Google Scholar] [CrossRef]
- Ekim Üstünel, B.; Friedrich, K.; Maida, A.; Wang, X.; Krones-Herzig, A.; Seibert, O.; Sommerfeld, A.; Jones, A.; Sijmonsma, T.P.; Sticht, C.; et al. Control of diabetic hyperglycaemia and insulin resistance through TSC22D4. Nat. Commun. 2016, 7, 13267. [Google Scholar] [CrossRef]
- Daams, R.; Massoumi, R. Nemo-Like Kinase in Development and Diseases: Insights from Mouse Studies. Int. J. Mol. Sci. 2020, 21, 9203. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, Y.; Huang, J.; Tan, Y.; Wei, J.; Xiao, J.; Zou, J.; Feng, H. NLK suppresses MAVS-mediated signaling in black carp antiviral innate immunity. Dev. Comp. Immunol. 2021, 122, 104105. [Google Scholar] [CrossRef]
- Zhu, X.; Song, G.; Zhang, S.; Chen, J.; Hu, X.; Zhu, H.; Jia, X.; Li, Z.; Song, W.; Chen, J.; et al. Asialoglycoprotein Receptor 1 Functions as a Tumor Suppressor in Liver Cancer via Inhibition of STAT3. Cancer Res. 2022, 82, 3987–4000. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Veeraraghavan, J.; Liu, C.-C.; Cao, X.; Qin, L.; Kim, J.-A.; Tan, Y.; Loo, S.K.; Hu, Y.; Lin, L.; et al. Therapeutic Targeting of Nemo-like Kinase in Primary and Acquired Endocrine-resistant Breast Cancer. Clin. Cancer Res. 2021, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.-X.; Wang, Y.; Li, P.-L.; Cai, L.; Wang, X.-M.; Bai, L.; Liu, Z.; Tian, H.; Tian, S.; Zhang, P.; et al. A Kinome screen reveals that Nemo-like kinase is a key suppressor of hepatic gluconeogenesis. Cell. Metab. 2021, 33, 1171–1186.e9. [Google Scholar] [CrossRef] [PubMed]
- Phan, P.; Saikia, B.B.; Sonnaila, S.; Agrawal, S.; Alraawi, Z.; Kumar, T.K.S.; Iyer, S. The Saga of Endocrine FGFs. Cells 2021, 10, 2418. [Google Scholar] [CrossRef]
- Geng, L.; Lam, K.S.L.; Xu, A. The therapeutic potential of FGF21 in metabolic diseases: From bench to clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Szczepańska, E.; Gietka-Czernel, M. FGF21: A Novel Regulator of Glucose and Lipid Metabolism and Whole-Body Energy Balance. Horm. Metab Res. 2022, 54, 203–211. [Google Scholar] [CrossRef]
- Flippo, K.H.; Potthoff, M.J. Metabolic Messengers: FGF21. Nat. Metab. 2021, 3, 309–317. [Google Scholar] [CrossRef]
- Aaldijk, A.S.; Verzijl, C.R.C.; Jonker, J.W.; Struik, D. Biological and pharmacological functions of the FGF19- and FGF21-coreceptor beta klotho. Front. Endocrinol. Lausanne 2023, 14, 1150222. [Google Scholar] [CrossRef]
- Spann, R.A.; Morrison, C.D.; den Hartigh, L.J. The Nuanced Metabolic Functions of Endogenous FGF21 Depend on the Nature of the Stimulus, Tissue Source, and Experimental Model. Front. Endocrinol. Lausanne 2022, 12, 802541. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyahisa, M.; Chikamatsu, M.; Nishida, K.; Minayoshi, Y.; Takano, M.; Ichimizu, S.; Kobashigawa, Y.; Morioka, H.; Maeda, H.; et al. Development of a long acting FGF21 analogue-albumin fusion protein and its anti-diabetic effects. J. Control. Release 2020, 324, 522–531. [Google Scholar] [CrossRef]
- Zhu, L.; Zhao, H.; Liu, J.; Cai, H.; Wu, B.; Liu, Z.; Zhou, S.; Liu, Q.; Li, X.; Bao, B.; et al. Dynamic folding modulation generates FGF21 variant against diabetes. EMBO Rep. 2021, 22, e51352. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhao, H.; Yin, C.; Lan, X.; Wu, L.; Du, X.; Griffiths, H.R.; Gao, D. Adipokines, Hepatokines and Myokines: Focus on Their Role and Molecular Mechanisms in Adipose Tissue Inflammation. Front. Endocrinol. Lausanne 2022, 13, 873699. [Google Scholar] [CrossRef]
