
Hyperandrogenism and Its Possible Effects on Endometrial Receptivity: A Review
 , , , and
, , , and
Abstract
1. Introduction
1.1. Normal Androgen Physiology
1.2. Hyperandrogenism
2. Hyperandrogenic Syndromes
2.1. Androgen-Secreting Tumours
2.2. Congenital Adrenal Hyperplasia
2.3. Hyperandrogenic Insulin-Resistant Acanthosis Nigricans (HAIR-AN) Syndrome
2.4. Hirsutism
3. Endometrial Receptivity
4. Assessment of Endometrial Receptivity
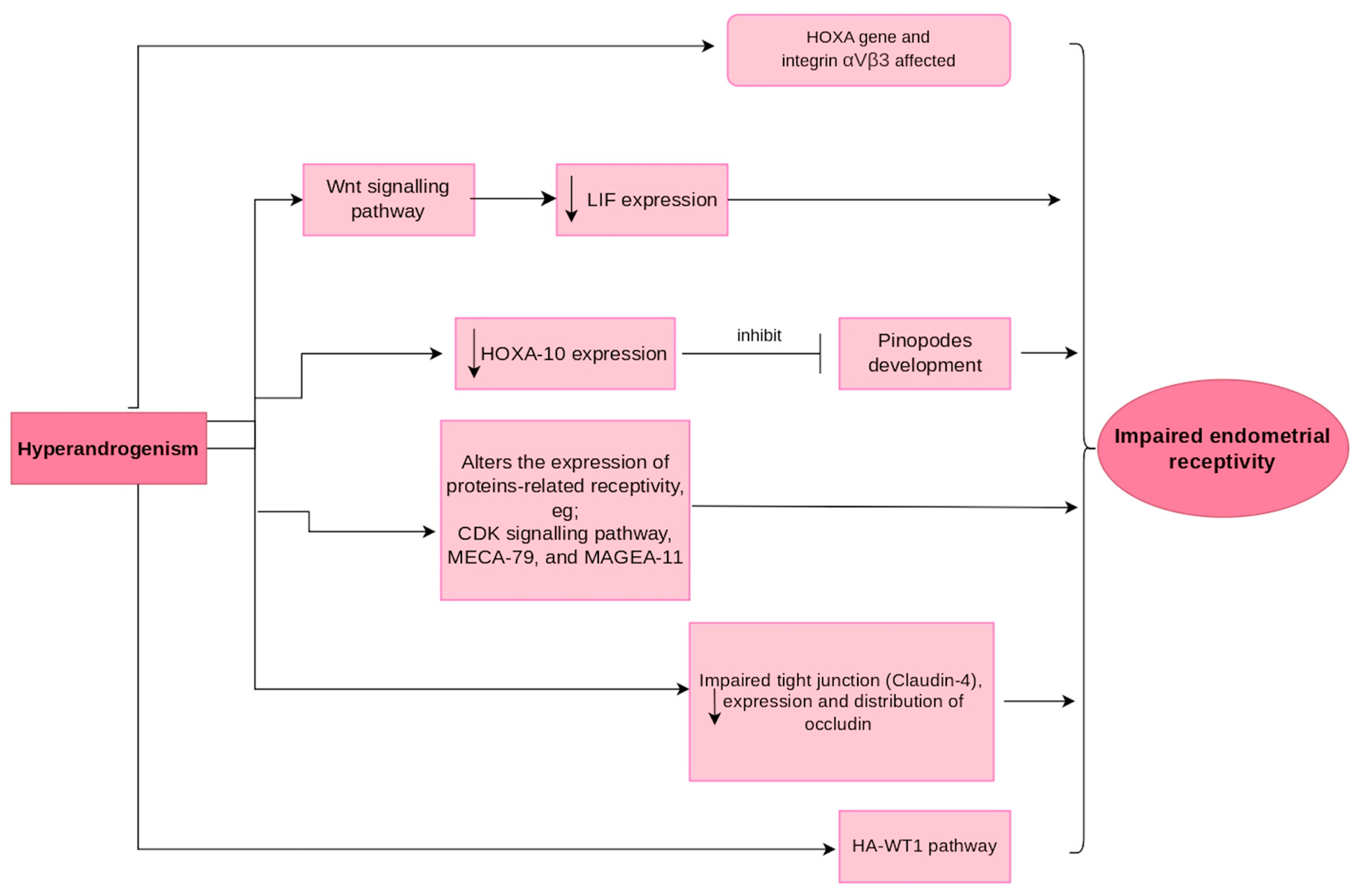
5. Mechanism of Hyperandrogenism Affecting Endometrial Receptivity
6. Impact of Uterine Anomalies in the Endometrial Molecular Expressions Affecting Endometrial Receptivity
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Starc, A.; Trampuš, M.; Pavan Jukić, D.; Grgas-Bile, C.; Jukić, T.; Polona Mivšek, A. Infertility and sexual dysfunctions: A systematic literature review. Acta Clin. Croat. 2019, 58, 508–515. [Google Scholar]
- Carson, S.A.; Kallen, A.N. Diagnosis and management of infertility: A review. JAMA 2021, 326, 65–76. [Google Scholar]
- Wang, C.; Wen, Y.-X.; Mai, Q.-Y. Impact of metabolic disorders on endometrial receptivity in patients with polycystic ovary syndrome. Exp. Ther. Med. 2022, 23, 221. [Google Scholar] [CrossRef]
- Horton, R.; Tait, J. Androstenedione production and interconversion rates measured in peripheral blood and studies on the possible site of its conversion to testosterone. J. Clin. Investig. 1966, 45, 301–313. [Google Scholar]
- Azzouni, F.; Godoy, A.; Li, Y.; Mohler, J. The 5 alpha-reductase isozyme family: A review of basic biology and their role in human diseases. Adv. Urol. 2011, 2012, 530121. [Google Scholar]
- Rosenfeld, R.; Hellman, L.; Roffwarg, H.; Weitzman, E.D.; Fukushima, D.K.; Gallagher, T. Dehydroisoandrosterone is secreted episodically and synchronously with cortisol by normal man. J. Clin. Endocrinol. Metab. 1971, 33, 87–92. [Google Scholar] [CrossRef]
- Marques, P.; Skorupskaite, K.; Rozario, K.S.; Anderson, R.A.; George, J.T. Physiology of GNRH and gonadotropin secretion. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2022. [Google Scholar]
- Iwasa, T.; Matsuzaki, T.; Yano, K.; Yanagihara, R.; Mayila, Y.; Irahara, M. The effects of chronic testosterone administration on hypothalamic gonadotropin-releasing hormone regulatory factors (Kiss1, NKB, pDyn and RFRP) and their receptors in female rats. Gynecol. Endocrinol. 2018, 34, 437–441. [Google Scholar]
- Auchus, R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. 2004, 15, 432–438. [Google Scholar] [CrossRef]
- Bird, C.E.; Green, R.N.; Clark, A.F. Effect of the administration of estrogen on the disappearance of 3H-testosterone in the plasma of human subjects. J. Clin. Endocrinol. Metab. 1969, 29, 123–126. [Google Scholar] [CrossRef]
- Southren, A.L.; Gordon, G.G.; Tochimoto, S.; Krikun, E.; Krieger, D.; Jacobson, M.; Kuntzman, R. Effect of N-phenylbarbital (phetharbital) on the metabolism of testosterone and cortisol in man. J. Clin. Endocrinol. Metab. 1969, 29, 251–256. [Google Scholar] [CrossRef]
- Gordon, G.G.; Southren, A.L.; Tochimoto, S.; Rand, J.J.; Olivo, J. Effect of hyperthyroidism and hypothyroidism on the metabolism of testosterone and androstenedione in man. J. Clin. Endocrinol. Metab. 1969, 29, 164–170. [Google Scholar] [CrossRef]
- Southren, A.L.; Gordon, G.G.; Tochimoto, S. Further study of factors affecting the metabolic clearance rate of testosterone in man. J. Clin. Endocrinol. Metab. 1968, 28, 1105–1112. [Google Scholar] [CrossRef]
- Coviello, A.D.; Lakshman, K.; Mazer, N.A.; Bhasin, S. Differences in the apparent metabolic clearance rate of testosterone in young and older men with gonadotropin suppression receiving graded doses of testosterone. J. Clin. Endocrinol. Metab. 2006, 91, 4669–4675. [Google Scholar] [CrossRef]
- Cordera, F.; Grant, C.; Van Heerden, J.; Thompson, G.; Young, W. Androgen-secreting adrenal tumors. Surgery 2003, 134, 874–880. [Google Scholar] [CrossRef]
