
Contribution of ADD3 and the HLA Genes to Biliary Atresia Risk in Chinese


Abstract
:1. Introduction
2. Results
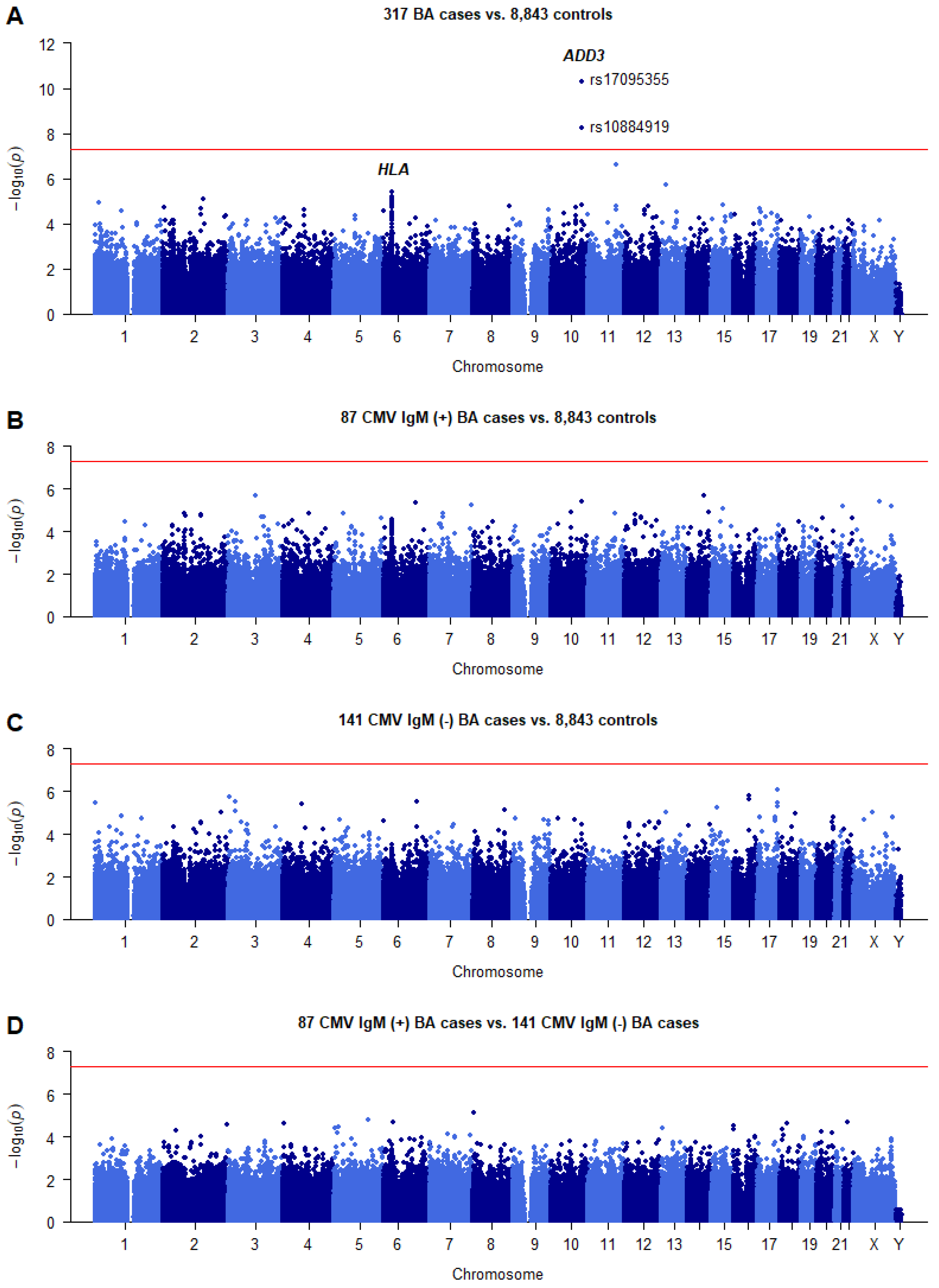
2.1. Summary of the GWAS Results
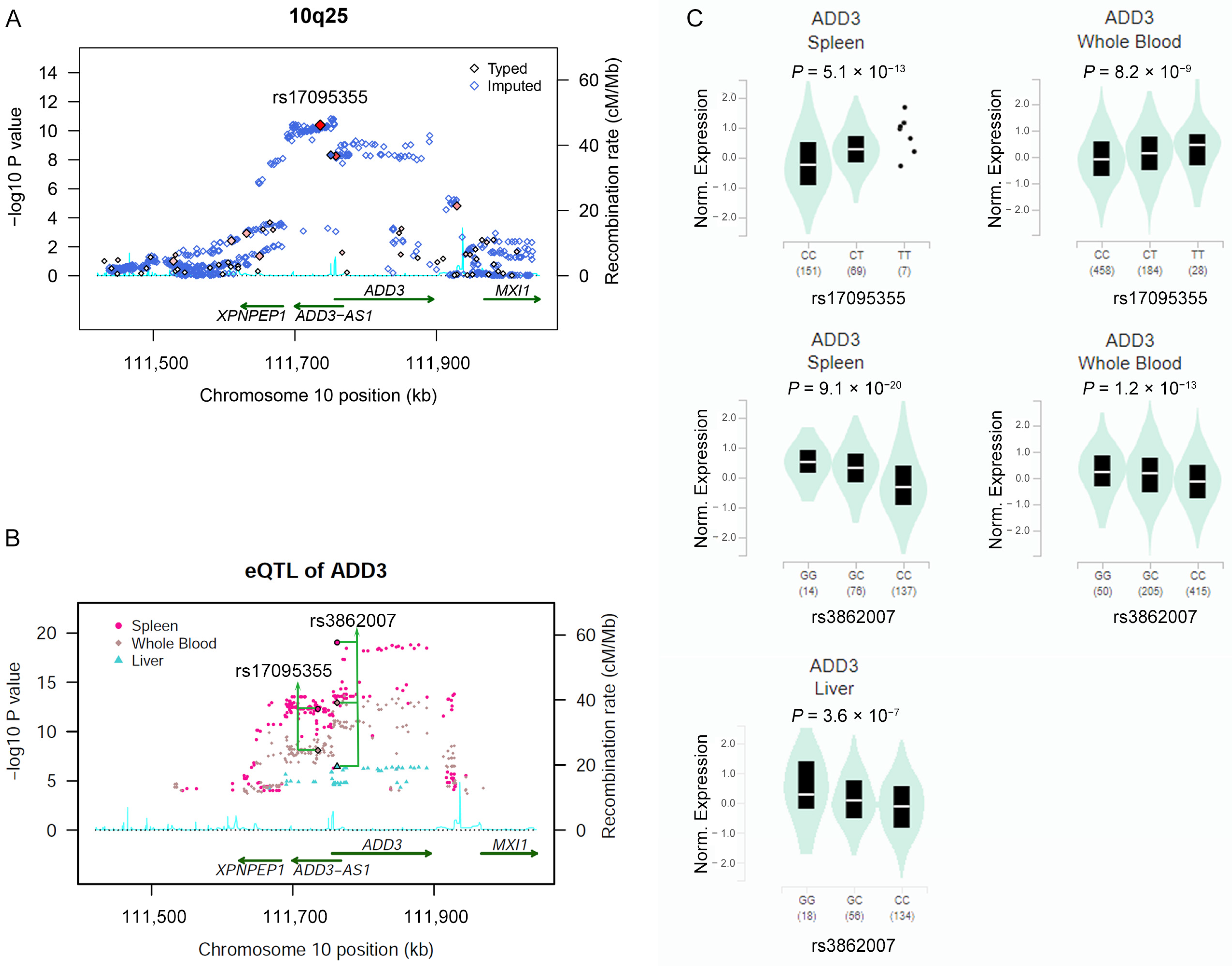
2.2. Association of SNPs in ADD3 Gene Region with BA Susceptibility
2.3. Risk Allele of rs17095355 Is Associated with Increased Expression of ADD3
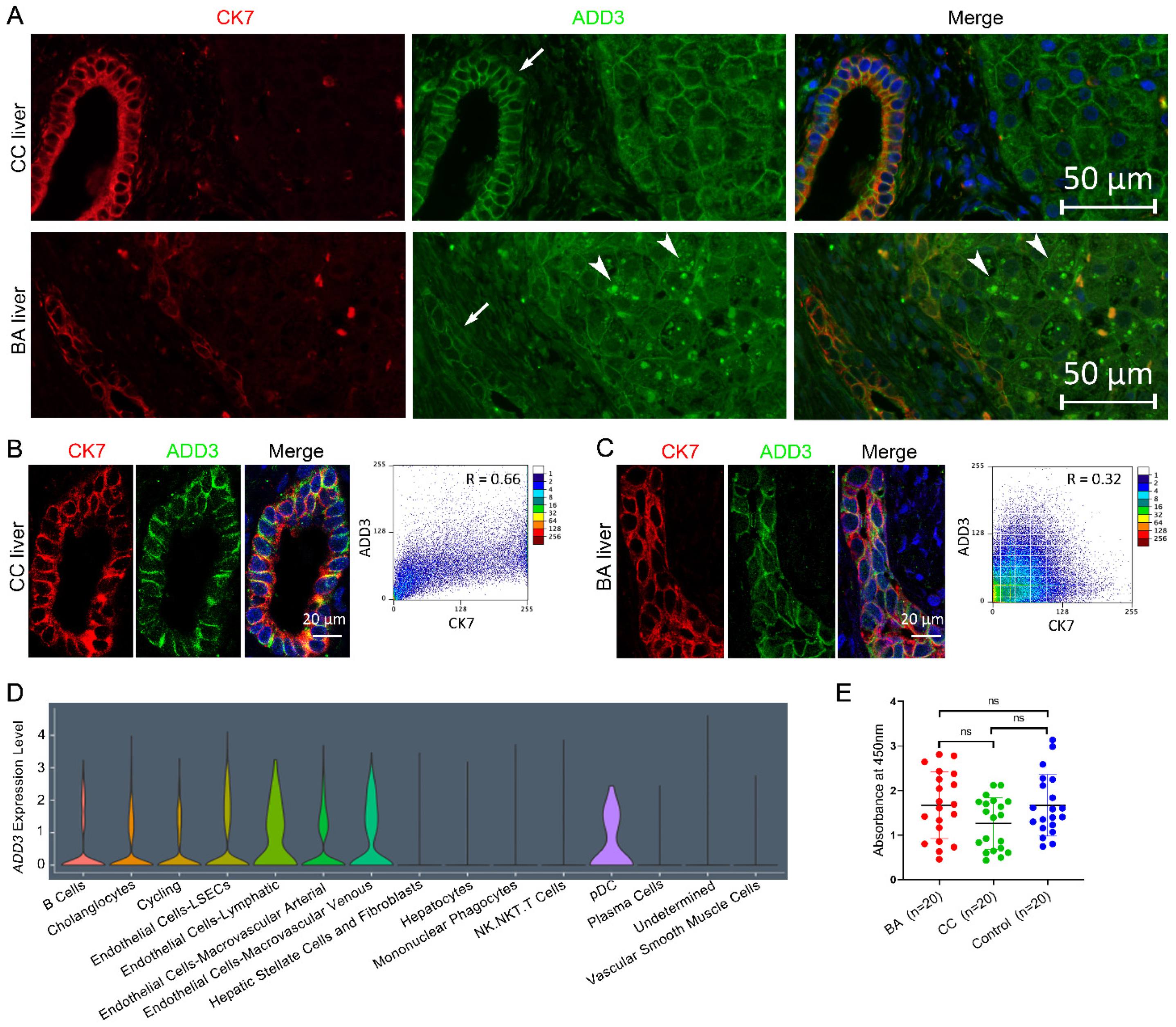
2.4. ADD3 Aberrantly Deposits in the Cytoplasm of BA Livers
2.5. Assay of Anti-ADD3 Antibodies
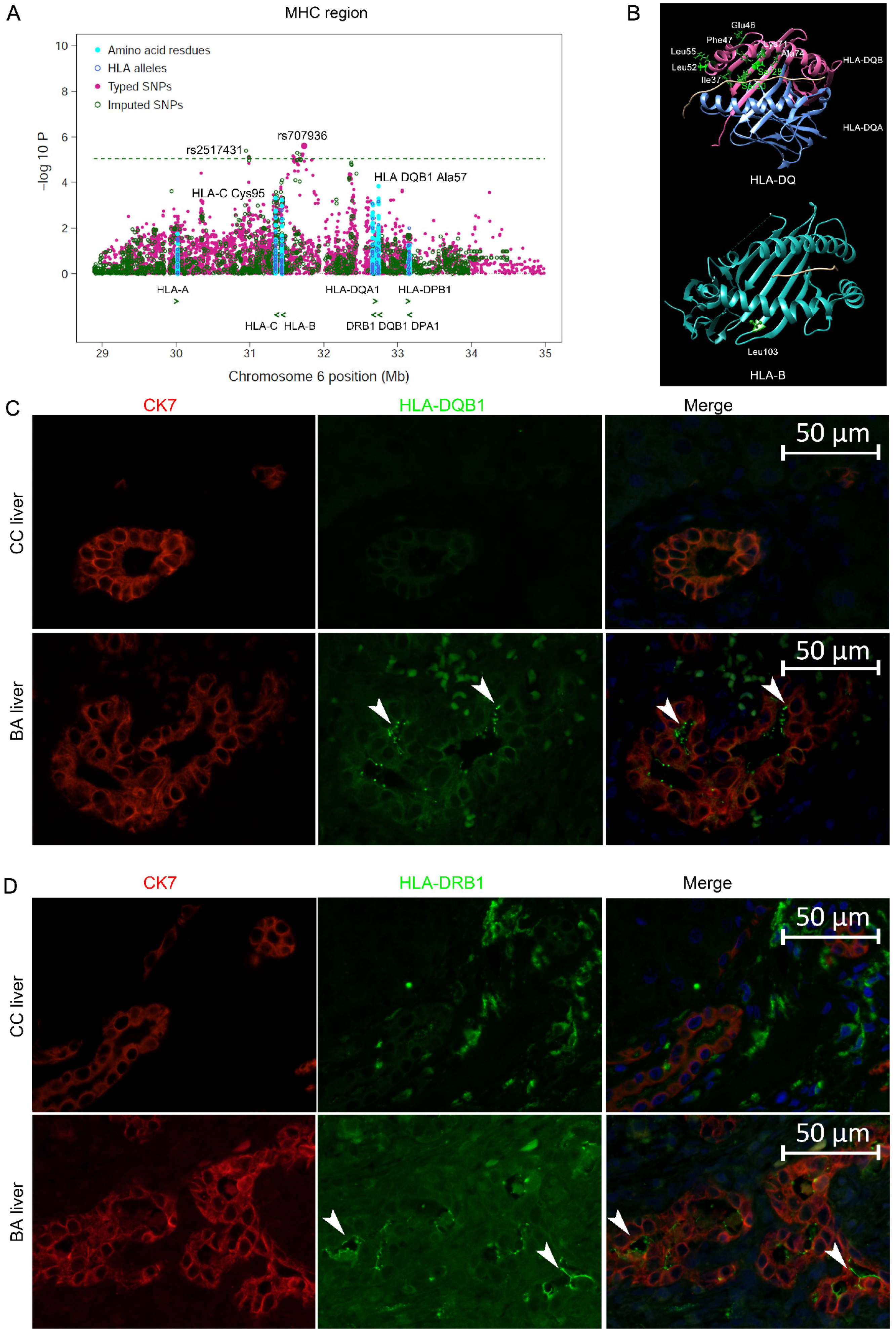
2.6. Summary of Imputation Results for the HLA Gene Region
2.7. Associations of Classical HLA Genes with BA
2.8. Stratification Analysis by CMV IgM
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Genotyping and Quality Control for GWAS Data
4.3. Imputation
4.4. In Silico Functional Annotation of Associated Loci
4.5. Single-Cell RNA-Sequencing Data Sets and Analysis
4.6. IF Analysis
4.7. Testing for the Autoantibody against ADD3
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berauer, J.P.; Mezina, A.I.; Okou, D.T.; Sabo, A.; Muzny, D.M.; Gibbs, R.A.; Hegde, M.R.; Chopra, P.; Cutler, D.J.; Perlmutter, D.H.; et al. Identification of Polycystic Kidney Disease 1 Like 1 Gene Variants in Children With Biliary Atresia Splenic Malformation Syndrome. Hepatology 2019, 70, 899–910. [Google Scholar] [CrossRef]
- Hartley, J.L.; Davenport, M.; Kelly, D.A. Biliary atresia. Lancet 2009, 374, 1704–1713. [Google Scholar] [CrossRef]
- Nizery, L.; Chardot, C.; Sissaoui, S.; Capito, C.; Henrion-Caude, A.; Debray, D.; Girard, M. Biliary atresia: Clinical advances and perspectives. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 281–287. [Google Scholar] [CrossRef]
- Wada, H.; Muraji, T.; Yokoi, A.; Okamoto, T.; Sato, S.; Takamizawa, S.; Tsugawa, J.; Nishijima, E. Insignificant seasonal and geographical variation in incidence of biliary atresia in Japan: A regional survey of over 20 years. J. Pediatr. Surg. 2007, 42, 2090–2092. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Chen, P.H.; Chan, C.F.; Chang, M.H.; Wu, T.C. Biliary atresia in preterm infants in Taiwan: A nationwide survey. J. Pediatr. 