
CXCR4 Expressed by Tumor-Infiltrating B Cells in Gastric Cancer Related to Survival in the Tumor Microenvironment: An Analysis Combining Single-Cell RNA Sequencing with Bulk RNA Sequencing

Abstract
:1. Introduction
2. Results
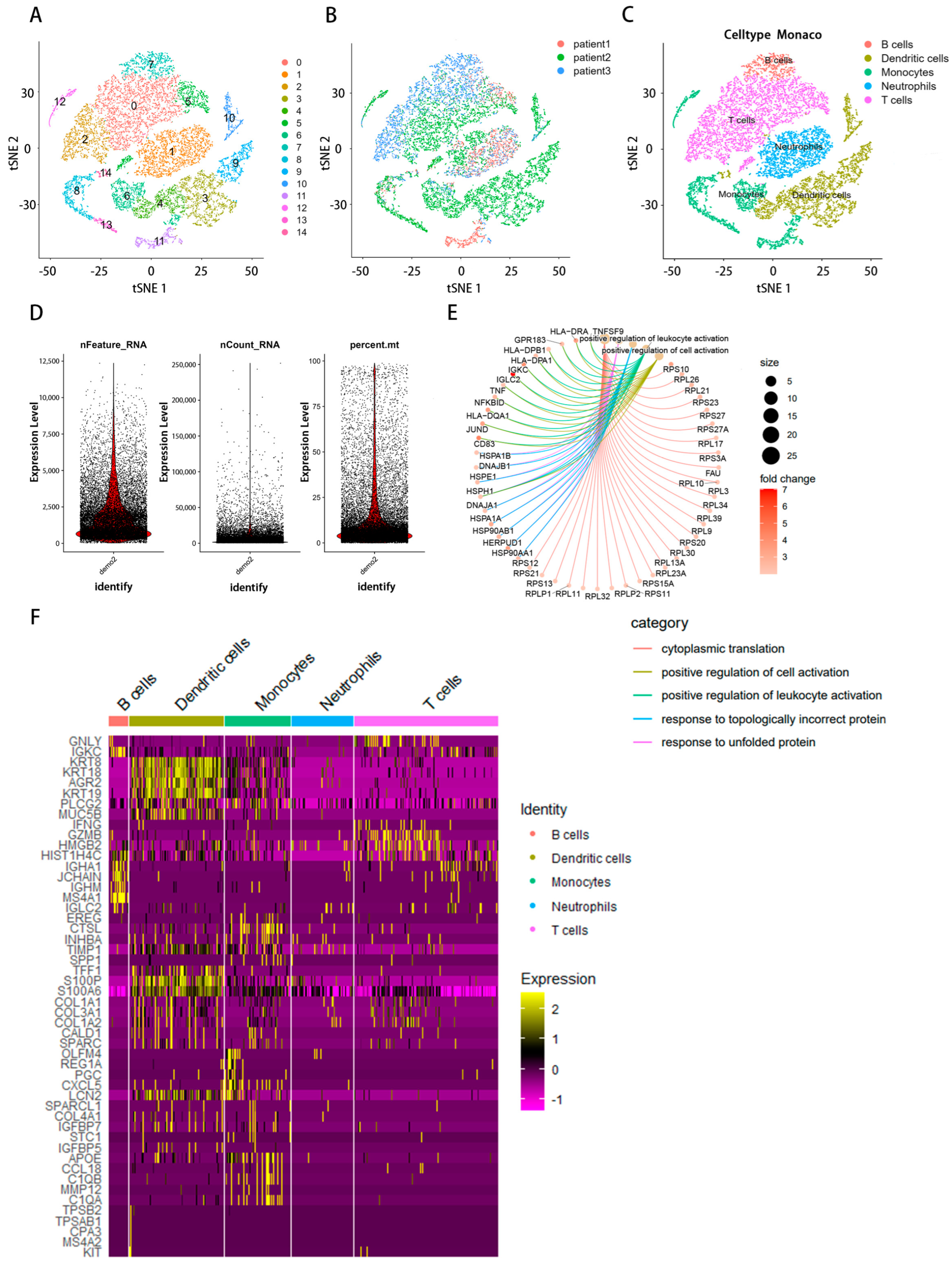
2.1. Examining Immunological Cells within scRNA-Seq Samples
2.2. Infiltrating Immune Cell Count
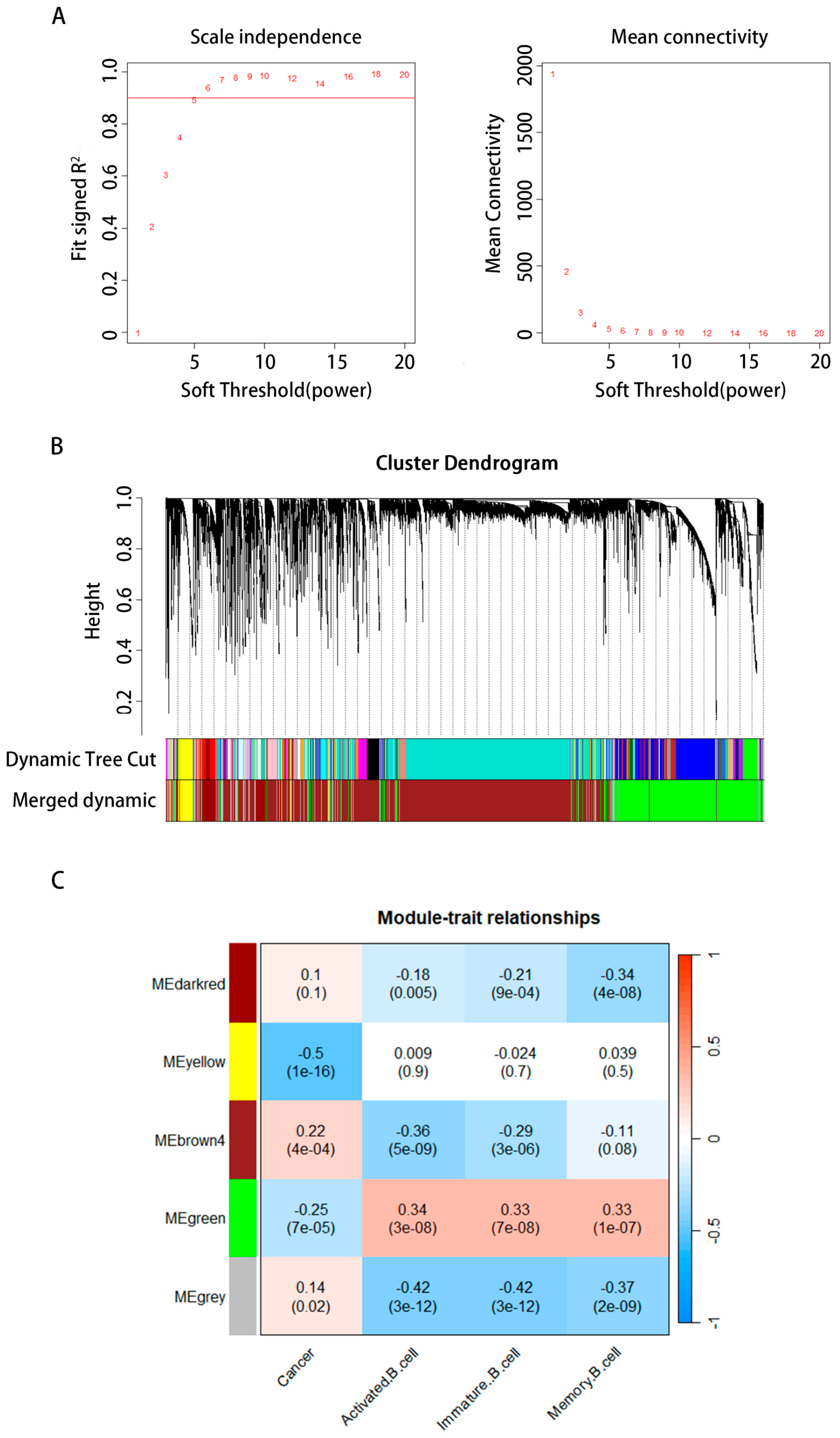
2.3. The WGCNA Structure and Identification of Essential Modules
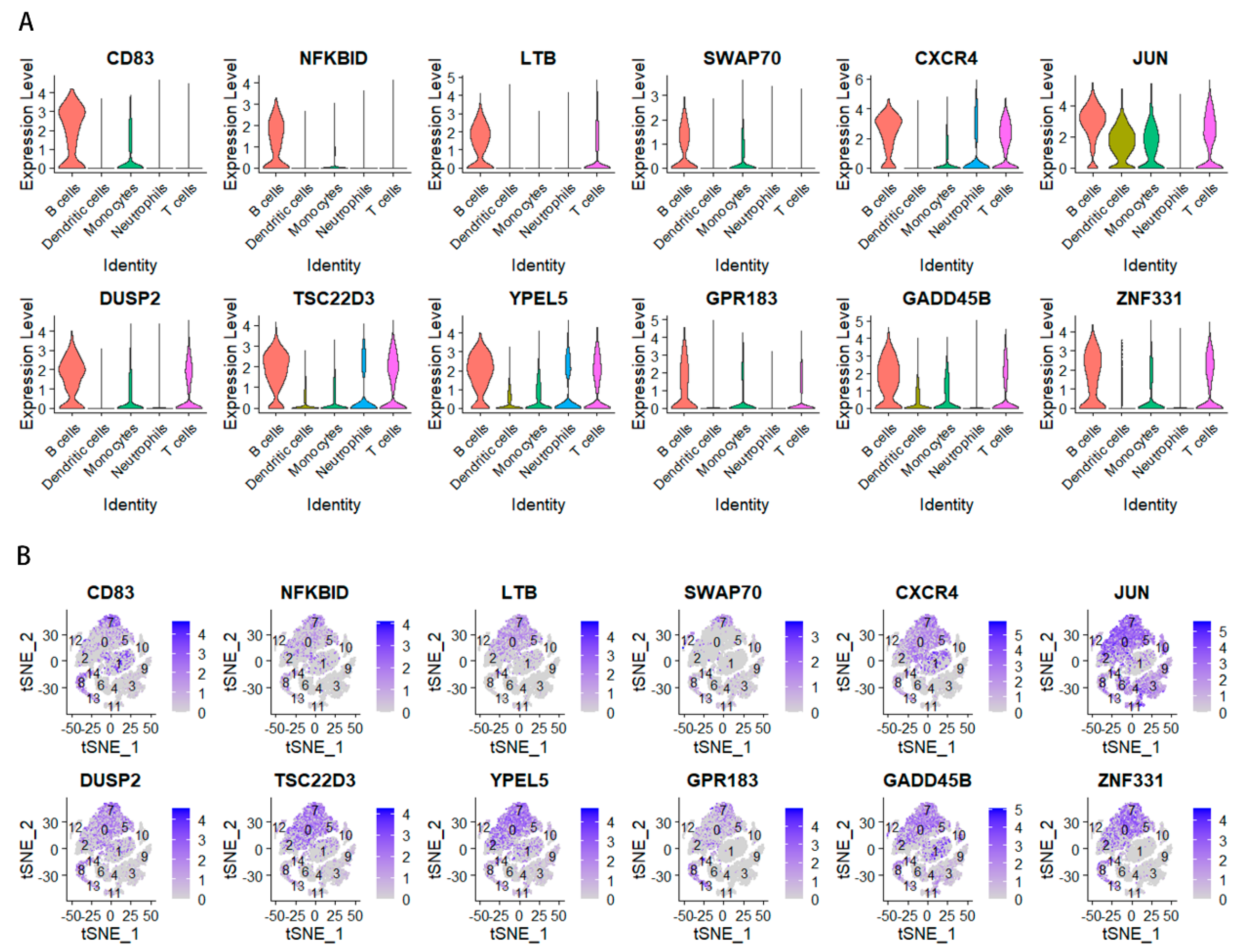
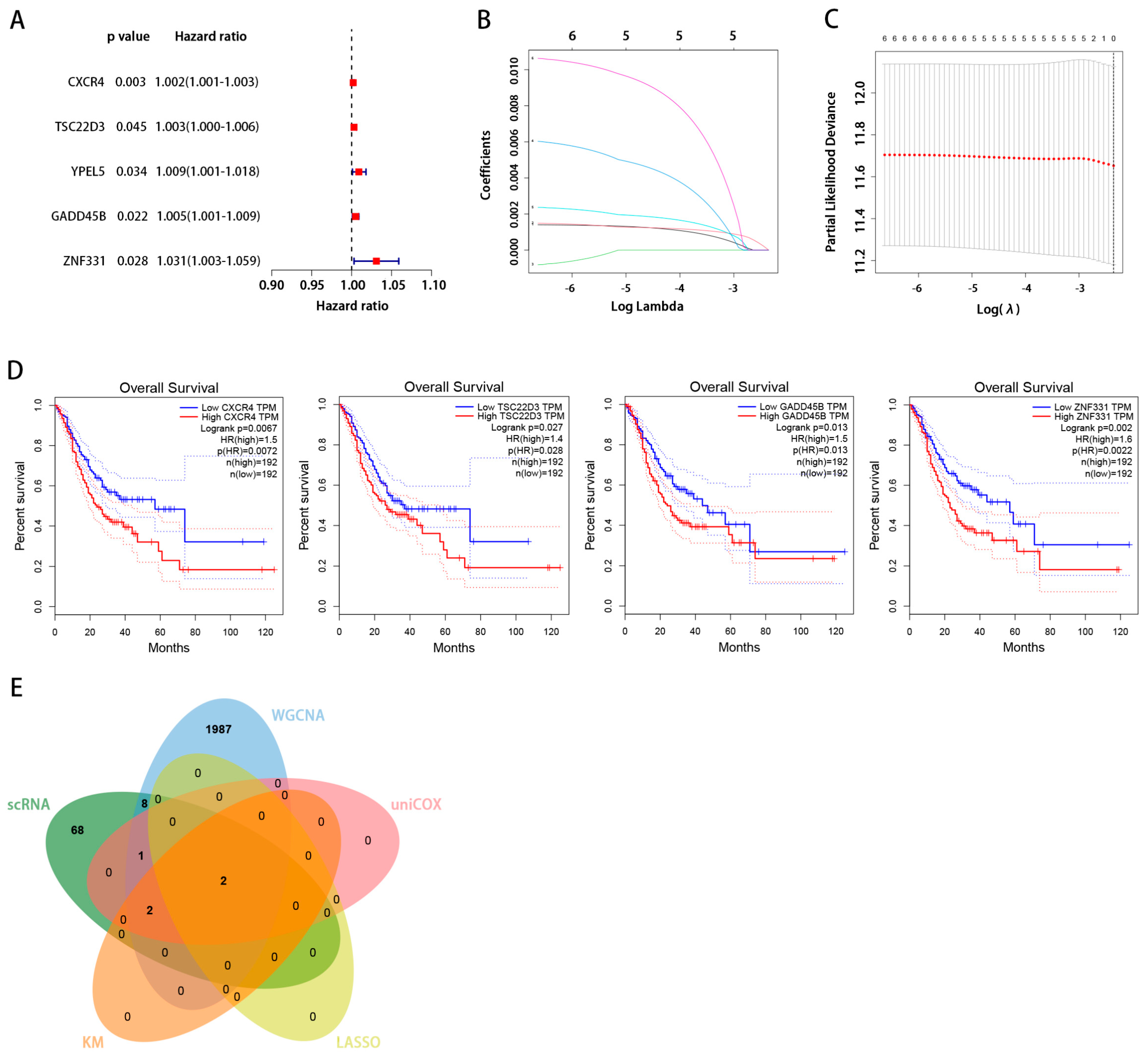
2.4. Analysis of the Relationship between Key Genes and Prognosis in Gastric Cancer
2.5. Screening the Key Genes with LASSO
2.6. GEPIA Database Analysis
2.7. TISIDB Database Verification of the Association between Key Genes and Immune Cell Invasion
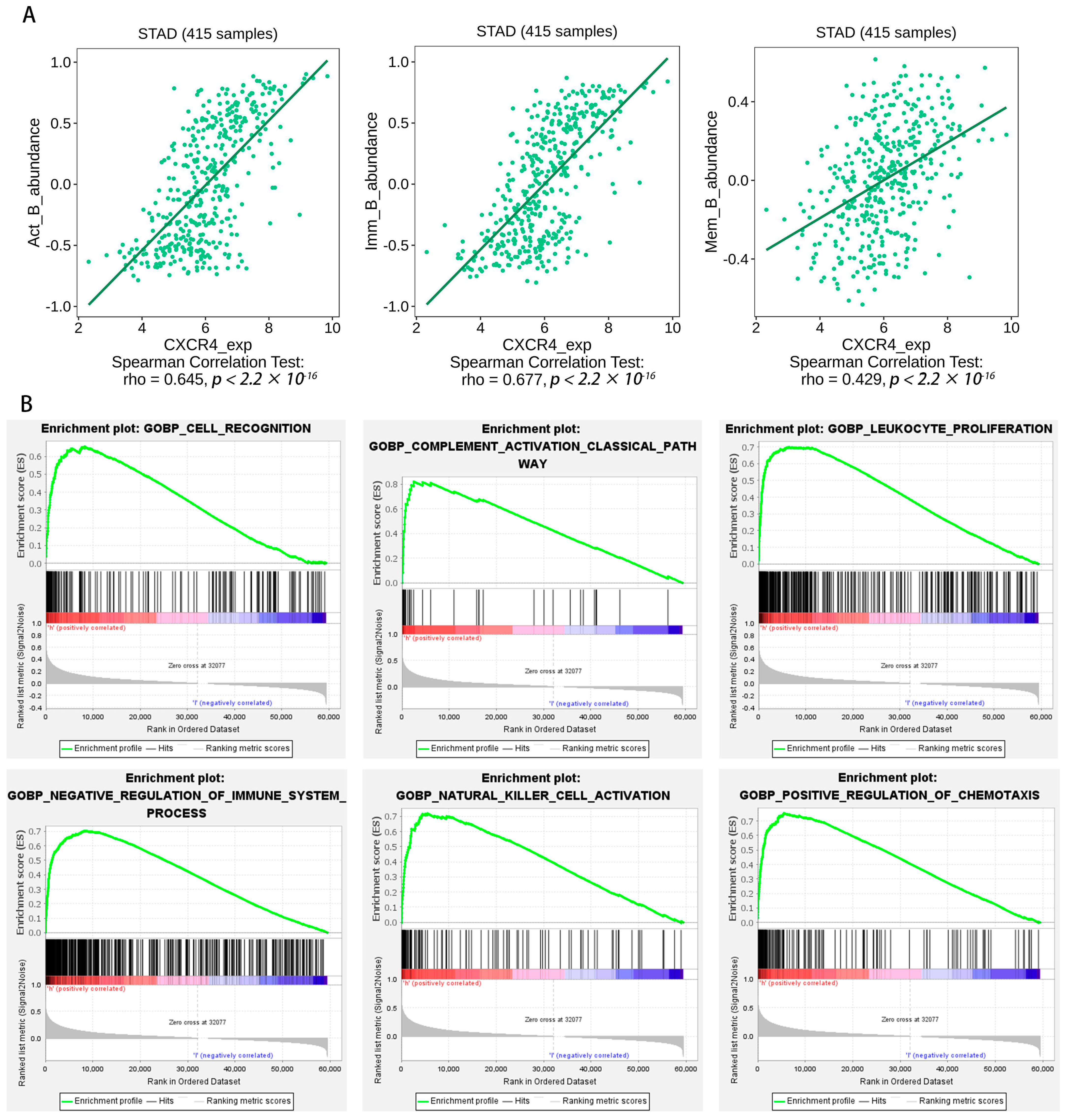
2.8. Gene Set Enrichment Analysis
2.9. CXCR4 Is Overexpressed in STAD
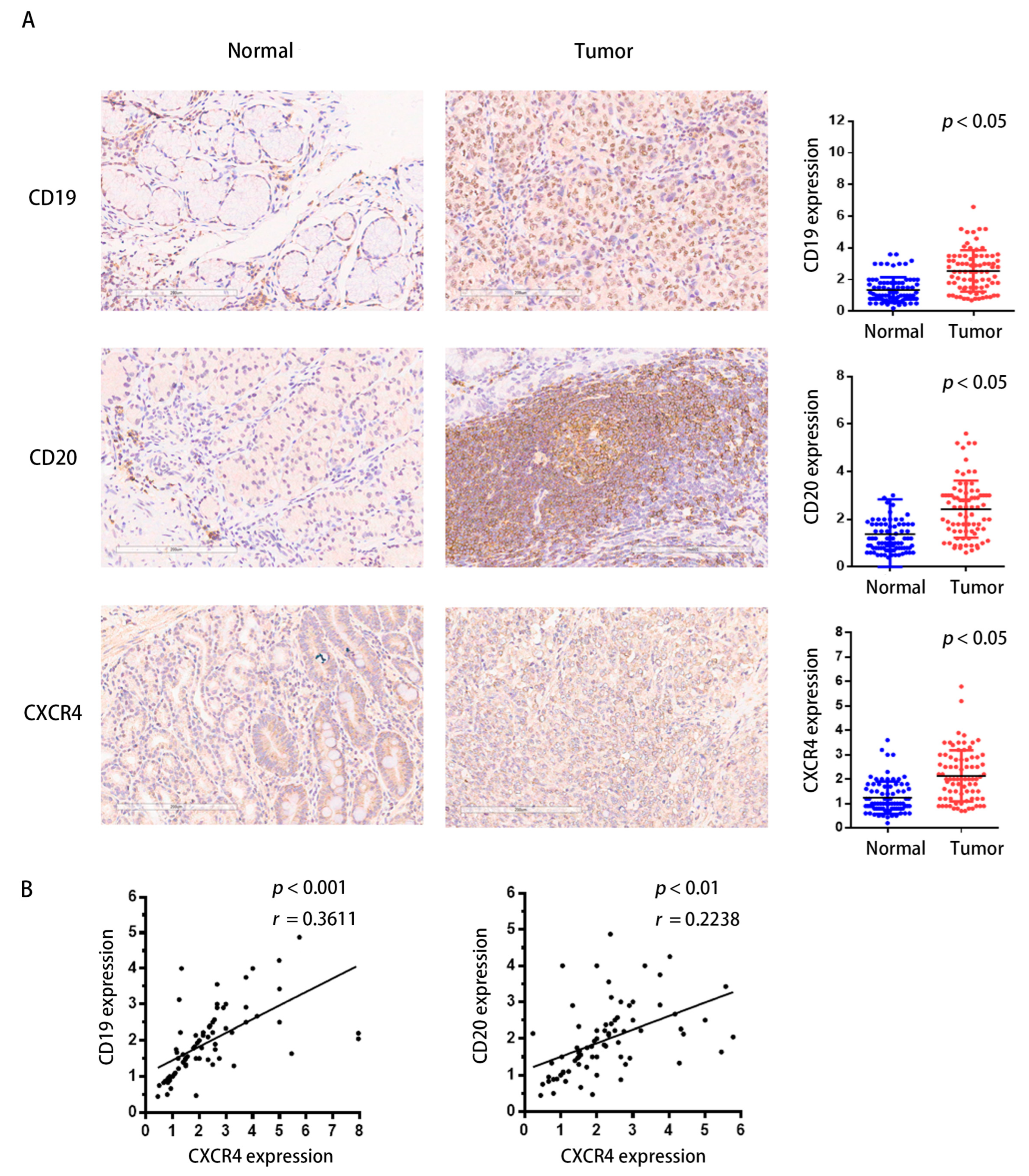
2.10. CXCR4 Expression Correlates Positively with TIBs in Clinical Samples of STAD
3. Discussion
4. Materials and Methods