- Critchley, H.O.; Saunders, P.T. Hormone receptor dynamics in a receptive human endometrium. Reprod. Sci. 2009, 16, 191–199. [Google Scholar] [CrossRef]
- Marshall, E.; Lowrey, J.; MacPherson, S.; Maybin, J.A.; Collins, F.; Critchley, H.O.; Saunders, P.T. In silico analysis identifies a novel role for androgens in the regulation of human endometrial apoptosis. J. Clin. Endocrinol. Metab. 2011, 96, E1746–E1755. [Google Scholar] [CrossRef]
- Giudice, L.C. Endometrium in PCOS: Implantation and predisposition to endocrine CA. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 235–244. [Google Scholar] [CrossRef]
- Milne, S.A.; Henderson, T.A.; Kelly, R.W.; Saunders, P.T.; Baird, D.T.; Critchley, H.O. Leukocyte populations and steroid receptor expression in human first-trimester decidua; regulation by antiprogestin and prostaglandin E analog. J. Clin. Endocrinol. Metab. 2005, 90, 4315–4321. [Google Scholar]
- Cloke, B.; Christian, M. The role of androgens and the androgen receptor in cycling endometrium. Mol. Cell. Endocrinol. 2012, 358, 166–175. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, M.; Yang, F.; Zhang, Y.; Ma, S.; Zhang, D.; Wang, X.; Sferruzzi-Perri, A.N.; Wu, X.; Brännström, M. Increased uterine androgen receptor protein abundance results in implantation and mitochondrial defects in pregnant rats with hyperandrogenism and insulin resistance. J. Mol. Med. 2021, 99, 1427–1446. [Google Scholar]
- Azziz, R.; Carmina, E.; Sawaya, M.E. Idiopathic hirsutism. Endocr. Rev. 2000, 21, 347–362. [Google Scholar]
- Luque-Ramírez, M.; F Escobar-Morreale, H. Adrenal hyperandrogenism and polycystic ovary syndrome. Curr. Pharm. Des. 2016, 22, 5588–5602. [Google Scholar] [CrossRef]
- Carmina, E. Ovarian and adrenal hyperandrogenism. Ann. N. Y. Acad. Sci. 2006, 1092, 130–137. [Google Scholar] [CrossRef]
- Baptiste, C.G.; Battista, M.-C.; Trottier, A.; Baillargeon, J.-P. Insulin and hyperandrogenism in women with polycystic ovary syndrome. J. Steroid Biochem. Mol. Biol. 2010, 122, 42–52. [Google Scholar]
- Barontini, M.; García-Rudaz, M.C.; Veldhuis, J.D. Mechanisms of hypothalamic-pituitary-gonadal disruption in polycystic ovarian syndrome. Arch. Med. Res. 2001, 32, 544–552. [Google Scholar] [CrossRef]
- Baskind, N.E.; Balen, A.H. Hypothalamic–pituitary, ovarian and adrenal contributions to polycystic ovary syndrome. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 37, 80–97. [Google Scholar] [CrossRef]
- Pretorius, E.; Arlt, W.; Storbeck, K.-H. A new dawn for androgens: Novel lessons from 11-oxygenated C19 steroids. Mol. Cell. Endocrinol. 2017, 441, 76–85. [Google Scholar] [CrossRef]
- Witchel, S.F.; Oberfield, S.E.; Peña, A.S. Polycystic ovary syndrome: Pathophysiology, presentation, and treatment with emphasis on adolescent girls. J. Endocr. Soc. 2019, 3, 1545–1573. [Google Scholar]
- Gonzalez, F. Adrenal involvement in polycystic ovary syndrome. Semin. Reprod. Endocrinol. 1997, 15, 137–157. [Google Scholar] [CrossRef]
- Kostakis, E.K.; Gkioni, L.N.; Macut, D.; Mastorakos, G. Androgens in menopausal women: Not only polycystic ovary syndrome. Hyperandrogenism Women 2019, 53, 135–161. [Google Scholar]
- Morton, N.M.; Seckl, J.R. 11β-hydroxysteroid dehydrogenase type 1 and obesity. Obes. Metab. 2008, 36, 146–164. [Google Scholar]
- Sandeep, T.C.; Walker, B.R. Pathophysiology of modulation of local glucocorticoid levels by 11β-hydroxysteroid dehydrogenases. Trends Endocrinol. Metab. 2001, 12, 446–453. [Google Scholar]
- Ding, H.; Zhang, J.; Zhang, F.; Zhang, S.; Chen, X.; Liang, W.; Xie, Q. Resistance to the insulin and elevated level of androgen: A major cause of polycystic ovary syndrome. Front. Endocrinol. 2021, 12, 741764. [Google Scholar]
