
The Molecular Mechanisms of Neuroinflammation in Alzheimer’s Disease, the Consequence of Neural Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Functional Consequences of Neural Cell Death in AD
2.1. Pathological Signaling Mechanisms Induced by Apoptosis in the AD Brain
2.2. Pathological Signaling Mechanisms Induced by Autophagic-Dependent Cell Death in the AD Brain
3. Functional Consequences of Neuroinflammation in AD
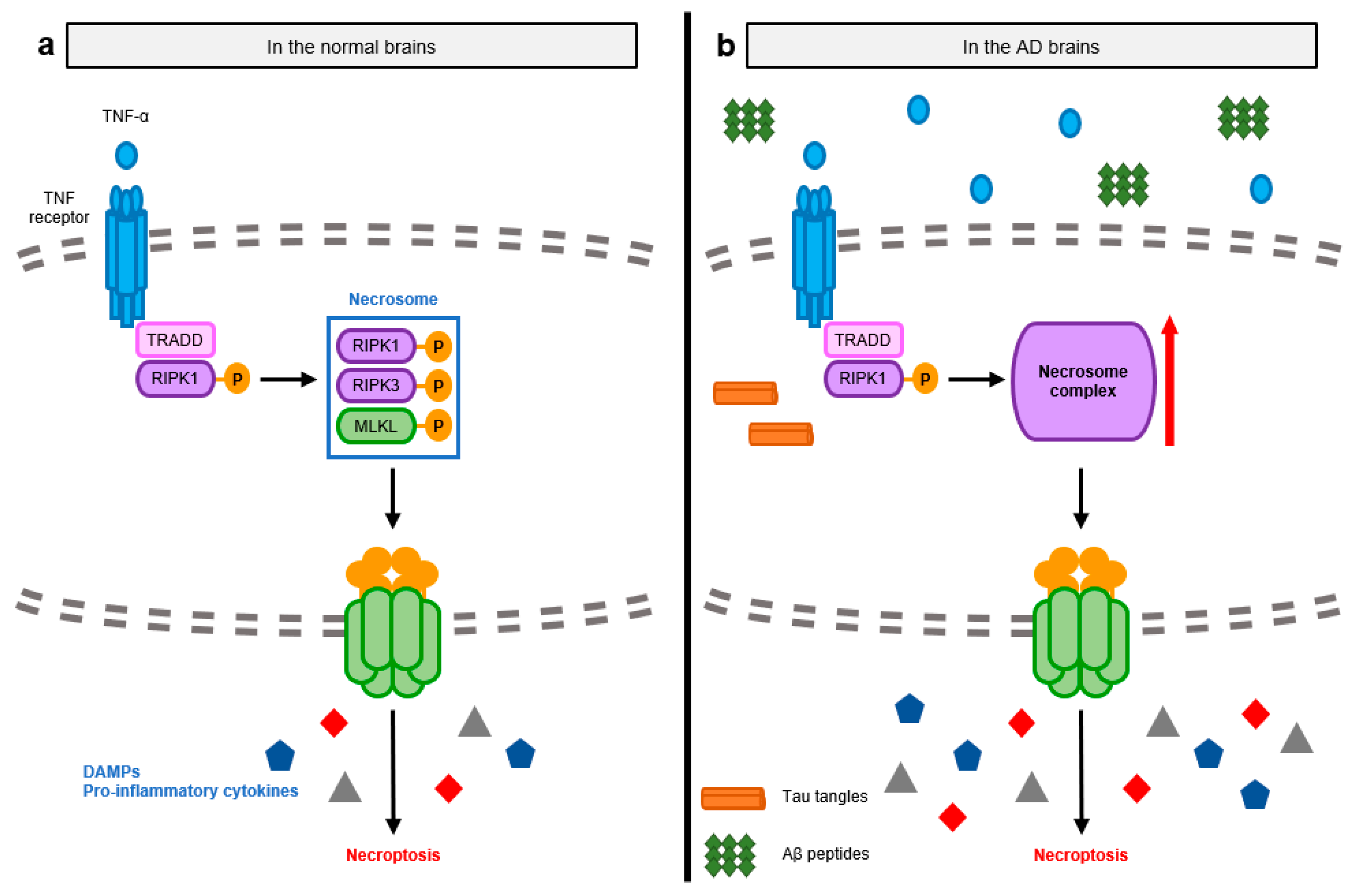
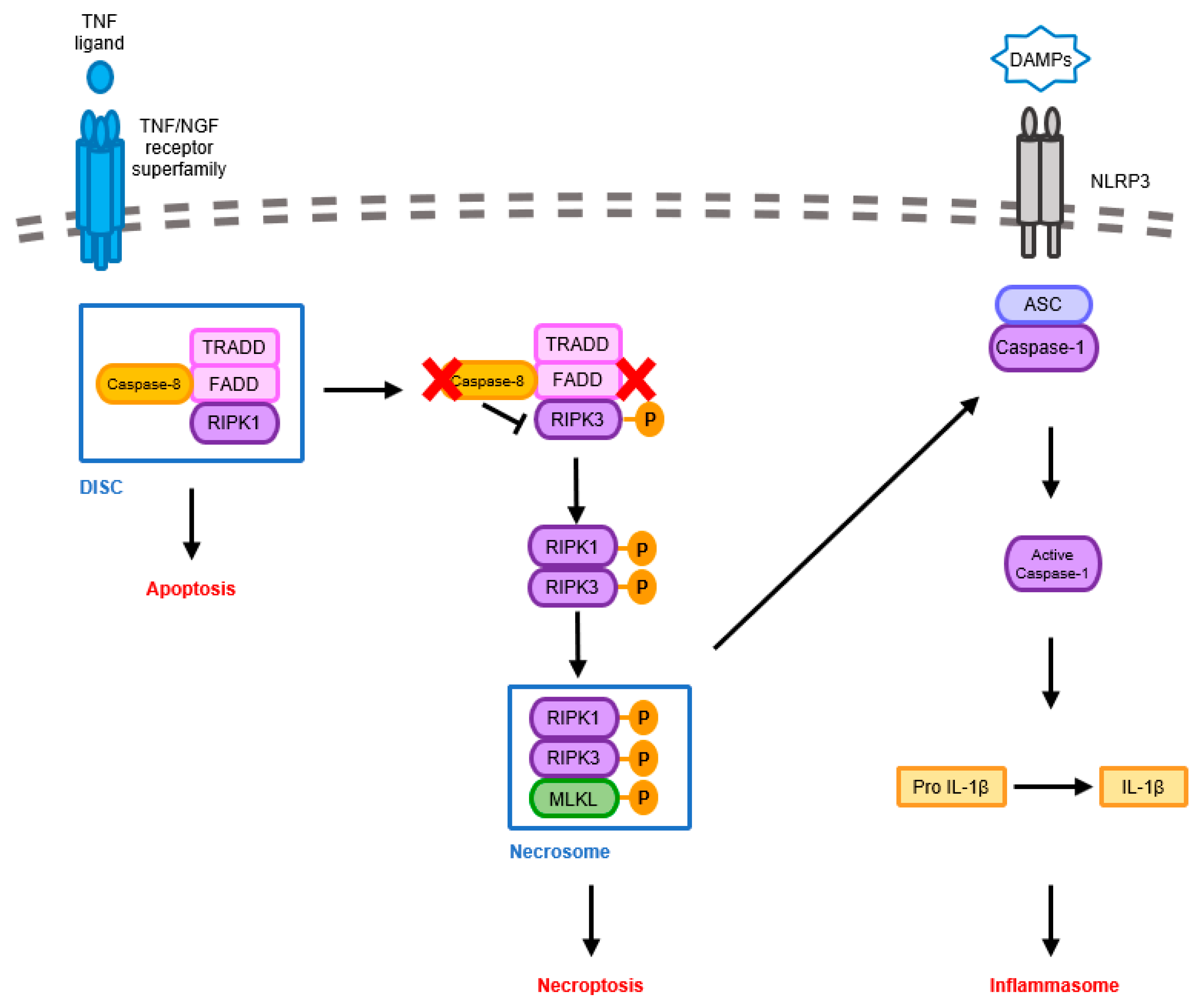
3.1. Pathological Signaling Mechanisms Induced by Necroptosis-Mediated Neuroinflammation in the AD Brain
3.2. Pathological Signaling Mechanisms Induced by Inflammasome-Mediated Neuroinflammation in the AD Brain
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schoonenboom, N.S.; Pijnenburg, Y.A.; Mulder, C.; Rosso, S.M.; Van Elk, E.J.; Van Kamp, G.J.; Van Swieten, J.C.; Scheltens, P. Amyloid beta(1-42) and phosphorylated tau in CSF as markers for early-onset Alzheimer disease. Neurology 2004, 62, 1580–1584. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In Alzheimer’s Disease: Drug Discovery; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, X.; Li, Y.; Xu, L.; Yu, X.; Ge, L.; Li, J.; Zhu, Y.; He, S. Necroptosis mediates TNF-induced toxicity of hippocampal neurons. BioMed Res. Int. 2014, 2014, 290182. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef]
- Luthi, A.U.; Martin, S.J. The CASBAH: A searchable database of caspase substrates. Cell Death Differ. 2007, 14, 641–650. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Smale, G.; Nichols, N.R.; Brady, D.R.; Finch, C.E.; Horton, W.E., Jr. Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol. 1995, 133, 225–230. [Google Scholar] [CrossRef]
- Chui, D.H.; Dobo, E.; Makifuchi, T.; Akiyama, H.; Kawakatsu, S.; Petit, A.; Checler, F.; Araki, W.; Takahashi, K.; Tabira, T. Apoptotic neurons in Alzheimer’s disease frequently show intracellular Abeta42 labeling. J. Alzheimers Dis. 2001, 3, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.S.; Esiri, M.M. Apoptosis-related protein expression in the hippocampus in Alzheimer’s disease. Neurobiol. Aging 1997, 18, 565–571. [Google Scholar] [CrossRef]
- Wu, C.K.; Thal, L.; Pizzo, D.; Hansen, L.; Masliah, E.; Geula, C. Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer’s disease. Exp. Neurol. 2005, 195, 484–496. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Sinha, M.; Ahmad, K.; Khalid, M.; Choi, I. Study of Caspase 8 Inhibition for the Management of Alzheimer’s Disease: A Molecular Docking and Dynamics Simulation. Molecules 2020, 25, 2071. [Google Scholar] [CrossRef]