4.1. Expression Profile Information on Genes
4.2. Clustering Dimension Reduction for scRNA Seq Data and Immune Cell Screening
4.3. Infiltration of Immune Cells
4.4. Weighted Gene Coexpression Network Analysis (WGCNA)
4.5. Analysis for Univariate Cox Regression
4.6. Screening Key Genes via LASSO
4.7. Prognosis Database Analysis
4.8. TISIDB Database Verification of the Association between Essential Genes and Immune Cell Infiltration
4.9. Gene Set Enrichment Analysis
4.10. Clinical Samples
4.11. Extraction of RNA via RT-PCR
4.12. IHC Staining
4.13. Analyses of Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kakinuma, D.; Arai, H.; Yasuda, T.; Kanazawa, Y.; Matsuno, K.; Sakurazawa, N.; Watanabe, M.; Suzuki, H.; Yoshida, H. Treatment of Gastric Cancer in Japan. J. Nippon Med. Sch. 2021, 88, 156–162. [Google Scholar] [CrossRef]
- Hou, J.; Wang, T.; Xie, Q.; Deng, W.; Yang, J.Y.; Zhang, S.Q.; Cai, J.C. N-Myc-interacting protein (NMI) negatively regulates epithelial-mesenchymal transition by inhibiting the acetylation of NF-κB/p65. Cancer Lett. 2016, 376, 22–33. [Google Scholar] [CrossRef]