- Holte, J.; Bergh, T.; Gennarelli, G.; Wide, L. The independent effects of polycystic ovary syndrome and obesity on serum concentrations of gonadotrophins and sex steroids in premenopausal women. Clin. Endocrinol. 1994, 41, 473–481. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Futterweit, W.; Janssen, O.E.; Legro, R.S.; Norman, R.J.; Taylor, A.E. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: The complete task force report. Fertil. Steril. 2009, 91, 456–488. [Google Scholar]
- Goodarzi, M.O.; Carmina, E.; Azziz, R. Dhea, dheas and pcos. J. Steroid Biochem. Mol. Biol. 2015, 145, 213–225. [Google Scholar] [CrossRef]
- Matheson, E.M.; Bain, J. Hirsutism in women. Am. Fam. Physician 2019, 100, 168–175. [Google Scholar]
- Macut, D.; Ilić, D.; Jovanović, A.M.; Bjekić-Macut, J. Androgen-secreting ovarian tumors. Hyperandrogenism Women 2019, 53, 100–107. [Google Scholar]
- Witchel, S.F. Congenital adrenal hyperplasia. J. Pediatr. Adolesc. Gynecol. 2017, 30, 520–534. [Google Scholar]
- Parsa, A.A.; New, M.I. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J. Steroid Biochem. Mol. Biol. 2017, 165, 2–11. [Google Scholar]
- Nandagopal, R.; Sinaii, N.; Avila, N.A.; Van Ryzin, C.; Chen, W.; Finkielstain, G.P.; Mehta, S.P.; McDonnell, N.B.; Merke, D.P. Phenotypic profiling of parents with cryptic nonclassic congenital adrenal hyperplasia: Findings in 145 unrelated families. Eur. J. Endocrinol. 2011, 164, 977–984. [Google Scholar] [PubMed]
- Pignatelli, D.; Pereira, S.S.; Pasquali, R. Androgens in congenital adrenal hyperplasia. Hyperandrogenism Women 2019, 53, 65–76. [Google Scholar]
- Pall, M.; Azziz, R.; Beires, J.; Pignatelli, D. The phenotype of hirsute women: A comparison of polycystic ovary syndrome and 21-hydroxylase–deficient nonclassic adrenal hyperplasia. Fertil. Steril. 2010, 94, 684–689. [Google Scholar]
- Raff, H.; Sharma, S.T.; Nieman, L.K. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing’s syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr. Physiol. 2014, 4, 739. [Google Scholar] [PubMed]
- Escobar-Morreale, H.; Carmina, E.; Dewailly, D.; Gambineri, A.; Kelestimur, F.; Moghetti, P.; Pugeat, M.; Qiao, J.; Wijeyaratne, C.; Witchel, S. Epidemiology, diagnosis and management of hirsutism: A consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum. Reprod. Update 2012, 18, 146–170. [Google Scholar] [PubMed]
- Binay, C.; Simsek, E.; Cilingir, O.; Yuksel, Z.; Kutlay, O.; Artan, S. Prevalence of nonclassic congenital adrenal hyperplasia in Turkish children presenting with premature pubarche, hirsutism, or oligomenorrhoea. Int. J. Endocrinol. 2014, 2014, 768506. [Google Scholar] [CrossRef]
- Armengaud, J.-B.; Charkaluk, M.-L.; Trivin, C.; Tardy, V.; Bréart, G.; Brauner, R.; Chalumeau, M. Precocious pubarche: Distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. J. Clin. Endocrinol. Metab. 2009, 94, 2835–2840. [Google Scholar]
- Azziz, R.; Waggoner, W.T.; Ochoa, T.; Knochenhauer, E.S.; Boots, L.R. Idiopathic hirsutism: An uncommon cause of hirsutism in Alabama. Fertil. Steril. 1998, 70, 274–278. [Google Scholar]
- Azziz, R.; Sanchez, L.; Knochenhauer, E.; Moran, C.; Lazenby, J.; Stephens, K.; Taylor, K.; Boots, L. Androgen excess in women: Experience with over 1000 consecutive patients. J. Clin. Endocrinol. Metab. 2004, 89, 453–462. [Google Scholar]
- Bidet, M.; Bellanne-Chantelot, C.; Galand-Portier, M.-B.; Golmard, J.-L.; Tardy, V.; Morel, Y.; Clauin, S.; Coussieu, C.; Boudou, P.; Mowzowicz, I. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 1182–1190. [Google Scholar]
- Lobo, R.A.; Goebelsmann, U. Adult manifestation of congenital adrenal hyperplasia due to incomplete 21-hydroxylase deficiency mimicking polycystic ovarian disease. Am. J. Obstet. Gynecol. 1980, 138, 720–726. [Google Scholar]
- Escobar-Morreale, H.F.; Sanchon, R.; San Millán, J.L. A prospective study of the prevalence of nonclassical congenital adrenal hyperplasia among women presenting with hyperandrogenic symptoms and signs. J. Clin. Endocrinol. Metab. 2008, 93, 527–533. [Google Scholar]