- Queen, N.J.; Bates, R.; Huang, W.; Xiao, R.; Appana, B.; Cao, L. Visceral adipose tissue-directed FGF21 gene therapy improves metabolic and immune health in BTBR mice. Mol. Ther. Methods Clin. Dev. 2020, 20, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Lin, S.; Li, Y.; Liu, L.; Li, X.; Gao, X.; Yan, J.; Gu, B.; Chen, X.; Li, W.; et al. A novel GLP-1 and FGF21 dual agonist has therapeutic potential for diabetes and non-alcoholic steatohepatitis. EBioMedicine 2021, 63, 103202. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Xiao, X.; Yu, T.; Xiao, X.; Xie, J.; Ji, Q.; Wang, L.; Na, T.; Meng, S.; Qian, L.; et al. Mesenchymal stem cells modified by FGF21 and GLP1 ameliorate lipid metabolism while reducing blood glucose in type 2 diabetic mice. Stem. Cell. Res. Ther. 2021, 12, 133. [Google Scholar] [CrossRef]
- Teimouri, M.; Hosseini, H.; ArabSadeghabadi, Z.; Babaei-Khorzoughi, R.; Gorgani-Firuzjaee, S.; Meshkani, R. The role of protein tyrosine phosphatase 1B (PTP1B) in the pathogenesis of type 2 diabetes mellitus and its complications. J. Physiol. Biochem. 2022, 78, 307–322. [Google Scholar] [CrossRef]
- Villamar-Cruz, O.; Loza-Mejía, M.A.; Arias-Romero, L.E.; Camacho-Arroyo, I. Recent advances in PTP1B signaling in metabolism and cancer. Biosci. Rep. 2021, 41, BSR20211994. [Google Scholar] [CrossRef]
- Sharma, B.; Xie, L.; Yang, F.; Wang, W.; Zhou, Q.; Xiang, M.; Zhou, S.; Lv, W.; Jia, Y.; Pokhrel, L.; et al. Recent advance on PTP1B inhibitors and their biomedical applications. Eur. J. Med. Chem. 2020, 199, 112376. [Google Scholar] [CrossRef]
- Liu, R.; Mathieu, C.; Berthelet, J.; Zhang, W.; Dupret, J.-M.; Rodrigues Lima, F. Human Protein Tyrosine Phosphatase 1B (PTP1B): From Structure to Clinical Inhibitor Perspectives. Int. J. Mol. Sci. 2022, 23, 7027. [Google Scholar] [CrossRef]
- Campos-Almazán, M.I.; Hernández-Campos, A.; Castillo, R.; Sierra-Campos, E.; Valdez-Solana, M.; Avitia-Domínguez, C.; Téllez-Valencia, A. Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century. Pharmaceuticals 2022, 15, 866. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R. Can Allostery Be a Key Strategy for Targeting PTP1B in Drug Discovery? A Lesson from Trodusquemine. Int. J. Mol. Sci. 2023, 24, 9621. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, H.; Zhao, Z.; Huang, M.; Wang, S.; Zhan, J. Status of research on natural protein tyrosine phosphatase 1B inhibitors as potential antidiabetic agents: Update. Biomed. Pharmacother. 2023, 157, 113990. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Page, R.; Peti, W. The mode of action of the Protein tyrosine phosphatase 1B inhibitor Ertiprotafib. PLoS ONE 2020, 15, e0240044. [Google Scholar] [CrossRef] [PubMed]
- Rath, P.; Ranjan, A.; Chauhan, A.; Verma, N.K.; Bhargava, A.; Prasad, R.; Jindal, T. A Critical Review on Role of Available Synthetic Drugs and Phytochemicals in Insulin Resistance Treatment by Targeting PTP1B. Appl. Biochem. Biotechnol. 2022, 194, 4683–4701. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Genovese, M.; Santi, A.; Pranzini, E.; Balestri, F.; Piazza, L.; Del Corso, A.; Avunduk, S.; Imperatore, C.; Menna, M.; et al. Evidence of Insulin-Sensitizing and Mimetic Activity of the Sesquiterpene Quinone Avarone, a Protein Tyrosine Phosphatase 1B and Aldose Reductase Dual Targeting Agent from the Marine Sponge Dysidea Avara. Pharmaceutics 2023, 15, 528. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Park, S.E.; Seong, S.H.; Zamponi, G.W.; Jung, H.A.; Choi, J.S. Ursonic acid from Artemisia montana exerts anti-diabetic effects through anti-glycating properties, and by inhibiting PTP1B and activating the PI3K/Akt signaling pathway in insulin-resistant C2C12 Cells. Chem. Biol. Interact. 2023, 376, 110452. [Google Scholar] [CrossRef]