- Hou, J.; Wang, L.; Zhao, J.; Zhuo, H.; Cheng, J.; Chen, X.; Zheng, W.; Hong, Z.; Cai, J. Inhibition of protein PMP22 enhances etoposide-induced cell apoptosis by p53 signaling pathway in Gastric Cancer. Int. J. Biol. Sci. 2021, 17, 3145–3157. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Tan, Y.; Su, C.; Wang, T.; Gao, Z.; Song, D.; Zhao, J.; Liao, Y.; Liu, X.; Jiang, Y.; et al. Inhibition of protein FAK enhances 5-FU chemosensitivity to gastric carcinoma via p53 signaling pathways. Comput. Struct. Biotechnol. J. 2020, 18, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef]
- Guo, J.N.; Chen, D.; Deng, S.H.; Huang, J.R.; Song, J.X.; Li, X.Y.; Cui, B.B.; Liu, Y.L. Identification and quantification of immune infiltration landscape on therapy and prognosis in left- and right-sided colon cancer. Cancer Immunol. Immunother. CII 2022, 71, 1313–1330. [Google Scholar] [CrossRef]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 34002. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Lin, Z.; Cui, Y.; Cai, J.C.; Hou, J. Identification of Essential Tumor-Infiltrating Immune Cells and Relevant Genes in Left-Sided and Right-Sided Colon Cancers. Cancers 2022, 14, 4713. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Phanthunane, C.; Wijers, R.; de Herdt, M.; Langeveld, T.P.M.; Koljenovic, S.; Dasgupta, S.; Sleijfer, S.; Baatenburg de Jong, R.J.; Hardillo, J.; Balcioglu, H.E.; et al. B-cell clusters at the invasive margin associate with longer survival in early-stage oral-tongue cancer patients. Oncoimmunology 2021, 10, 1882743. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Richter, G.; Schüler, T.; Ibe, S.; Cao, X.; Blankenstein, T. B cells inhibit induction of T cell-dependent tumor immunity. Nat. Med. 1998, 4, 627–630. [Google Scholar] [CrossRef]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; DeNardo, D.G.; et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Kinker, G.S.; Greenwald, A.C.; Tal, R.; Orlova, Z.; Cuoco, M.S.; McFarland, J.M.; Warren, A.; Rodman, C.; Roth, J.A.; Bender, S.A.; et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 2020, 52, 1208–1218. [Google Scholar] [CrossRef]