2013, 163, 100–103.e101. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.L.; Feldman, A.G.; Sokol, R.J. Clues to the etiology of bile duct injury in biliary atresia. Semin. Liver Dis. 2012, 32, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Lorent, K.; Gong, W.; Koo, K.A.; Waisbourd-Zinman, O.; Karjoo, S.; Zhao, X.; Sealy, I.; Kettleborough, R.N.; Stemple, D.L.; Windsor, P.A.; et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci. Transl. Med. 2015, 7, 286ra267. [Google Scholar] [CrossRef] [PubMed]
- Zani, A.; Quaglia, A.; Hadzić, N.; Zuckerman, M.; Davenport, M. Cytomegalovirus-associated biliary atresia: An aetiological and prognostic subgroup. J. Pediatr. Surg. 2015, 50, 1739–1745. [Google Scholar] [CrossRef]
- Davenport, M. Biliary atresia: From Australia to the zebrafish. J. Pediatr. Surg. 2016, 51, 200–205. [Google Scholar] [CrossRef]
- Garcia-Barceló, M.M.; Yeung, M.Y.; Miao, X.P.; Tang, C.S.; Cheng, G.; So, M.T.; Ngan, E.S.; Lui, V.C.; Chen, Y.; Liu, X.L.; et al. Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum. Mol. Genet. 2010, 19, 2917–2925. [Google Scholar] [CrossRef]
- Cheng, G.; Tang, C.S.; Wong, E.H.; Cheng, W.W.; So, M.T.; Miao, X.; Zhang, R.; Cui, L.; Liu, X.; Ngan, E.S.; et al. Common genetic variants regulating ADD3 gene expression alter biliary atresia risk. J. Hepatol. 2013, 59, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Kaewkiattiyot, S.; Honsawek, S.; Vejchapipat, P.; Chongsrisawat, V.; Poovorawan, Y. Association of X-prolyl aminopeptidase 1 rs17095355 polymorphism with biliary atresia in Thai children. Hepatol. Res. 2011, 41, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Sun, P.; Chen, Z.; Mao, J.; Wang, J.; Wang, B.; Liu, L. Association between single nucleotide polymorphisms in the ADD3 gene and susceptibility to biliary atresia. PLoS ONE 2014, 9, e107977. [Google Scholar] [CrossRef]