- Pignatelli, D. Non-classic adrenal hyperplasia due to the deficiency of 21-hydroxylase and its relation to polycystic ovarian syndrome. Polycystic Ovary Syndr. 2013, 40, 158–170. [Google Scholar]
- Roland, A.V.; Moenter, S.M. Reproductive neuroendocrine dysfunction in polycystic ovary syndrome: Insight from animal models. Front. Neuroendocrinol. 2014, 35, 494–511. [Google Scholar] [PubMed]
- Pache, T.; Chadha, S.; Gooren, L.; Hop, W.; Jaarsma, K.; Dommerholt, H.; Fauser, B. Ovarian morphology in long-term androgen-treated female to male transsexuals. A human model for the study of polycystic ovarian syndrome? Histopathology 1991, 19, 445–452. [Google Scholar]
- Lucis, O.; Hobkirk, R.; Hollenberg, C.; MacDonald, S.; Blahey, P. Polycystic ovaries associated with congenital adrenal hyperplasia. Can. Med. Assoc. J. 1966, 94, 1. [Google Scholar] [PubMed]
- Sen, A.; Hammes, S.R. Granulosa cell-specific androgen receptors are critical regulators of ovarian development and function. Mol. Endocrinol. 2010, 24, 1393–1403. [Google Scholar] [PubMed]
- Walters, K.A. Role of androgens in normal and pathological ovarian function. Reproduction 2015, 149, R193–R218. [Google Scholar] [PubMed]
- Walters, K.; Handelsman, D. Role of androgens in the ovary. Mol. Cell. Endocrinol. 2018, 465, 36–47. [Google Scholar]
- Gleicher, N.; Weghofer, A.; Barad, D.H. The role of androgens in follicle maturation and ovulation induction: Friend or foe of infertility treatment? Reprod. Biol. Endocrinol. 2011, 9, 116. [Google Scholar] [CrossRef]
- Lebbe, M.; Woodruff, T. Involvement of androgens in ovarian health and disease. Mol. Hum. Reprod. 2013, 19, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Mulaikal, R.M.; Migeon, C.J.; Rock, J.A. Fertility rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N. Engl. J. Med. 1987, 316, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Anwar, A. Infertility: A review on causes, treatment and management. Womens Health Gynecol. 2016, 5, 2–5. [Google Scholar]
- Dédjan, A.H.; Chadli, A.; El Aziz, S.; Farouqi, A. Hyperandrogenism-Insulin Resistance-Acanthosis Nigricans Syndrome. Case Rep. Endocrinol. 2015, 2015, 193097. [Google Scholar] [CrossRef]
- Elmer, K.B.; George, R.M. HAIR-AN syndrome: A multisystem challenge. Am. Fam. Physician 2001, 63, 2385. [Google Scholar]
- Esperanza, L.E.; Fenske, N.A. Hyperandrogenism, insulin resistance, and acanthosis nigricans (HAIR-AN) syndrome: Spontaneous remission in a 15-year-old girl. J. Am. Acad. Dermatol. 1996, 34, 892–897. [Google Scholar]
- Brady, M.F.; Rawla, P. Acanthosis Nigricans. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rager, K.M.; Omar, H.A. Androgen excess disorders in women: The severe insulin-resistant hyperandrogenic syndrome, HAIR-AN. Sci. World J. 2006, 6, 116–121. [Google Scholar]
- Hermanns-Lê, T.; Scheen, A.; Piérard, G.E. Acanthosis nigricans associated with insulin resistance. Am. J. Clin. Dermatol. 2004, 5, 199–203. [Google Scholar] [CrossRef]
- Ferriman, D.; Gallwey, J. Clinical assessment of body hair growth in women. J. Clin. Endocrinol. Metab. 1961, 21, 1440–1447. [Google Scholar] [CrossRef]
- Lessey, B.A.; Young, S.L. What exactly is endometrial receptivity? Fertil. Steril. 2019, 111, 611–617. [Google Scholar] [CrossRef]
- Rock, J.; Bartlett, M.K. Biopsy studies of human endometrium: Criteria of dating and information about amenorrhea, menorrhagia and time of ovulation. J. Am. Med. Assoc. 1937, 108, 2022–2028. [Google Scholar] [PubMed]
- Tan, J.; Paria, B.C.; Dey, S.K.; Das, S.K. Differential uterine expression of estrogen and progesterone receptors correlates with uterine preparation for implantation and decidualization in the mouse. Endocrinology 1999, 140, 5310–5321. [Google Scholar] [PubMed]
- Gibson, D.A.; Simitsidellis, I.; Cousins, F.L.; Critchley, H.O.; Saunders, P.T. Intracrine androgens enhance decidualization and modulate expression of human endometrial receptivity genes. Sci. Rep. 2016, 6, 19970. [Google Scholar]