- Ilicic, T.; Kim, J.K.; Kolodziejczyk, A.A.; Bagger, F.O.; McCarthy, D.J.; Marioni, J.C.; Teichmann, S.A. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016, 17, 29. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Laumont, C.M.; Banville, A.C.; Gilardi, M.; Hollern, D.P.; Nelson, B.H. Tumour-infiltrating B cells: Immunological mechanisms, clinical impact and therapeutic opportunities. Nat. Rev. Cancer 2022, 22, 414–430. [Google Scholar] [CrossRef]
- Fridman, W.H.; Petitprez, F.; Meylan, M.; Chen, T.W.; Sun, C.M.; Roumenina, L.T.; Sautès-Fridman, C. B cells and cancer: To B or not to B? J. Exp. Med. 2021, 218, e20200851. [Google Scholar] [CrossRef]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Scotton, C.J.; Wilson, J.L.; Milliken, D.; Stamp, G.; Balkwill, F.R. Epithelial cancer cell migration: A role for chemokine receptors? Cancer Res. 2001, 61, 4961–4965. [Google Scholar] [PubMed]
- Yasumoto, K.; Koizumi, K.; Kawashima, A.; Saitoh, Y.; Arita, Y.; Shinohara, K.; Minami, T.; Nakayama, T.; Sakurai, H.; Takahashi, Y.; et al. Role of the CXCL12/CXCR4 axis in peritoneal carcinomatosis of gastric cancer. Cancer Res. 2006, 66, 2181–2187. [Google Scholar] [CrossRef]
- Ying, J.; Xu, Q.; Zhang, G.; Liu, B.; Zhu, L. The expression of CXCL12 and CXCR4 in gastric cancer and their correlation to lymph node metastasis. Med. Oncol. 2012, 29, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Lv, S.; Zhang, Y.; Yi, R.; Huang, B.; Fu, H.; Bian, R.; Li, X. The prognosis and clinicopathology of CXCR4 in gastric cancer patients: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 4589–4597. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ouyang, J.; Wienand, K.; Bojarczuk, K.; Hao, Y.; Chapuy, B.; Neuberg, D.; Juszczynski, P.; Lawton, L.N.; Rodig, S.J.; et al. CXCR4 upregulation is an indicator of sensitivity to B-cell receptor/PI3K blockade and a potential resistance mechanism in B-cell receptor-dependent diffuse large B-cell lymphomas. Haematologica 2020, 105, 1361–1368. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Rosenfeld, R.; Simon, I.; Nau, G.J.; Bar-Joseph, Z. A probabilistic generative model for GO enrichment analysis. Nucleic Acids Res. 2008, 36, e109. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Parameters | Expression | p-Value | |
|---|---|---|---|
| High | Low | ||
| Age, years | |||
| ≤60 | 8 | 1 | 0.374 |
| >60 | 14 | 7 | |
| Gender | |||
| Male | 17 | 7 | 0.655 |
| Female | 5 | 1 | |
| Location | |||
| Cardiac | 3 | 5 | 0.024 |
| Antrum | 6 | 1 | |
| Pylorus | 13 | 2 | |
| Tumor size | |||
| ≤5 cm | 16 | 4 | 0.384 |
| >5 cm | 6 | 4 | |
| Pathology grade | |||
| II | 9 | 3 | 1 |
| III | 13 | 5 | |
| TNM stage | |||
| I + II | 12 | 1 | 0.024 |
| III + IV | 10 | 9 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.; Yu, R.; Hong, X.; Zhang, P.; Guo, Y.; Cai, J.-C.; Hou, J. CXCR4 Expressed by Tumor-Infiltrating B Cells in Gastric Cancer Related to Survival in the Tumor Microenvironment: An Analysis Combining Single-Cell RNA Sequencing with Bulk RNA Sequencing. Int. J. Mol. Sci. 2023, 24, 12890. https://doi.org/10.3390/ijms241612890