- Tsai, E.A.; Grochowski, C.M.; Loomes, K.M.; Bessho, K.; Hakonarson, H.; Bezerra, J.A.; Russo, P.A.; Haber, B.A.; Spinner, N.B.; Devoto, M. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum. Genet. 2014, 133, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.R.; Niu, W.B.; Zhou, Y.; Gong, Y.M.; Lu, Y.J.; Yu, X.X.; Wei, Z.L.; Wu, W.; Song, H.L.; Yu, W.W.; et al. Association of common variation in ADD3 and GPC1 with biliary atresia susceptibility. Aging 2020, 12, 7163–7182. [Google Scholar] [CrossRef]
- Tang, V.; Cofer, Z.C.; Cui, S.; Sapp, V.; Loomes, K.M.; Matthews, R.P. Loss of a Candidate Biliary Atresia Susceptibility Gene, add3a, Causes Biliary Developmental Defects in Zebrafish. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 524–530. [Google Scholar] [CrossRef]
- Leyva-Vega, M.; Gerfen, J.; Thiel, B.D.; Jurkiewicz, D.; Rand, E.B.; Pawlowska, J.; Kaminska, D.; Russo, P.; Gai, X.; Krantz, I.D.; et al. Genomic alterations in biliary atresia suggest region of potential disease susceptibility in 2q37.3. Am. J. Med. Genet. A 2010, 152a, 886–895. [Google Scholar] [CrossRef]
- Cui, S.; Leyva-Vega, M.; Tsai, E.A.; EauClaire, S.F.; Glessner, J.T.; Hakonarson, H.; Devoto, M.; Haber, B.A.; Spinner, N.B.; Matthews, R.P. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 2013, 144, 1107–1115.e1103. [Google Scholar] [CrossRef]
- Ke, J.; Zeng, S.; Mao, J.; Wang, J.; Lou, J.; Li, J.; Chen, X.; Liu, C.; Huang, L.M.; Wang, B.; et al. Common genetic variants of GPC1 gene reduce risk of biliary atresia in a Chinese population. J. Pediatr. Surg. 2016, 51, 1661–1664. [Google Scholar] [CrossRef]
- Ningappa, M.; So, J.; Glessner, J.; Ashokkumar, C.; Ranganathan, S.; Min, J.; Higgs, B.W.; Sun, Q.; Haberman, K.; Schmitt, L.; et al. The Role of ARF6 in Biliary Atresia. PLoS ONE 2015, 10, e0138381. [Google Scholar] [CrossRef]
- Chen, Y.; Gilbert, M.A.; Grochowski, C.M.; McEldrew, D.; Llewellyn, J.; Waisbourd-Zinman, O.; Hakonarson, H.; Bailey-Wilson, J.E.; Russo, P.; Wells, R.G.; et al. A genome-wide association study identifies a susceptibility locus for biliary atresia on 2p16.1 within the gene EFEMP1. PLoS Genet. 2018, 14, e1007532. [Google Scholar] [CrossRef] [PubMed]
- Silveira, T.R.; Salzano, F.M.; Donaldson, P.T.; Mieli-Vergani, G.; Howard, E.R.; Mowat, A.P. Association between HLA and extrahepatic biliary atresia. J. Pediatr. Gastroenterol. Nutr. 1993, 16, 114–117. [Google Scholar] [CrossRef]