- Picone, P.; Carrotta, R.; Montana, G.; Nobile, M.R.; San Biagio, P.L.; Di Carlo, M. Abeta oligomers and fibrillar aggregates induce different apoptotic pathways in LAN5 neuroblastoma cell cultures. Biophys. J. 2009, 96, 4200–4211. [Google Scholar] [CrossRef] [PubMed]
- Stoppelkamp, S.; Bell, H.S.; Palacios-Filardo, J.; Shewan, D.A.; Riedel, G.; Platt, B. In vitro modelling of Alzheimer’s disease: Degeneration and cell death induced by viral delivery of amyloid and tau. Exp. Neurol. 2011, 229, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Kienlen-Campard, P.; Miolet, S.; Tasiaux, B.; Octave, J.N. Intracellular amyloid-beta 1-42, but not extracellular soluble amyloid-beta peptides, induces neuronal apoptosis. J. Biol. Chem. 2002, 277, 15666–15670. [Google Scholar] [CrossRef]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [CrossRef]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Selznick, L.A.; Zheng, T.S.; Flavell, R.A.; Rakic, P.; Roth, K.A. Amyloid beta-induced neuronal death is bax-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 2000, 59, 271–279. [Google Scholar] [CrossRef]
- Paradis, E.; Douillard, H.; Koutroumanis, M.; Goodyer, C.; LeBlanc, A. Amyloid beta peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neurons. J. Neurosci. 1996, 16, 7533–7539. [Google Scholar] [CrossRef]
- Kamelia, E.; Islam, A.; Hatta, M.; Kaelan, C.; Patellongi, I. Evaluation of Caspase-3 mRNA Gene Expression Activity in Amyloid Beta-induced Alzheimer’s Disease Rats. J. Med. Sci. 2017, 17, 117–125. [Google Scholar] [CrossRef]
- Kang, H.J.; Yoon, W.J.; Moon, G.J.; Kim, D.Y.; Sohn, S.; Kwon, H.J.; Gwag, B.J. Caspase-3-mediated cleavage of PHF-1 tau during apoptosis irrespective of excitotoxicity and oxidative stress: An implication to Alzheimer’s disease. Neurobiol. Dis. 2005, 18, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Griffin, W.S.; Mattson, M.P. Evidence for caspase-mediated cleavage of AMPA receptor subunits in neuronal apoptosis and Alzheimer’s disease. J. Neurosci. Res. 1999, 57, 315–323. [Google Scholar] [CrossRef]
- Higgins, G.C.; Devenish, R.J.; Beart, P.M.; Nagley, P. Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell. Mol. Life Sci. 2011, 68, 3725–3740. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.C.; Lin, J.W.; Liu, J.M.; Liu, S.H.; Fang, K.M.; Su, C.C.; Hsu, R.J.; Wu, C.C.; Huang, C.F.; Lee, K.I.; et al. Arsenic induces autophagy-dependent apoptosis via Akt inactivation and AMPK activation signaling pathways leading to neuronal cell death. Neurotoxicology 2021, 85, 133–144. [Google Scholar] [CrossRef]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, 215152. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef]