Su C, Yu R, Hong X, Zhang P, Guo Y, Cai J-C, Hou J. CXCR4 Expressed by Tumor-Infiltrating B Cells in Gastric Cancer Related to Survival in the Tumor Microenvironment: An Analysis Combining Single-Cell RNA Sequencing with Bulk RNA Sequencing. International Journal of Molecular Sciences. 2023; 24(16):12890. https://doi.org/10.3390/ijms241612890
Chicago/Turabian StyleSu, Chen, Rong Yu, Xiaoquan Hong, Panpan Zhang, Yingying Guo, Jian-Chun Cai, and Jingjing Hou. 2023. "CXCR4 Expressed by Tumor-Infiltrating B Cells in Gastric Cancer Related to Survival in the Tumor Microenvironment: An Analysis Combining Single-Cell RNA Sequencing with Bulk RNA Sequencing" International Journal of Molecular Sciences 24, no. 16: 12890. https://doi.org/10.3390/ijms241612890
APA StyleSu, C., Yu, R., Hong, X., Zhang, P., Guo, Y., Cai, J.-C., & Hou, J. (2023). CXCR4 Expressed by Tumor-Infiltrating B Cells in Gastric Cancer Related to Survival in the Tumor Microenvironment: An Analysis Combining Single-Cell RNA Sequencing with Bulk RNA Sequencing. International Journal of Molecular Sciences, 24(16), 12890. https://doi.org/10.3390/ijms241612890