- Donaldson, P.T.; Clare, M.; Constantini, P.K.; Hadzic, N.; Mieli-Vergani, G.; Howard, E.; Kelley, D. HLA and cytokine gene polymorphisms in biliary atresia. Liver 2002, 22, 213–219. [Google Scholar] [CrossRef]
- Yuasa, T.; Tsuji, H.; Kimura, S.; Niwa, N.; Yurugi, K.; Egawa, H.; Tanaka, K.; Maruya, E.; Saji, H.O.; Asano, H.; et al. Human leukocyte antigens in Japanese patients with biliary atresia: Retrospective analysis of patients who underwent living donor liver transplantation. Hum. Immunol. 2005, 66, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.L.; Anderson, K.M.; Aubrey, M.T.; Rosenthal, P.; Sokol, R.J.; Freed, B.M. Lack of HLA predominance and HLA shared epitopes in biliary Atresia. Springerplus 2013, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, D.M.; Lozovatsky, L.; Gwynn, B.; Brugnara, C.; Mohandas, N.; Peters, L.L. Targeted disruption of the beta adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 10717–10722. [Google Scholar] [CrossRef]
- Ye, Y.; Li, Z.; Feng, Q.; Chen, Z.; Wu, Z.; Wang, J.; Ye, X.; Zhang, D.; Liu, L.; Gao, W.; et al. Downregulation of microRNA-145 may contribute to liver fibrosis in biliary atresia by targeting ADD3. PLoS ONE 2017, 12, e0180896. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Dai, Y.M.; Zhang, R.Z.; Chen, Y.H.; Peng, X.F.; Fu, J.; Chen, Z.R.; Liu, Y.F.; Yang, L.Y.; Wen, Z.; et al. Autoimmune liver disease-related autoantibodies in patients with biliary atresia. World J. Gastroenterol. 2018, 24, 387–396. [Google Scholar] [CrossRef]
- Luo, Y.; Brigham, D.; Bednarek, J.; Torres, R.; Wang, D.; Ahmad, S.; Mack, C.L. Unique Cholangiocyte-Targeted IgM Autoantibodies Correlate With Poor Outcome in Biliary Atresia. Hepatology 2021, 73, 1855–1867. [Google Scholar] [CrossRef]
- Jia, X.; Han, B.; Onengut-Gumuscu, S.; Chen, W.-M.; Concannon, P.J.; Rich, S.S.; Raychaudhuri, S.; de Bakker, P.I.W. Imputing Amino Acid Polymorphisms in Human Leukocyte Antigens. PLoS ONE 2013, 8, e64683. [Google Scholar] [CrossRef] [PubMed]
