
The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer’s Disease Mouse Model

 ,
,  , ,
, ,  ,
,  ,
,  ,
, 
Abstract
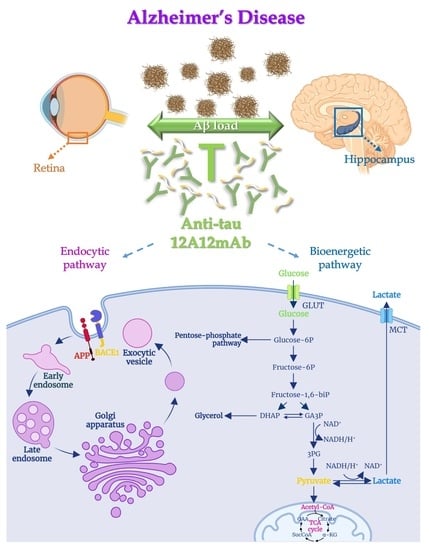
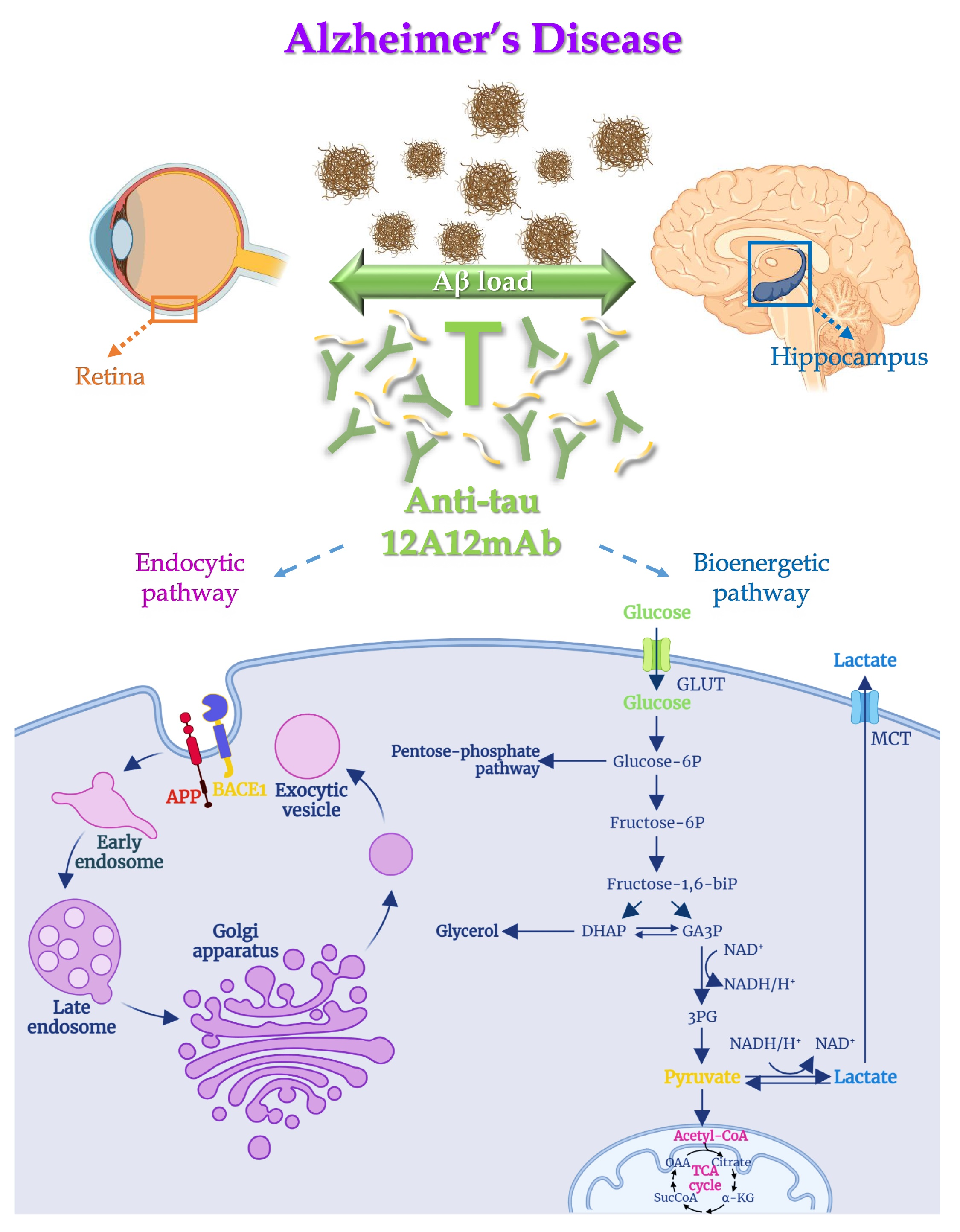{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
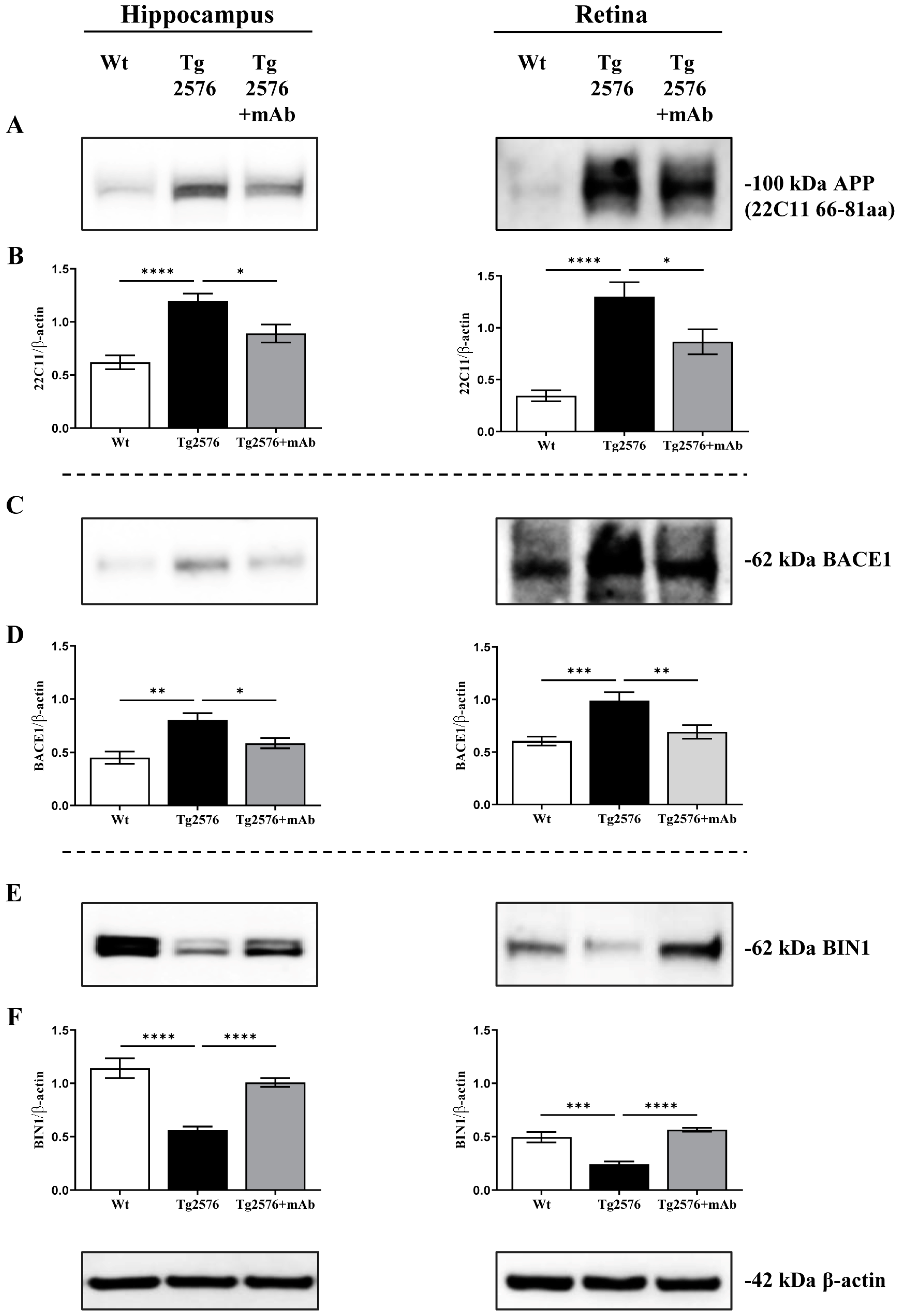
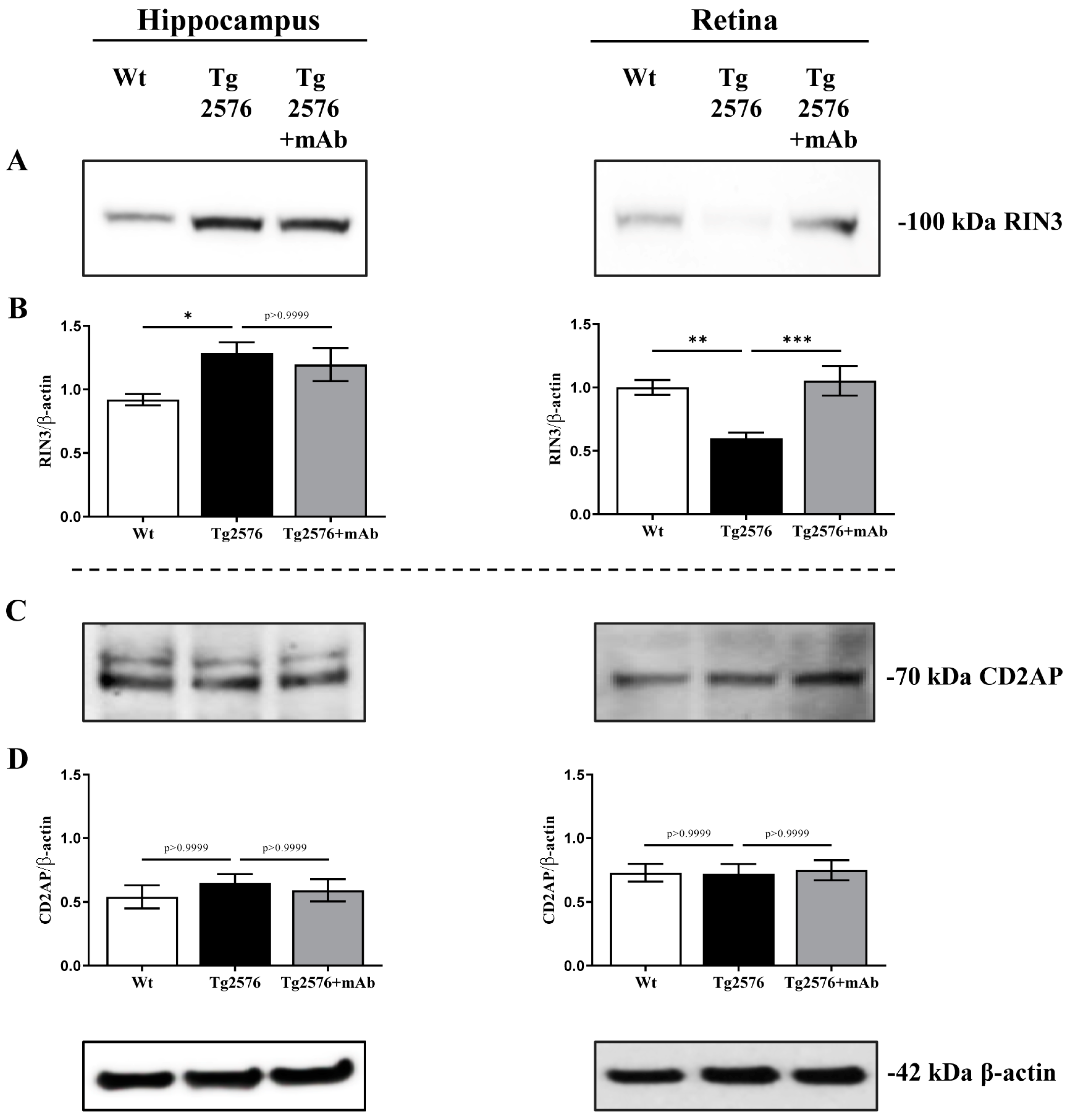