- Gao, Y.-M.; Feng, S.-T.; Wen, Y.; Tang, T.-T.; Wang, B.; Liu, B.-C. Cardiorenal protection of SGLT2 inhibitors—Perspectives from metabolic reprogramming. EBioMedicine 2022, 83, 104215. [Google Scholar] [CrossRef]
- Rizzo, M.R.; Di Meo, I.; Polito, R.; Auriemma, M.C.; Gambardella, A.; Mauro, G.; Capuano, A.; Paolisso, G. Cognitive impairment and type 2 diabetes mellitus: Focus of SGLT2 inhibitors treatment. Pharmacol. Res. 2022, 176, 106062. [Google Scholar] [CrossRef]
- Scheen, A.J. Efficacy/safety balance of DPP-4 inhibitors versus SGLT2 inhibitors in elderly patients with type 2 diabetes. Diabetes Metab. 2021, 47, 101275. [Google Scholar] [CrossRef]
- Kaneto, H.; Obata, A.; Kimura, T.; Shimoda, M.; Kinoshita, T.; Matsuoka, T.-A.; Kaku, K. Unexpected Pleiotropic Effects of SGLT2 Inhibitors: Pearls and Pitfalls of This Novel Antidiabetic Class. Int. J. Mol. Sci. 2021, 22, 3062. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Ha, K.H.; Kim, D.J. Cardiovascular Safety of Sodium Glucose Cotransporter 2 Inhibitors as Add-on to Metformin Monotherapy in Patients with Type 2 Diabetes Mellitus. Diabetes Metab. J. 2021, 45, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Elahi, A.; Hussain, A.; Iqbal, J.; Akhtar, L.; Majid, A. Sodium-Glucose Cotransporter-2 (SGLT-2) Attenuates Serum Uric Acid (SUA) Level in Patients with Type 2 Diabetes. J. Diabetes Res. 2021, 2021, 9973862. [Google Scholar] [CrossRef]
- La Grotta, R.; Candia, P.; Olivieri, F.; Matacchione, G.; Giuliani, A.; Rippo, M.R.; Tagliabue, E.; Mancino, M.; Rispoli, F.; Ferroni, S.; et al. Anti-inflammatory effect of SGLT-2 inhibitors via uric acid and insulin. Cell. Mol. Life Sci. 2022, 79, 273. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Wakabayashi, M.; Bhalla, A.; Chopra, N.; Miyashita, H.; Mikami, T.; Ueyama, H.; Fujisaki, T.; Saigusa, Y.; Yamaji, T.; et al. Cardiovascular and renal outcomes with SGLT-2 inhibitors versus GLP-1 receptor agonists in patients with type 2 diabetes mellitus and chronic kidney disease: A systematic review and network meta-analysis. Cardiovasc. Diabetol. 2021, 20, 14. [Google Scholar] [CrossRef]
- Wiegley, N.; So, P.N. Sodium-Glucose Cotransporter 2 Inhibitors and Urinary Tract Infection: Is There Room for Real Concern? Kidney360 2022, 3, 1991–1993. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Li, S.; Wang, Y.; Qin, X.; Deng, K.; Liu, Y.; Zou, K.; Sun, X. Sodium-glucose co-transporter-2 inhibitors and the risk of diabetic ketoacidosis in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2020, 22, 1619–1627. [Google Scholar] [CrossRef]
- Zhang, Y.-S.; Zheng, Y.-D.; Yuan, Y.; Chen, S.-C.; Xie, B.-C. Effects of Anti-Diabetic Drugs on Fracture Risk: A Systematic Review and Network Meta-Analysis. Front. Endocrinol. Lausanne 2021, 12, 735824. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C.; Cavender, M.A.; Fioretto, P.; Fowler, K.J.; Heerspink, H.J.L.; Manley, T.; McGuire, D.K.; Molitch, M.E.; Mottl, A.K.; et al. SGLT2 Inhibition for CKD and Cardiovascular Disease in Type 2 Diabetes: Report of a Scientific Workshop Sponsored by the National Kidney Foundation. Am. J. Kidney Dis. 2021, 77, 94–109. [Google Scholar] [CrossRef]