- Wang, H.; Ma, J.; Tan, Y.; Wang, Z.; Sheng, C.; Chen, S.; Ding, J. Amyloid-beta1-42 induces reactive oxygen species-mediated autophagic cell death in U87 and SH-SY5Y cells. J. Alzheimers Dis. 2010, 21, 597–610. [Google Scholar] [CrossRef]
- Ulamek-Koziol, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Saha, A.; Saleem, S.; Paidi, R.K.; Biswas, S.C. BH3-only proteins Puma and Beclin1 regulate autophagic death in neurons in response to Amyloid-beta. Cell Death Discov. 2021, 7, 356. [Google Scholar] [CrossRef]
- Soura, V.; Stewart-Parker, M.; Williams, T.L.; Ratnayaka, A.; Atherton, J.; Gorringe, K.; Tuffin, J.; Darwent, E.; Rambaran, R.; Klein, W.; et al. Visualization of co-localization in Abeta42-administered neuroblastoma cells reveals lysosome damage and autophagosome accumulation related to cell death. Biochem. J. 2012, 441, 579–590. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Theofilas, P.; Ehrenberg, A.J.; Nguy, A.; Thackrey, J.M.; Dunlop, S.; Mejia, M.B.; Alho, A.T.; Paraizo Leite, R.E.; Rodriguez, R.D.; Suemoto, C.K.; et al. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: A quantitative study in humans. Neurobiol. Aging 2018, 61, 1–12. [Google Scholar] [CrossRef]
- Ye, J.; Jiang, Z.; Chen, X.; Liu, M.; Li, J.; Liu, N. The role of autophagy in pro-inflammatory responses of microglia activation via mitochondrial reactive oxygen species in vitro. J. Neurochem. 2017, 142, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Houtman, J.; Freitag, K.; Gimber, N.; Schmoranzer, J.; Heppner, F.L.; Jendrach, M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. 2019, 38, e99430. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.; Whalen, D.M.; Mengistu, M.; Swanson, C.; Quinn, J.G.; Taylor, S.S.; Webster, J.D.; Newton, K.; Shaw, A.S. Kinase domain dimerization drives RIPK3-dependent necroptosis. Sci. Signal. 2018, 11, eaar2188. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef] [PubMed]
- Saleh, D.; Najjar, M.; Zelic, M.; Shah, S.; Nogusa, S.; Polykratis, A.; Paczosa, M.K.; Gough, P.J.; Bertin, J.; Whalen, M.; et al. Kinase Activities of RIPK1 and RIPK3 Can Direct IFN-beta Synthesis Induced by Lipopolysaccharide. J. Immunol. 2017, 198, 4435–4447. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Erlich, Z.; Speir, M.; Zargarian, S.; Baram, N.; Engler, M.; Edry-Botzer, L.; Munitz, A.; Croker, B.A.; Gerlic, M. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J. 2019, 286, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Liang, W.; Ma, Z.; Xu, D.; Cao, S.; Lu, X.; Liu, N.; Shan, B.; Qian, L.; Yuan, J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018, 9, 500. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Ruggiero, E.A.; Crawford, D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J. Alzheimers Dis. 2012, 30, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Shin, H.Y.; Park, J.H.; Koo, S.Y.; Kim, S.M.; Yang, S.H. Fucoxanthin from microalgae Phaeodactylum tricornutum inhibits pro-inflammatory cytokines by regulating both NF-kappaB and NLRP3 inflammasome activation. Sci. Rep. 2021, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef]