- Piltonen, T.T. Polycystic ovary syndrome: Endometrial markers. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 37, 66–79. [Google Scholar]
- Gellersen, B.; Brosens, I.A.; Brosens, J.J. Decidualization of the human endometrium: Mechanisms, functions, and clinical perspectives. Semin. Reprod. Med. 2007, 25, 445–453. [Google Scholar] [CrossRef]
- Governini, L.; Luongo, F.P.; Haxhiu, A.; Piomboni, P.; Luddi, A. Main actors behind the endometrial receptivity and successful implantation. Tissue Cell 2021, 73, 101656. [Google Scholar]
- Usadi, R.S.; Murray, M.J.; Bagnell, R.C.; Fritz, M.A.; Kowalik, A.I.; Meyer, W.R.; Lessey, B.A. Temporal and morphologic characteristics of pinopod expression across the secretory phase of the endometrial cycle in normally cycling women with proven fertility. Fertil. Steril. 2003, 79, 970–974. [Google Scholar]
- Fitzgerald, H.C.; Salamonsen, L.A.; Rombauts, L.J.; Vollenhoven, B.J.; Edgell, T.A. The proliferative phase underpins endometrial development: Altered cytokine profiles in uterine lavage fluid of women with idiopathic infertility. Cytokine 2016, 88, 12–19. [Google Scholar]
- Zhao, Y.; Garcia, J.; Kolp, L.; Cheadle, C.; Rodriguez, A.; Vlahos, N.F. The impact of luteal phase support on gene expression of extracellular matrix protein and adhesion molecules in the human endometrium during the window of implantation following controlled ovarian stimulation with a GnRH antagonist protocol. Fertil. Steril. 2010, 94, 2264–2271. [Google Scholar]
- Robertshaw, I.; Bian, F.; Das, S. Mechanisms of uterine estrogen signaling during early pregnancy in mice: An update. J. Mol. Endocrinol. 2016, 56, R127. [Google Scholar] [PubMed]
- Wang, X.; Wu, S.-P.; DeMayo, F.J. Hormone dependent uterine epithelial-stromal communication for pregnancy support. Placenta 2017, 60, S20–S26. [Google Scholar]
- Kodaman, P.H.; Taylor, H.S. Hormonal regulation of implantation. Obstet. Gynecol. Clin. 2004, 31, 745–766. [Google Scholar]
- Bulmer, J.N.; Morrison, L.; Longfellow, M.; Ritson, A.; Pace, D. Granulated lymphocytes in human endometrium: Histochemical and immunohistochemical studies. Hum. Reprod. 1991, 6, 791–798. [Google Scholar] [PubMed]
- Adams, S.; Gayer, N.; Hosie, M.; Murphy, C. Human uterodomes (pinopods) do not display pinocytotic function. Hum. Reprod. 2002, 17, 1980–1986. [Google Scholar]
- Zhao, J.; Zhang, Q.; Li, Y. The effect of endometrial thickness and pattern measured by ultrasonography on pregnancy outcomes during IVF-ET cycles. Reprod. Biol. Endocrinol. 2012, 10, 100. [Google Scholar]
- Craciunas, L.; Gallos, I.; Chu, J.; Bourne, T.; Quenby, S.; Brosens, J.J.; Coomarasamy, A. Conventional and modern markers of endometrial receptivity: A systematic review and meta-analysis. Hum. Reprod. Update 2019, 25, 202–223. [Google Scholar]
- Zollner, U.; Zollner, K.-P.; Specketer, M.-T.; Blissing, S.; Müller, T.; Steck, T.; Dietl, J. Endometrial volume as assessed by three-dimensional ultrasound is a predictor of pregnancy outcome after in vitro fertilization and embryo transfer. Fertil. Steril. 2003, 80, 1515–1517. [Google Scholar] [PubMed]
- Wang, L.; Qiao, J.; Li, R.; Zhen, X.; Liu, Z. Role of endometrial blood flow assessment with color Doppler energy in predicting pregnancy outcome of IVF-ET cycles. Reprod. Biol. Endocrinol. 2010, 8, 122. [Google Scholar] [CrossRef]
- Chung, C.H.S.; Wong, A.W.Y.; Chan, C.P.S.; Saravelos, S.H.; Kong, G.W.S.; Cheung, L.P.; Chung, J.P.W.; Li, T.C. The changing pattern of uterine contractions before and after fresh embryo transfer and its relation to clinical outcome. Reprod. Biomed. Online 2017, 34, 240–247. [Google Scholar]
- Sharma, S. Natural killer cells and regulatory T cells in early pregnancy loss. Int. J. Dev. Biol. 2014, 58, 219. [Google Scholar] [PubMed]