- Marilly, E.; Cottin, J.; Cabrera, N.; Cornu, C.; Boussageon, R.; Moulin, P.; Lega, J.-C.; Gueyffier, F.; Cucherat, M.; Grenet, G. SGLT2 inhibitors in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials balancing their risks and benefits. Diabetologia 2022, 65, 2000–2010. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Sattar, N.; Brueckmann, M.; Jamal, W.; Cotton, D.; et al. Empagliflozin in Patients With Heart Failure, Reduced Ejection Fraction, and Volume Overload: EMPEROR-Reduced Trial. J. Am. Coll. Cardiol. 2021, 77, 1381–1392. [Google Scholar] [CrossRef]
- Packer, M.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Carson, P.; Anand, I.; Doehner, W.; Haass, M.; et al. Effect of Empagliflozin on Worsening Heart Failure Events in Patients With Heart Failure and Preserved Ejection Fraction: EMPEROR-Preserved Trial. Circulation 2021, 144, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Kameda, A.; Terauchi, Y. An evaluation of canagliflozin for the treatment of type 2 diabetes: An update. Expert Opin. Pharmacother. 2021, 22, 2087–2094. [Google Scholar] [CrossRef]
- He, X.; Liu, G.; Chen, X.; Wang, Y.; Liu, R.; Wang, C.; Huang, Y.; Shen, J.; Jia, Y. Pharmacokinetic and Pharmacodynamic Interactions Between Henagliflozin, a Novel Selective SGLT-2 Inhibitor, and Warfarin in Healthy Chinese Subjects. Clin. Ther. 2023, S0149-2918(23)00193-5. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Zeng, L.; Zhang, Y.; Qu, S.; Wang, X.; Li, P.; Fu, L.; Ma, B.; Ye, S.; Sun, J.; et al. Henagliflozin as add-on therapy to metformin in patients with type 2 diabetes inadequately controlled with metformin: A multicentre, randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Obes. Metab. 2021, 23, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Fu, L.; Li, Y.; Geng, J.; Qin, L.; Li, P.; Zheng, H.; Sun, Z.; Li, Y.; Zhang, L.; et al. Henagliflozin monotherapy in patients with type 2 diabetes inadequately controlled on diet and exercise: A randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Obes. Metab. 2021, 23, 1111–1120. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Sun, J.; Yu, L.; Qu, X.; Huang, T. The role of peroxisome proliferator-activated receptors in the tumor microenvironment, tumor cell metabolism, and anticancer therapy. Front. Pharmacol. 2023, 14, 1184794. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Kaur, P.; Sahu, S.K.; Mittal, A. A new insight into the treatment of diabetes by means of pan PPAR agonists. Chem. Biol. Drug Des. 2022, 100, 947–967. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jiang, C.; Kim, M.; Xiao, Y.; Richter, H.J.; Guan, D.; Zhu, K.; Krusen, B.M.; Roberts, A.N.; Miller, J.; et al. Isoform-specific functions of PPARγ in gene regulation and metabolism. Genes Dev. 2022, 36, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-Mediated Toxicology and Applied Pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhang, W.; Yin, L.; Shi, Z.; Luan, J.; Chen, L.; Liu, L. The Potential Roles of Post-Translational Modifications of PPARγ in Treating Diabetes. Biomolecules 2022, 12, 1832. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Hao, P.; Du, H. Regulatory T cells differentiation in visceral adipose tissues contributes to insulin resistance by regulating JAZF-1/PPAR-γ pathway. J. Cell. Mol. Med. 2023, 27, 553–562. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Sabatini, S.; Carli, F.; Gaggini, M.; Bril, F.; Belfort-DeAguiar, R.; Positano, V.; Barb, D.; Kadiyala, S.; Harrison, S.; et al. PPAR-γ-induced changes in visceral fat and adiponectin levels are associated with improvement of steatohepatitis in patients with NASH. Liver Int. 2021, 41, 2659–2670. [Google Scholar] [CrossRef]
- Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.; Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571–1580. [Google Scholar] [CrossRef]
- Jia, W.; Ma, J.; Miao, H.; Wang, C.; Wang, X.; Li, Q.; Lu, W.; Yang, J.; Zhang, L.; Yang, J.; et al. Chiglitazar monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: A randomized, double-blind, phase 3 trial (CMAS). Sci. Bull. 2021, 66, 1581–1590. [Google Scholar] [CrossRef]
- Iatcu, C.O.; Steen, A.; Covasa, M. Gut Microbiota and Complications of Type-2 Diabetes. Nutrients. 2021, 14, 166. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- Yang, G.; Wei, J.; Liu, P.; Zhang, Q.; Tian, Y.; Hou, G.; Meng, L.; Xin, Y.; Jiang, X. Role of the gut microbiota in type 2 diabetes and related diseases. Metabolism. 2021, 117, 154712. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Panagiotou, G. Recent advances in diabetes and microbiota. Sci. Bull. Beijing 2022, 67, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Stephens, J.W.; Harris, D.A. Gut microbiota influence in type 2 diabetes mellitus (T2DM). Gut Pathog. 2021, 13, 50. [Google Scholar] [CrossRef]
- Khin, P.P.; Lee, J.H.; Jun, H.-S. Pancreatic Beta-Cell Dysfunction in Type 2 Diabetes. Eur. J. Inflamm. 2023, 21, 1–13. [Google Scholar] [CrossRef]
- Mayorga-Ramos, A.; Barba-Ostria, C.; Simancas-Racines, D.; Guamán, L.P. Protective role of butyrate in obesity and diabetes: New insights. Front. Nutr. 2022, 9, 1067647. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, B.; Yu, D.; Zhu, C. Gut Microbiota: An Important Player in Type 2 Diabetes Mellitus. Front. Cell. Infect. Microbiol. 2022, 12, 834485. [Google Scholar] [CrossRef]
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- De Bandt, J.-P.; Coumoul, X.; Barouki, R. Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity. Nutrients 2022, 15, 68. [Google Scholar] [CrossRef]
- Vanweert, F.; Schrauwen, P.; Phielix, E. Role of branched-chain amino acid metabolism in the pathogenesis of obesity and type 2 diabetes-related metabolic disturbances BCAA metabolism in type 2 diabetes. Nutr. Diabetes 2022, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; Mannerås-Holm, L.; Yunn, N.-O.; Nilsson, P.M.; Ryu, S.H.; Molinaro, A.; Perkins, R.; Smith, J.G.; Bäckhed, F. Microbial Imidazole Propionate Affects Responses to Metformin through P38γ-Dependent Inhibitory AMPK Phosphorylation. Cell. Metab. 2020, 32, 643–653.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gu, Y.; Ren, H.; Wang, S.; Zhong, H.; Zhao, X.; Ma, J.; Gu, X.; Xue, Y.; Huang, S.; et al. Gut microbiome-related effects of berberine and probiotics on type 2 diabetes (the PREMOTE study). Nat. Commun. 2020, 11, 5015. [Google Scholar] [CrossRef] [PubMed]
- Palacios, T.; Vitetta, L.; Coulson, S.; Madigan, C.D.; Lam, Y.Y.; Manuel, R.; Briskey, D.; Hendy, C.; Kim, J.-N.; Ishoey, T.; et al. Targeting the Intestinal Microbiota to Prevent Type 2 Diabetes and Enhance the Effect of Metformin on Glycaemia: A Randomised Controlled Pilot Study. Nutrients 2020, 12, 2041. [Google Scholar] [CrossRef]
- Liu, W.; Luo, Z.; Zhou, J.; Sun, B. Gut Microbiota and Antidiabetic Drugs: Perspectives of Personalized Treatment in Type 2 Diabetes Mellitus. Front. Cell. Infect. Microbiol. 2022, 12, 853771. [Google Scholar] [CrossRef]
- Center for Biologics Evaluation and Research. Important Safety Alert Regarding Use of Fecal Microbiota for Transplantation and Risk of Serious Adverse Reactions Due to Transmission of Multi-Drug Resistant Organisms; FDA: Silver Spring, MD, USA, 2020. [Google Scholar]