- Telegina, D.V.; Suvorov, G.K.; Kozhevnikova, O.S.; Kolosova, N.G. Mechanisms of Neuronal Death in the Cerebral Cortex during Aging and Development of Alzheimer’s Disease-Like Pathology in Rats. Int. J. Mol. Sci. 2019, 20, 5632. [Google Scholar] [CrossRef]
- Koper, M.J.; Van Schoor, E.; Ospitalieri, S.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Tousseyn, T.; Balusu, S.; De Strooper, B.; Thal, D.R. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 2020, 139, 463–484. [Google Scholar] [CrossRef]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 194. [Google Scholar] [CrossRef]
- Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Jin, S.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Oligomeric amyloid β facilitates microglial excitotoxicity by upregulating tumor necrosis factor-α and downregulating excitatory amino acid transporter 2 in astrocytes. Clin. Exp. Neuroimmunol. 2015, 6, 183–190. [Google Scholar] [CrossRef]
- Salvadores, N.; Moreno-Gonzalez, I.; Gamez, N.; Quiroz, G.; Vegas-Gomez, L.; Escandon, M.; Jimenez, S.; Vitorica, J.; Gutierrez, A.; Soto, C.; et al. Abeta oligomers trigger necroptosis-mediated neurodegeneration via microglia activation in Alzheimer’s disease. Acta Neuropathol. Commun. 2022, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Mangiarotti, F.; Angeretti, N.; Lucca, E.; De Simoni, M.G. Beta-amyloid fragment potentiates IL-6 and TNF-alpha secretion by LPS in astrocytes but not in microglia. Cytokine 1997, 9, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef]
- Zhao, M.; Zhou, A.; Xu, L.; Zhang, X. The role of TLR4-mediated PTEN/PI3K/AKT/NF-kappaB signaling pathway in neuroinflammation in hippocampal neurons. Neuroscience 2014, 269, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mao, J.; Zhang, Y.; Zhou, A. Role of Toll-like receptor 4 in inflammatory reactions of hippocampal neurons. Neural. Regen. Res. 2013, 8, 1465–1472. [Google Scholar] [CrossRef]
- Cao, Z.; Xiong, J.; Takeuchi, M.; Kurama, T.; Goeddel, D.V. TRAF6 is a signal transducer for interleukin-1. Nature 1996, 383, 443–446. [Google Scholar] [CrossRef]
- Dick, M.S.; Sborgi, L.; Ruhl, S.; Hiller, S.; Broz, P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 2016, 7, 11929. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Terai, K.; Matsuo, A.; McGeer, P.L. Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 1996, 735, 159–168. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk with NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-kappaB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Lee, S.; Cho, H.J.; Ryu, J.H. Innate Immunity and Cell Death in Alzheimer’s Disease. ASN Neuro. 2021, 13, 17590914211051908. [Google Scholar] [CrossRef]