- Sauerbrun-Cutler, M.T.; Huber, W.J.; Krueger, P.M.; Sung, C.J.; Has, P.; Sharma, S. Do endometrial natural killer and regulatory T cells differ in infertile and clinical pregnancy patients? An analysis in patients undergoing frozen embryo transfer cycles. Am. J. Reprod. Immunol. 2021, 85, e13393. [Google Scholar] [CrossRef]
- Fu, B.; Li, X.; Sun, R.; Tong, X.; Ling, B.; Tian, Z.; Wei, H. Natural killer cells promote immune tolerance by regulating inflammatory TH17 cells at the human maternal–fetal interface. Proc. Natl. Acad. Sci. USA 2013, 110, E231–E240. [Google Scholar] [CrossRef]
- Gaynor, L.M.; Colucci, F. Uterine natural killer cells: Functional distinctions and influence on pregnancy in humans and mice. Front. Immunol. 2017, 8, 467. [Google Scholar]
- Díaz-Gimeno, P.; Horcajadas, J.A.; Martínez-Conejero, J.A.; Esteban, F.J.; Alamá, P.; Pellicer, A.; Simón, C. A genomic diagnostic tool for human endometrial receptivity based on the transcriptomic signature. Fertil. Steril. 2011, 95, 50–60.e15. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.; Nikas, G.; Hsiu, J.-G.; Díaz, J.; Oehninger, S. Gene expression profiles and structural/functional features of the peri-implantation endometrium in natural and gonadotropin-stimulated cycles. J. Clin. Endocrinol. Metab. 2004, 89, 5742–5752. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gimeno, P.; Ruiz-Alonso, M.; Sebastian-Leon, P.; Pellicer, A.; Valbuena, D.; Simón, C. Window of implantation transcriptomic stratification reveals different endometrial subsignatures associated with live birth and biochemical pregnancy. Fertil. Steril. 2017, 108, 703–710.e703. [Google Scholar]
- Ruiz-Alonso, M.; Blesa, D.; Díaz-Gimeno, P.; Gómez, E.; Fernández-Sánchez, M.; Carranza, F.; Carrera, J.; Vilella, F.; Pellicer, A.; Simón, C. The endometrial receptivity array for diagnosis and personalized embryo transfer as a treatment for patients with repeated implantation failure. Fertil. Steril. 2013, 100, 818–824. [Google Scholar] [CrossRef]
- Ruiz-Alonso, M.; Galindo, N.; Pellicer, A.; Simón, C. What a difference two days make:“personalized” embryo transfer (pET) paradigm: A case report and pilot study. Hum. Reprod. 2014, 29, 1244–1247. [Google Scholar] [PubMed]
- Tan, J.; Kan, A.; Hitkari, J.; Taylor, B.; Tallon, N.; Warraich, G.; Yuzpe, A.; Nakhuda, G. The role of the endometrial receptivity array (ERA) in patients who have failed euploid embryo transfers. J. Assist. Reprod. Genet. 2018, 35, 683–692. [Google Scholar] [CrossRef]
- Hashimoto, T.; Koizumi, M.; Doshida, M.; Toya, M.; Sagara, E.; Oka, N.; Nakajo, Y.; Aono, N.; Igarashi, H.; Kyono, K. Efficacy of the endometrial receptivity array for repeated implantation failure in Japan: A retrospective, two-centers study. Reprod. Med. Biol. 2017, 16, 290–296. [Google Scholar]
- Mahajan, N. Endometrial receptivity array: Clinical application. J. Hum. Reprod. Sci. 2015, 8, 121. [Google Scholar] [CrossRef]
- Ma, L.; Cao, Y.; Ma, Y.; Zhai, J. Association between hyperandrogenism and adverse pregnancy outcomes in patients with different polycystic ovary syndrome phenotypes undergoing in vitro fertilization/intracytoplasmic sperm injection: A systematic review and meta-analysis. Gynecol. Endocrinol. 2021, 37, 694–701. [Google Scholar] [PubMed]
- Yang, W.; Yang, R.; Yang, S.; Li, J.; Tu, B.; Gao, C.; Wang, Y. Infertile polycystic ovary syndrome patients undergoing in vitro fertilization with the gonadotropin-releasing hormone-antagonist protocol: Role of hyperandrogenism. Gynecol. Endocrinol. 2018, 34, 715–718. [Google Scholar] [PubMed]
- Pan, D.; Shi, J.; Zhou, H.; Li, N.; Qu, P. Predictive value of basal androgen levels on ongoing pregnancy rates during in vitro fertilization cycles. Gynecol. Endocrinol. 2018, 34, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.; Johnson, I.; Raine-Fenning, N. Endometrial blood flow is impaired in women with polycystic ovarian syndrome who are clinically hyperandrogenic. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2009, 34, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gimeno, P.; Ruíz-Alonso, M.; Blesa, D.; Simón, C. Transcriptomics of the human endometrium. Int. J. Dev. Biol. 2014, 58, 127–137. [Google Scholar] [CrossRef]