- Natalicchio, A.; Montagnani, M.; Gallo, M.; Marrano, N.; Faggiano, A.; Zatelli, M.C.; Mazzilli, R.; Argentiero, A.; Danesi, R.; D’Oronzo, S.; et al. MiRNA dysregulation underlying common pathways in type 2 diabetes and cancer development: An Italian Association of Medical Oncology (AIOM)/Italian Association of Medical Diabetologists (AMD)/Italian Society of Diabetology (SID)/Italian Society of Endocrinology (SIE)/Italian Society of Pharmacology (SIF) Multidisciplinary Critical View. ESMO Open 2023, 8, 101573. [Google Scholar] [CrossRef]
- Hu, H.; Zhao, M.; Li, Z.; Nie, H.; He, J.; Chen, Z.; Yuan, J.; Guo, H.; Zhang, X.; Yang, H.; et al. Plasma MiR-193b-3p Is Elevated in Type 2 Diabetes and Could Impair Glucose Metabolism. Front. Endocrinol. Lausanne 2022, 13, 814347. [Google Scholar] [CrossRef]
- Liu, R.; Liu, C.; He, X.; Sun, P.; Zhang, B.; Yang, H.; Shi, W.; Ruan, Q. MicroRNA-21 promotes pancreatic β cell function through modulating glucose uptake. Nat. Commun. 2022, 13, 3545. [Google Scholar] [CrossRef]
- Ying, W.; Gao, H.; Dos Reis, F.C.G.; Bandyopadhyay, G.; Ofrecio, J.M.; Luo, Z.; Ji, Y.; Jin, Z.; Ly, C.; Olefsky, J.M. MiR-690, an exosomal-derived miRNA from M2-polarized macrophages, improves insulin snsitivity in obese mice. Cell. Metab. 2021, 33, 781–790.e5. [Google Scholar] [CrossRef]
- Ali, H.S.; Kamel, M.M.; Agwa, S.H.A.; Hakeem, M.S.A.; Meteini, M.S.E.; Matboli, M. Analysis of mRNA-miRNA-LncRNA differential expression in prediabetes/type 2 diabetes mellitus patients as potential players in insulin resistance. Front. Endocrinol. Lausanne 2023, 14, 1131171. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Leung, S.-W. MicroRNA biomarkers of type 2 diabetes: Evidence synthesis from meta-analyses and pathway modelling. Diabetologia 2023, 66, 288–299. [Google Scholar] [CrossRef]
- Kraczkowska, W.; Stachowiak, L.; Pławski, A.; Jagodziński, P.P. Circulating miRNA as potential biomarkers for diabetes mellitus type 2: Should we focus on searching for sex differences? J. Appl. Genet. 2022, 63, 293–303. [Google Scholar] [CrossRef]
- He, Y.; Xu, P.; Wang, C.; Xia, Y.; Yu, M.; Yang, Y.; Yu, K.; Cai, X.; Qu, N.; Saito, K.; et al. Estrogen receptor-α expressing neurons in the ventrolateral VMH regulate glucose balance. Nat. Commun. 2020, 11, 2165. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, W.; Tang, Y.; Tu, H.; Chen, T.; Zhou, J.; Wang, D.; Xu, Q.; Niu, J.; Dong, W.; et al. Sustained remission of type 2 diabetes in rodents by centrally administered fibroblast growth factor 4. Cell. Metab. 2023, 35, 1022–1037.e6. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Brill, G.; Zhang, P.; Kumar, A.; Baumel-Alterzon, S.; Honig, L.B.; Gómez-Banoy, N.; Karakose, E.; Tanase, M.; Doridot, L.; et al. Maladaptive positive feedback production of ChREBPβ underlies glucotoxic β-Cell failure. Nat. Commun. 2022, 13, 4423. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Lamarche-Vane, N.; Richard, S. Microexon alternative splicing of small GTPase regulators: Implication in central nervous system diseases. Wiley Interdiscip. Rev. RNA 2022, 13, e1678. [Google Scholar] [CrossRef]
- Juan-Mateu, J.; Bajew, S.; Miret-Cuesta, M.; Íñiguez, L.P.; Lopez-Pascual, A.; Bonnal, S.; Atla, G.; Bonàs-Guarch, S.; Ferrer, J.; Valcárcel, J.; et al. Pancreatic microexons regulate islet function and glucose homeostasis. Nat. Metab. 2023, 5, 219–236. [Google Scholar] [CrossRef]