- Pillai, N.E.; Okada, Y.; Saw, W.Y.; Ong, R.T.; Wang, X.; Tantoso, E.; Xu, W.; Peterson, T.A.; Bielawny, T.; Ali, M.; et al. Predicting HLA alleles from high-resolution SNP data in three Southeast Asian populations. Hum. Mol. Genet. 2014, 23, 4443–4451. [Google Scholar] [CrossRef]
- Okada, Y.; Kim, K.; Han, B.; Pillai, N.E.; Ong, R.T.; Saw, W.Y.; Luo, M.; Jiang, L.; Yin, J.; Bang, S.Y.; et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum. Mol. Genet. 2014, 23, 6916–6926. [Google Scholar] [CrossRef] [PubMed]
- Broomé, U.; Nemeth, A.; Hultcrantz, R.; Scheynius, A. Different expression of HLA-DR and ICAM-1 in livers from patients with biliary atresia and Byler’s disease. J. Hepatol. 1997, 26, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, M.; Gu, W.; Tang, H.; Yu, S. The aberrant expression of HLA-DR in intrahepatic bile ducts in patients with biliary atresia: An immunohistochemistry and immune electron microscopy study. J. Pediatr. Surg. 2004, 39, 1658–1662. [Google Scholar] [CrossRef]
- Naydenov, N.G.; Ivanov, A.I. Adducins regulate remodeling of apical junctions in human epithelial cells. Mol. Biol. Cell 2010, 21, 3506–3517. [Google Scholar] [CrossRef]
- Khandekar, G.; Llewellyn, J.; Kriegermeier, A.; Waisbourd-Zinman, O.; Johnson, N.; Du, Y.; Giwa, R.; Liu, X.; Kisseleva, T.; Russo, P.A.; et al. Coordinated development of the mouse extrahepatic bile duct: Implications for neonatal susceptibility to biliary injury. J. Hepatol. 2020, 72, 135–145. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Tsai, E.A.; Grochowski, C.M.; Kelly, S.M.; Loomes, K.M.; Spinner, N.B.; Devoto, M. Exome Sequencing in Individuals with Isolated Biliary Atresia. Sci. Rep. 2020, 10, 2709. [Google Scholar] [CrossRef]
- Lam, W.Y.; Tang, C.S.; So, M.T.; Yue, H.; Hsu, J.S.; Chung, P.H.; Nicholls, J.M.; Yeung, F.; Lee, C.D.; Ngo, D.N.; et al. Identification of a wide spectrum of ciliary gene mutations in nonsyndromic biliary atresia patients implicates ciliary dysfunction as a novel disease mechanism. EBioMedicine 2021, 71, 103530. [Google Scholar] [CrossRef]