- Jiang, N.X.; Li, X.L. The Disorders of Endometrial Receptivity in PCOS and Its Mechanisms. Reprod. Sci. 2022, 29, 2465–2476. [Google Scholar] [CrossRef]
- Mills, L.J.; Gutjahr-Gobell, R.E.; Zaroogian, G.E.; Horowitz, D.B.; Laws, S.C. Modulation of aromatase activity as a mode of action for endocrine disrupting chemicals in a marine fish. Aquat. Toxicol. 2014, 147, 140–150. [Google Scholar] [CrossRef]
- Bulun, S.E.; Sebastian, S.; Takayama, K.; Suzuki, T.; Sasano, H.; Shozu, M. The human CYP19 (aromatase P450) gene: Update on physiologic roles and genomic organization of promoters. J. Steroid Biochem. Mol. Biol. 2003, 86, 219–224. [Google Scholar] [CrossRef]
- Ma, C.X.; Adjei, A.A.; Salavaggione, O.E.; Coronel, J.; Pelleymounter, L.; Wang, L.; Eckloff, B.W.; Schaid, D.; Wieben, E.D.; Adjei, A.A. Human aromatase: Gene resequencing and functional genomics. Cancer Res. 2005, 65, 11071–11082. [Google Scholar] [CrossRef]
- Chen, J.; Shen, S.; Tan, Y.; Xia, D.; Xia, Y.; Cao, Y.; Wang, W.; Wu, X.; Wang, H.; Yi, L. The correlation of aromatase activity and obesity in women with or without polycystic ovary syndrome. J. Ovarian Res. 2015, 8, 1–6. [Google Scholar] [CrossRef]
- Gonzalez, D.; Thackeray, H.; Lewis, P.; Mantani, A.; Brook, N.; Ahuja, K.; Margara, R.; Joels, L.; White, J.; Conlan, R.S. Loss of WT1 expression in the endometrium of infertile PCOS patients: A hyperandrogenic effect? J. Clin. Endocrinol. Metab. 2012, 97, 957–966. [Google Scholar] [CrossRef]
- Apparao, K.; Lovely, L.P.; Gui, Y.; Lininger, R.A.; Lessey, B.A. Elevated endometrial androgen receptor expression in women with polycystic ovarian syndrome. Biol. Reprod. 2002, 66, 297–304. [Google Scholar] [CrossRef]
- Cakmak, H.; Taylor, H.S. Implantation failure: Molecular mechanisms and clinical treatment. Hum. Reprod. Update 2011, 17, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Cermik, D.; Selam, B.; Taylor, H.S. Regulation of HOXA-10 expression by testosterone in vitro and in the endometrium of patients with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2003, 88, 238–243. [Google Scholar] [CrossRef]
- Gregory, C.W.; Wilson, E.M.; Apparao, K.; Lininger, R.A.; Meyer, W.R.; Kowalik, A.; Fritz, M.A.; Lessey, B.A. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. 2002, 87, 2960–2966. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Celik, O.; Dalkilic, S.; Hatirnaz, S.; Celik, N.; Unlu, C.; Otlu, B. Laparoscopic ovarian drilling improves endometrial Homeobox gene expression in PCOS. Reprod. Sci. 2020, 27, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.U.; Ullah, K.; Guo, M.-X.; Pan, H.-T.; Liu, J.; Ren, J.; Jin, L.-Y.; Zhou, Y.-Z.; Cheng, Y.; Sheng, J.-Z. Androgen-induced alterations in endometrial proteins crucial in recurrent miscarriages. Oncotarget 2018, 9, 24627. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, M.H.; Giribabu, N.; Salleh, N. Testosterone decreases the number of implanting embryos, expression of pinopode and L-selectin ligand (MECA-79) in the endometrium of early pregnant rats. Int. J. Environ. Res. Public Health 2020, 17, 2293. [Google Scholar] [CrossRef]
- Brenner, R.M.; Slayden, O.D.; Critchley, H. Anti-proliferative effects of progesterone antagonists in the primate endometrium: A potential role for the androgen receptor. Reproduction 2002, 124, 167–172. [Google Scholar] [CrossRef]