- Papakonstantinou, E.; Oikonomou, C.; Nychas, G.; Dimitriadis, G.D. Effects of Diet, Lifestyle, Chrononutrition and Alternative Dietary Interventions on Postprandial Glycemia and Insulin Resistance. Nutrients 2022, 14, 823. [Google Scholar] [CrossRef]
- Evert, A.B.; Dennison, M.; Gardner, C.D.; Garvey, W.T.; Lau, K.H.K.; MacLeod, J.; Mitri, J.; Pereira, R.F.; Rawlings, K.; Robinson, S.; et al. Nutrition Therapy for Adults With Diabetes or Prediabetes: A Consensus Report. Diabetes Care 2019, 42, 731–754. [Google Scholar] [CrossRef]
- Dyńka, D.; Kowalcze, K.; Ambrozkiewicz, F.; Paziewska, A. Effect of the Ketogenic Diet on the Prophylaxis and Treatment of Diabetes Mellitus: A Review of the Meta-Analyses and Clinical Trials. Nutrients 2023, 15, 500. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, M.; Liang, J.; He, G.; Chen, N. Ketogenic Diet Benefits to Weight Loss, Glycemic Control, and Lipid Profiles in Overweight Patients with Type 2 Diabetes Mellitus: A Meta-Analysis of Randomized Controlled Trails. Int. J. Environ. Res. Public Health 2022, 19, 10429. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.D.; Landry, M.J.; Perelman, D.; Petlura, C.; Durand, L.R.; Aronica, L.; Crimarco, A.; Cunanan, K.M.; Chang, A.; Dant, C.C.; et al. Effect of a ketogenic diet versus Mediterranean diet on glycated hemoglobin in individuals with prediabetes and type 2 diabetes mellitus: The interventional Keto-Med randomized crossover trial. Am. J. Clin. Nutr. 2022, 116, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.; Dupuis, N.; Auvin, S. Ketogenic diet and Neuroinflammation. Epilepsy Res. 2020, 167, 106454. [Google Scholar] [CrossRef]
- Paoli, A.; Cerullo, G. Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review. Antioxidants 2023, 12, 1065. [Google Scholar] [CrossRef]
- Cannataro, R.; Caroleo, M.C.; Fazio, A.; La Torre, C.; Plastina, P.; Gallelli, L.; Lauria, G.; Cione, E. Ketogenic Diet and microRNAs Linked to Antioxidant Biochemical Homeostasis. Antioxidants 2019, 8, 269. [Google Scholar] [CrossRef]
- Peng, F.; Zhang, Y.-H.; Zhang, L.; Yang, M.; Chen, C.; Yu, H.; Li, T. Ketogenic diet attenuates post-cardiac arrest brain injury by upregulation of pentose phosphate pathway-mediated antioxidant defense in a mouse model of cardiac arrest. Nutrition 2022, 103–104, 111814. [Google Scholar] [CrossRef]
- Churuangsuk, C.; Hall, J.; Reynolds, A.; Griffin, S.J.; Combet, E.; Lean, M.E.J. Diets for weight management in adults with type 2 diabetes: An umbrella review of published meta-analyses and systematic review of trials of diets for diabetes remission. Diabetologia 2022, 65, 14–36. [Google Scholar] [CrossRef]
- Jing, T.; Zhang, S.; Bai, M.; Chen, Z.; Gao, S.; Li, S.; Zhang, J. Effect of Dietary Approaches on Glycemic Control in Patients with Type 2 Diabetes: A Systematic Review with Network Meta-Analysis of Randomized Trials. Nutrients 2023, 15, 3156. [Google Scholar] [CrossRef]
- Pittas, A.G.; Kawahara, T.; Jorde, R.; Dawson-Hughes, B.; Vickery, E.M.; Angellotti, E.; Nelson, J.; Trikalinos, T.A.; Balk, E.M. Vitamin D and Risk for Type 2 Diabetes in People With Prediabetes: A Systematic Review and Meta-analysis of Individual Participant Data From 3 Randomized Clinical Trials. Ann. Intern. Med. 2023, 176, 355–363. [Google Scholar] [CrossRef]