- Tran, K.T.; Le, V.S.; Dao, L.T.M.; Nguyen, H.K.; Mai, A.K.; Nguyen, H.T.; Ngo, M.D.; Tran, Q.A.; Nguyen, L.T. Novel findings from family-based exome sequencing for children with biliary atresia. Sci. Rep. 2021, 11, 21815. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.; Gonde, C.; Redkar, R.; Koukoulis, G.; Tredger, M.; Mieli-Vergani, G.; Portmann, B.; Howard, E.R. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J. Pediatr. Surg. 2001, 36, 1017–1025. [Google Scholar] [CrossRef]
- Hu, X.; Deutsch, A.J.; Lenz, T.L.; Onengut-Gumuscu, S.; Han, B.; Chen, W.-M.; Howson, J.M.M.; Todd, J.A.; de Bakker, P.I.W.; Rich, S.S.; et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet. 2015, 47, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Yang, M.; Song, Z.J.; Dong, Y.; Li, C.; Shen, M.; Zhu, Y.Q.; Song, H.D.; Chen, S.J.; Chen, Z.; et al. Fine mapping MHC associations in Graves’ disease and its clinical subtypes in Han Chinese. J. Med. Genet. 2018, 55, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Muraji, T.; Hosaka, N.; Takada, Y.; Sakamoto, S.; Tanaka, K. Maternal HLA class I compatibility in patients with biliary atresia. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 488–492. [Google Scholar] [CrossRef]
- Muraji, T.; Tanaka, H.; Ieiri, S. Ethnic variation in the incidence of biliary atresia correlates with the frequency of the most prevalent haplotype in its population. Hum. Immunol. 2018, 79, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Miethke, A.; Bezerra, J.A. Pathogenesis of biliary atresia: Defining biology to understand clinical phenotypes. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 342–352. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904. [Google Scholar] [CrossRef]
- Howie, B.; Fuchsberger, C.; Stephens, M.; Marchini, J.; Abecasis, G.R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012, 44, 955. [Google Scholar] [CrossRef]
- Marchini, J.; Howie, B.; Myers, S.; McVean, G.; Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007, 39, 906. [Google Scholar] [CrossRef]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | SNP | Genotype/Allele | Genotype/Allele Distribution N (%) | p Value | ||
|---|---|---|---|---|---|---|