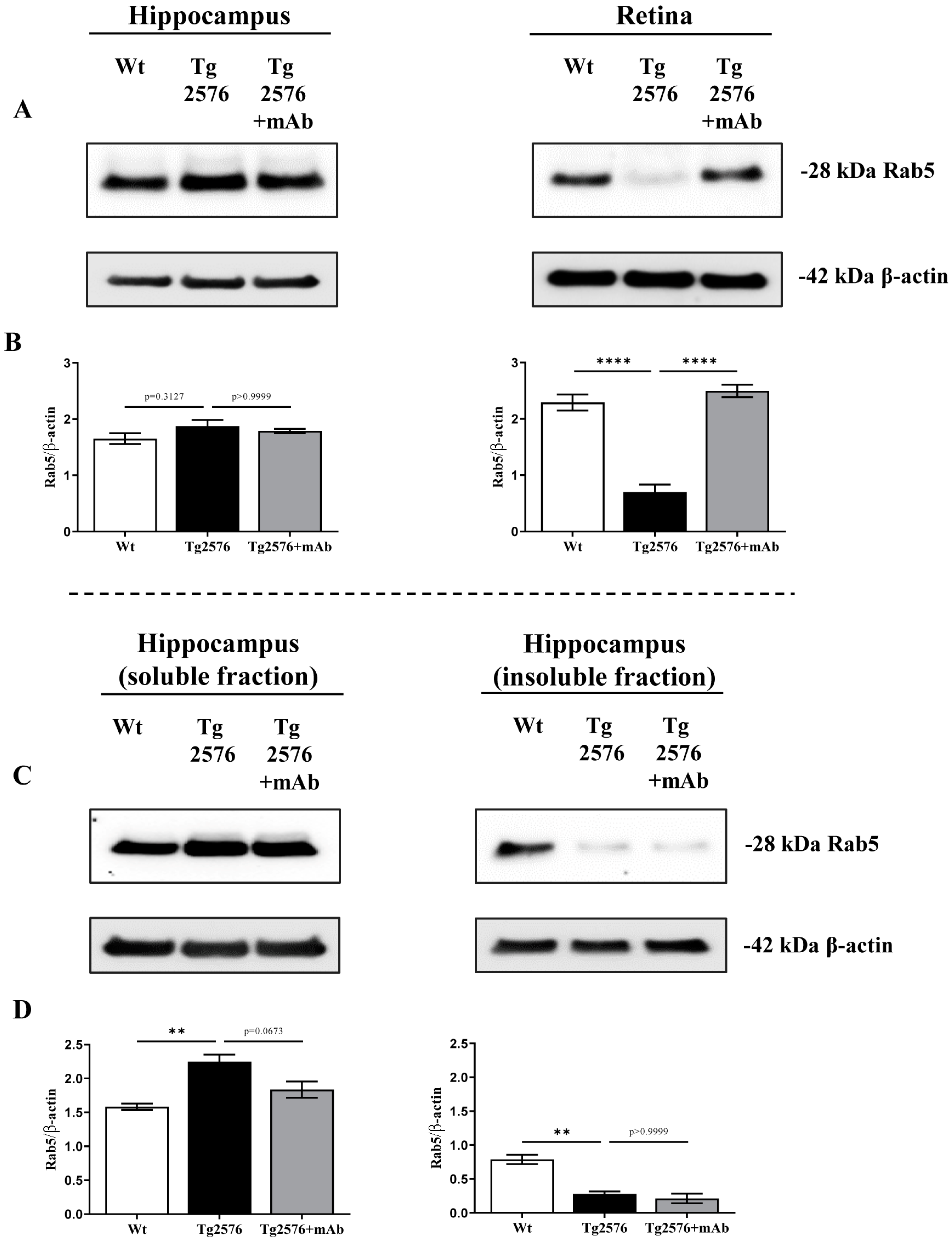
2.1. 12A12mAb Immunization Antagonizes the BACE1-Initiated Amyloidogenic Processing of APP by Altering the Protein Expression of Neuron-Specific