- Naz, R.; Saqib, F.; Awadallah, S.; Wahid, M.; Latif, M.F.; Iqbal, I.; Mubarak, M.S. Food Polyphenols and Type II Diabetes Mellitus: Pharmacology and Mechanisms. Molecules 2023, 28, 3996. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Ardisson Korat, A.V.; Imamura, F.; Marklund, M.; Tintle, N.; Virtanen, J.K.; Zhou, X.; Bassett, J.K.; Lai, H.; Hirakawa, Y.; et al. n-3 Fatty Acid Biomarkers and Incident Type 2 Diabetes: An Individual Participant-Level Pooling Project of 20 Prospective Cohort Studies. Diabetes Care 2021, 44, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhou, Y.; Wu, X.; Jia, X.; Zhu, Y.; Zheng, R.; Wang, S.; Lin, L.; Qi, H.; Lin, H.; et al. Evaluating the distinct pleiotropic effects of omega-3 fatty acids on type 2 diabetes mellitus: A mendelian randomization study. J. Transl. Med. 2023, 21, 370. [Google Scholar] [CrossRef] [PubMed]
- Hulett, N.A.; Scalzo, R.L.; Reusch, J.E.B. Glucose Uptake by Skeletal Muscle within the Contexts of Type 2 Diabetes and Exercise: An Integrated Approach. Nutrients 2022, 14, 647. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Sun, Y.; Zhang, Z.Y.; Aboelela, Z.; Qiu, X.; Meng, Z.-X. β-cell dynamics in type 2 diabetes and in dietary and exercise interventions. J. Mol. Cell. Biol. 2022, 14, mjac046. [Google Scholar] [CrossRef]
- Cai, L.; Gonzales, T.; Wheeler, E.; Kerrison, N.D.; Day, F.R.; Langenberg, C.; Perry, J.R.B.; Brage, S.; Wareham, N.J. Causal associations between cardiorespiratory fitness and type 2 diabetes. Nat. Commun. 2023, 14, 3904. [Google Scholar] [CrossRef]
- Kanaley, J.A.; Colberg, S.R.; Corcoran, M.H.; Malin, S.K.; Rodriguez, N.R.; Crespo, C.J.; Kirwan, J.P.; Zierath, J.R. Exercise/Physical Activity in Individuals with Type 2 Diabetes: A Consensus Statement from the American College of Sports Medicine. Med. Sci. Sports Exerc. 2022, 54, 353–368. [Google Scholar] [CrossRef]
- Kong, L.; Ren, J.; Fang, S.; He, T.; Zhou, X.; Fang, M. Effects of traditional Chinese mind-body exercise-baduanjin for type 2 diabetes on psychological well-being: A systematic review and meta-analysis. Front. Public Health 2022, 10, 923411. [Google Scholar] [CrossRef]
- Khan, T.A.; Field, D.; Chen, V.; Ahmad, S.; Mejia, S.B.; Kahleová, H.; Rahelić, D.; Salas-Salvadó, J.; Leiter, L.A.; Uusitupa, M.; et al. Combination of Multiple Low-Risk Lifestyle Behaviors and Incident Type 2 Diabetes: A Systematic Review and Dose-Response Meta-Analysis of Prospective Cohort Studies. Diabetes Care 2023, 46, 643–656. [Google Scholar] [CrossRef]
- Cannataro, R.; Cione, E.; Cerullo, G.; Rondanelli, M.; Micheletti, P.; Crisafulli, O.; Micheli, M.L.; D’Antona, G. Type 1 diabetes management in a competitive athlete: A five-year case report. Physiol. Rep. 2023, 11, e15740. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J.; Luo, Y.; Hu, S.; Tang, L.; Ouyang, S. Advances in Research on Type 2 Diabetes Mellitus Targets and Therapeutic Agents. Int. J. Mol. Sci. 2023, 24, 13381. https://doi.org/10.3390/ijms241713381
Su J, Luo Y, Hu S, Tang L, Ouyang S. Advances in Research on Type 2 Diabetes Mellitus Targets and Therapeutic Agents. International Journal of Molecular Sciences. 2023; 24(17):13381. https://doi.org/10.3390/ijms241713381
Chicago/Turabian StyleSu, Jingqian, Yingsheng Luo, Shan Hu, Lu Tang, and Songying Ouyang. 2023. "Advances in Research on Type 2 Diabetes Mellitus Targets and Therapeutic Agents" International Journal of Molecular Sciences 24, no. 17: 13381. https://doi.org/10.3390/ijms241713381
APA StyleSu, J., Luo, Y., Hu, S., Tang, L., & Ouyang, S. (2023). Advances in Research on Type 2 Diabetes Mellitus Targets and Therapeutic Agents. International Journal of Molecular Sciences, 24(17), 13381. https://doi.org/10.3390/ijms241713381