| Case | Control | Logistic | Genotype | |||
| ADD3 | rs17095355 | TT | 83 (26.2) | 1328 (15.0) | 4.83 × 10−11 | 3.16 × 10−10 |
| TC | 161 (50.8) | 4164 (47.1) | ||||
| CC | 73 (23.0) | 3351 (37.9) | ||||
| T | 327 (51.6) | 6820 (38.6) | ||||
| C | 307 (48.4) | 10,866 (61.4) | ||||
| ADD3 | rs10884919 | TT | 73 (23.1) | 1212 (13.7) | 5.74 × 10−9 | 3.54 × 10−8 |
| TC | 160 (50.6) | 4117 (46.6) | ||||
| CC | 83 (26.3) | 3511 (39.7) | ||||
| T | 306 (48.4) | 6541 (37.0) | ||||
| C | 326 (51.6) | 11,139 (63.0) | ||||
| HLA | rs707936 | AA | 7 (2.2) | 88 (1.0) | 3.46 × 10−6 | 2.8 × 10−5 |
| AG | 82 (25.9) | 1517 (17.2) | ||||
| GG | 228 (71.9) | 7232 (81.8) | ||||
| A | 96 (15.1) | 1693 (9.6) | ||||
| G | 538 (84.9) | 15,981 (90.4) | ||||
| Genes | Effective Residue/Allele | PLINK INFO | ERF (%) | OR (95% CI) | p Value | |
|---|---|---|---|---|---|---|
| Cases (n = 317) | Controls (n = 8843) | |||||
| HLA-DQB1 | Ala57 | 0.97 | 0.24 | 0.18 | 1.44 (1.20–1.74) | 1.23 × 10−4 |
| HLA-DQB1 | Asp57 | 0.99 | 0.62 | 0.69 | 0.75 (0.63–0.88) | 4.32 × 10−4 |
| HLA-DQB1 | Val57 | 1.00 | 0.08 | 0.07 | 1.10 (0.83–1.47) | 5.09 × 10−1 |
| HLA-DQB1 | Ser57 | 1.02 | 0.06 | 0.06 | 0.99 (0.71–1.38) | 9.58 × 10−1 |
| HLA-DQB1 | Ala74 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Glu74 | 0.99 | 0.63 | 0.68 | 0.82 (0.70–0.97) | 1.83 × 10−2 |
| HLA-DQB1 | Ser74 | 0.99 | 0.20 | 0.20 | 0.99 (0.81–1.21) | 9.11 × 10−1 |
| HLA-DQB1 | Lys71 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Thr71 | 0.99 | 0.63 | 0.68 | 0.82 (0.70–0.97) | 1.83 × 10−2 |
| HLA-DQB1 | Asp71 | 0.99 | 0.05 | 0.06 | 0.91 (0.64–1.31) | 6.22 × 10−1 |
| HLA-DQB1 | Ala71 | 1.00 | 0.15 | 0.15 | 1.02 (0.82–1.28) | 8.48 × 10−1 |
| HLA-DQB1 | Leu52 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Phe47 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Glu46 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Ile37 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Tyr37 | 0.95 | 0.75 | 0.80 | 0.77 (0.64–0.93) | 5.18 × 10−2 |
| HLA-DQB1 | Asp37 | 0.90 | 0.08 | 0.08 | 0.99 (0.73–1.32) | 9.25 × 10−1 |