- Rajesh, Y.; Kanneganti, T.D. Innate Immune Cell Death in Neuroinflammation and Alzheimer’s Disease. Cells 2022, 11, 1885. [Google Scholar] [CrossRef]
- Kumar, S.; Budhathoki, S.; Oliveira, C.B.; Kahle, A.D.; Calhan, O.Y.; Lukens, J.R.; Deppmann, C.D. Role of the caspase-8/RIPK3 axis in Alzheimer’s disease pathogenesis and Abeta-induced NLRP3 inflammasome activation. JCI Insight 2023, 8, e157433. [Google Scholar] [CrossRef]
- Li, S.; Qu, L.; Wang, X.; Kong, L. Novel insights into RIPK1 as a promising target for future Alzheimer’s disease treatment. Pharmacol. Ther. 2022, 231, 107979. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dong, Y.; Chu, Y.; Guo, Y.; Li, L. The mechanisms of ferroptosis and its role in alzheimer’s disease. Front. Mol. Biosci. 2022, 9, 965064. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yang, Y.; Guan, Q.; Zhang, X.; Shen, H.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Guo, L.; et al. New mechanism of nerve injury in Alzheimer’s disease: Beta-amyloid-induced neuronal pyroptosis. J. Cell. Mol. Med. 2020, 24, 8078–8090. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Nam, Y.W.; Kim, S.; Oh, D.B.; Song, J. Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators. Exp. Mol. Med. 2021, 53, 1007–1017. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.-B.; Kwon, S.; Kim, J.-H.; Ahn, N.-H.; Lee, J.-H.; Yang, S.-H. The Molecular Mechanisms of Neuroinflammation in Alzheimer’s Disease, the Consequence of Neural Cell Death. Int. J. Mol. Sci. 2023, 24, 11757. https://doi.org/10.3390/ijms241411757
Choi S-B, Kwon S, Kim J-H, Ahn N-H, Lee J-H, Yang S-H. The Molecular Mechanisms of Neuroinflammation in Alzheimer’s Disease, the Consequence of Neural Cell Death. International Journal of Molecular Sciences. 2023; 24(14):11757. https://doi.org/10.3390/ijms241411757
Chicago/Turabian StyleChoi, Su-Bin, Sehee Kwon, Ji-Hye Kim, Na-Hyun Ahn, Joo-Hee Lee, and Seung-Hoon Yang. 2023. "The Molecular Mechanisms of Neuroinflammation in Alzheimer’s Disease, the Consequence of Neural Cell Death" International Journal of Molecular Sciences 24, no. 14: 11757. https://doi.org/10.3390/ijms241411757
APA StyleChoi, S.-B., Kwon, S., Kim, J.-H., Ahn, N.-H., Lee, J.-H., & Yang, S.-H. (2023). The Molecular Mechanisms of Neuroinflammation in Alzheimer’s Disease, the Consequence of Neural Cell Death. International Journal of Molecular Sciences, 24(14), 11757. https://doi.org/10.3390/ijms241411757