- Younas, K.; Quintela, M.; Thomas, S.; Garcia-Parra, J.; Blake, L.; Whiteland, H.; Bunkheila, A.; Francis, L.W.; Margarit, L.; Gonzalez, D. Delayed endometrial decidualisation in polycystic ovary syndrome; the role of AR-MAGEA11. J. Mol. Med. 2019, 97, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, H.M.; Giribabu, N.; Salleh, N. Testosterone down-regulates expression of αVβ3-integrin, E-cadherin and mucin-1 during uterine receptivity period in rats. Sains Malays. 2018, 47, 2509–2517. [Google Scholar] [CrossRef]
- Mokhtar, M.H.; Giribabu, N.; Salleh, N. Testosterone reduces tight junction complexity and down-regulates expression of claudin-4 and occludin in the endometrium in ovariectomized, sex-steroid replacement rats. In Vivo 2020, 34, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, M.; Jia, W.; Liu, G.; Zhang, J.; Wang, B.; Li, J.; Cui, P.; Li, X.; Lager, S. Hyperandrogenism and insulin resistance modulate gravid uterine and placental ferroptosis in PCOS-like rats. J. Endocrinol. 2020, 246, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Munro, M.G. Uterine polyps, adenomyosis, leiomyomas, and endometrial receptivity. Fertil. Steril. 2019, 111, 629–640. [Google Scholar]
- Vlahos, N.F.; Theodoridis, T.D.; Partsinevelos, G.A. Myomas and adenomyosis: Impact on reproductive outcome. BioMed Res. Int. 2017, 2017, 5926470. [Google Scholar] [CrossRef]
- Lisiecki, M.; Paszkowski, M.; Woźniak, S. Fertility impairment associated with uterine fibroids–a review of literature. Menopause Rev./Przegląd Menopauzalny 2017, 16, 137–140. [Google Scholar] [CrossRef]
- Antero, M.F.; Ayhan, A.; Segars, J.; Shih, I.-M. Pathology and pathogenesis of adenomyosis. Semin. Reprod. Med. 2020, 38, 108–118. [Google Scholar] [CrossRef]
- Nijkang, N.P.; Anderson, L.; Markham, R.; Manconi, F. Endometrial polyps: Pathogenesis, sequelae, and treatment. SAGE Open Med. 2019, 7, 2050312119848247. [Google Scholar] [CrossRef]
- Bosteels, J.; Kasius, J.; Weyers, S.; Broekmans, F.J.; Mol, B.W.J.; D’Hooghe, T.M. Hysteroscopy for treating subfertility associated with suspected major uterine cavity abnormalities Cochrane Database of Systematic Reviews. Cochrane Database Syst. Rev. 2018, 12, CD009461. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Endometrial Receptivity Marker | Receptive Endometrium | Less Receptive Endometrium | Accuracy | References |
|---|---|---|---|---|
| Endometrial thickness | >7 mm | <7 mm | Sensitivity: 99% Specificity: 3% | [88] |
| Endometrial volume | >2 mL | <2 mL | Sensitivity: 93% Specificity: 7% | [88] |
| Endometrial pattern | Presence of triple line pattern | Absence of triple line pattern | Sensitivity: 87% Specificity: 15% | [88] |
| Endometrial blood flow | Presence of flow | Absence of flow | Sensitivity: 100% Specificity: 8% | [88] |
| Endometrial contractions | Contractions absent | Contractions present | Sensitivity: 7% Specificity: 94% | [88] |
| Endometrial receptivity array (ERA) |  |   | Insufficient data available | [88,108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yusuf, A.N.M.; Amri, M.F.; Ugusman, A.; Hamid, A.A.; Wahab, N.A.; Mokhtar, M.H. Hyperandrogenism and Its Possible Effects on Endometrial Receptivity: A Review. Int. J. Mol. Sci. 2023, 24, 12026. https://doi.org/10.3390/ijms241512026
Yusuf ANM, Amri MF, Ugusman A, Hamid AA, Wahab NA, Mokhtar MH. Hyperandrogenism and Its Possible Effects on Endometrial Receptivity: A Review. International Journal of Molecular Sciences. 2023; 24(15):12026. https://doi.org/10.3390/ijms241512026
Chicago/Turabian StyleYusuf, Allia Najmie Muhammad, Mohd Fariz Amri, Azizah Ugusman, Adila A. Hamid, Norhazlina Abdul Wahab, and Mohd Helmy Mokhtar. 2023. "Hyperandrogenism and Its Possible Effects on Endometrial Receptivity: A Review" International Journal of Molecular Sciences 24, no. 15: 12026. https://doi.org/10.3390/ijms241512026
APA StyleYusuf, A. N. M., Amri, M. F., Ugusman, A., Hamid, A. A., Wahab, N. A., & Mokhtar, M. H. (2023). Hyperandrogenism and Its Possible Effects on Endometrial Receptivity: A Review. International Journal of Molecular Sciences, 24(15), 12026. https://doi.org/10.3390/ijms241512026