| HLA-DQB1 | Ser30 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | Tyr30 | 1.00 | 0.67 | 0.70 | 0.84 (0.71–0.99) | 3.72 × 10−2 |
| HLA-DQB1 | His30 | 0.99 | 0.17 | 0.18 | 0.95 (0.76–1.17) | 6.06 × 10−1 |
| HLA-DQB1 | Ser28 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.58 × 10−4 |
| HLA-DQB1 | 0201 | 0.99 | 0.17 | 0.12 | 1.46 (1.18–1.81) | 4.75 × 10−4 |
| HLA-B | Leu103 | 0.98 | 0.34 | 0.28 | 1.34 (1.13–1.58) | 6.76 × 10−4 |
| HLA-B | 5801 | 1.00 | 0.09 | 0.06 | 1.58 (1.19–2.10) | 1.62 × 10−3 |
| HLA-DRB1 | Ala71 | 1.03 | 0.11 | 0.15 | 0.65 (0.50–0.84) | 9.29 × 10−4 |
| HLA-DRB1 | Lys71 | 0.99 | 0.09 | 0.06 | 1.49 (1.12–1.98) | 6.03 × 10−3 |
| HLA-DRB1 | Arg71 | 1.00 | 0.77 | 0.74 | 1.16 (0.96–1.39) | 1.32 × 10−1 |
| HLA-DRB1 | Glu71 | 1.00 | 0.04 | 0.05 | 0.89 (0.60–1.32) | 5.62 × 10−1 |
| HLA-DRB1 | 15 | 1.02 | 0.11 | 0.15 | 0.65 (0.50–0.84) | 9.41 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, M.-M.; Gong, Y.-M.; Pan, W.-H.; Pei, H.-Y.; Bai, M.-R.; Song, H.-L.; Han, X.-R.; Wu, W.-J.; Yu, W.-W.; Gu, B.-L.; et al. Contribution of ADD3 and the HLA Genes to Biliary Atresia Risk in Chinese. Int. J. Mol. Sci. 2023, 24, 14719. https://doi.org/10.3390/ijms241914719
Cui M-M, Gong Y-M, Pan W-H, Pei H-Y, Bai M-R, Song H-L, Han X-R, Wu W-J, Yu W-W, Gu B-L, et al. Contribution of ADD3 and the HLA Genes to Biliary Atresia Risk in Chinese. International Journal of Molecular Sciences. 2023; 24(19):14719. https://doi.org/10.3390/ijms241914719
Chicago/Turabian StyleCui, Meng-Meng, Yi-Ming Gong, Wei-Hua Pan, Hao-Yue Pei, Mei-Rong Bai, Huan-Lei Song, Xin-Ru Han, Wen-Jie Wu, Wen-Wen Yu, Bei-Lin Gu, and et al. 2023. "Contribution of ADD3 and the HLA Genes to Biliary Atresia Risk in Chinese" International Journal of Molecular Sciences 24, no. 19: 14719. https://doi.org/10.3390/ijms241914719
APA StyleCui, M.-M., Gong, Y.-M., Pan, W.-H., Pei, H.-Y., Bai, M.-R., Song, H.-L., Han, X.-R., Wu, W.-J., Yu, W.-W., Gu, B.-L., Cai, W., Zhou, Y., & Chu, X. (2023). Contribution of ADD3 and the HLA Genes to Biliary Atresia Risk in Chinese. International Journal of Molecular Sciences, 24(19), 14719. https://doi.org/10.3390/ijms241914719