BIN1 and RIN3 Endocytic Adaptors, Both in Hippocampus and Retina from Tg2576 AD Mice
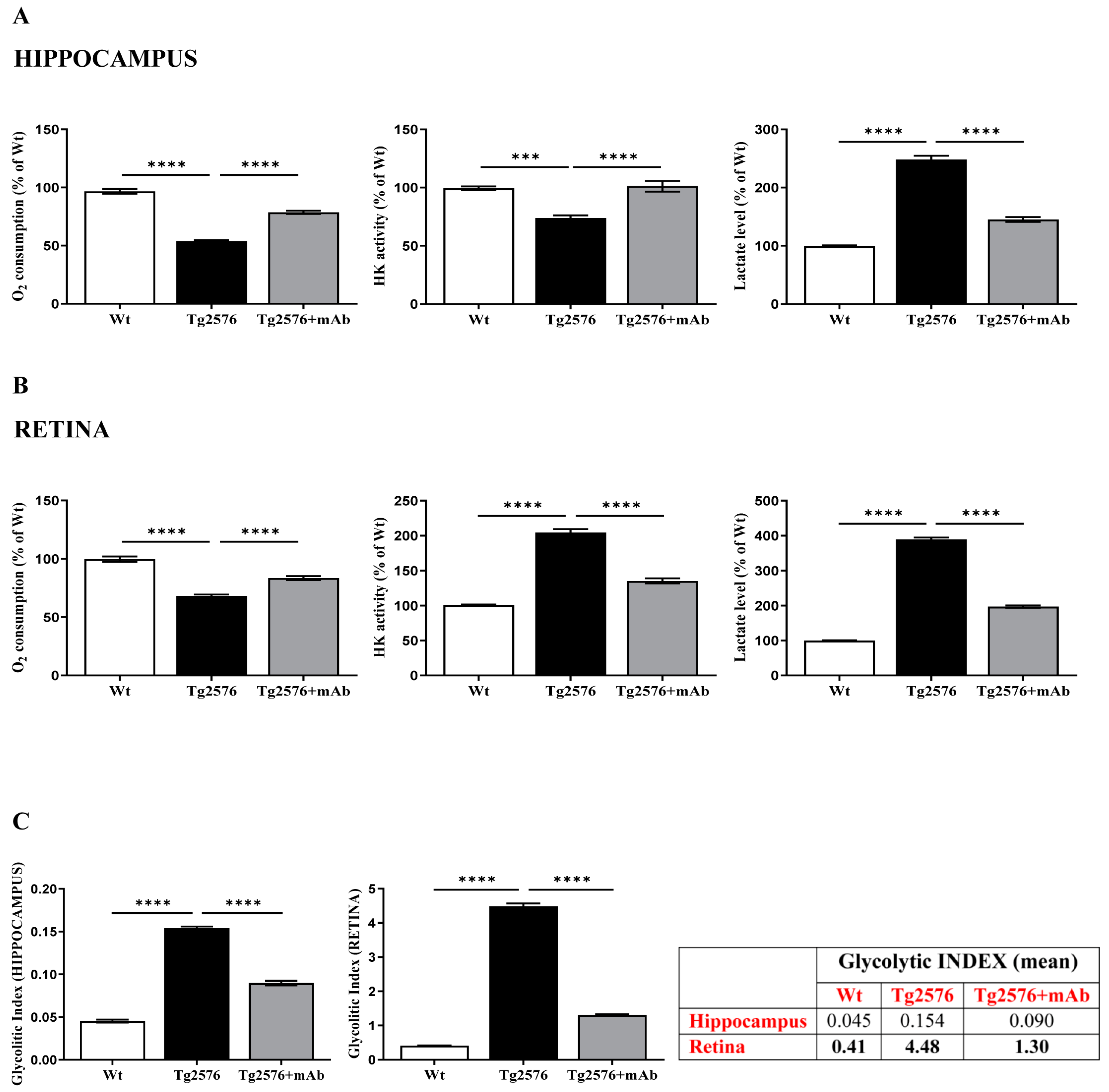
2.2. Energetic Alterations of Glucose Utilization That Are Strictly Linked with the Aβ Generation Are Recovered by 12A12mAb Treatment Both in Retina and Hippocampus in Concomitance with Its Local Anti-Amyloidogenic Action
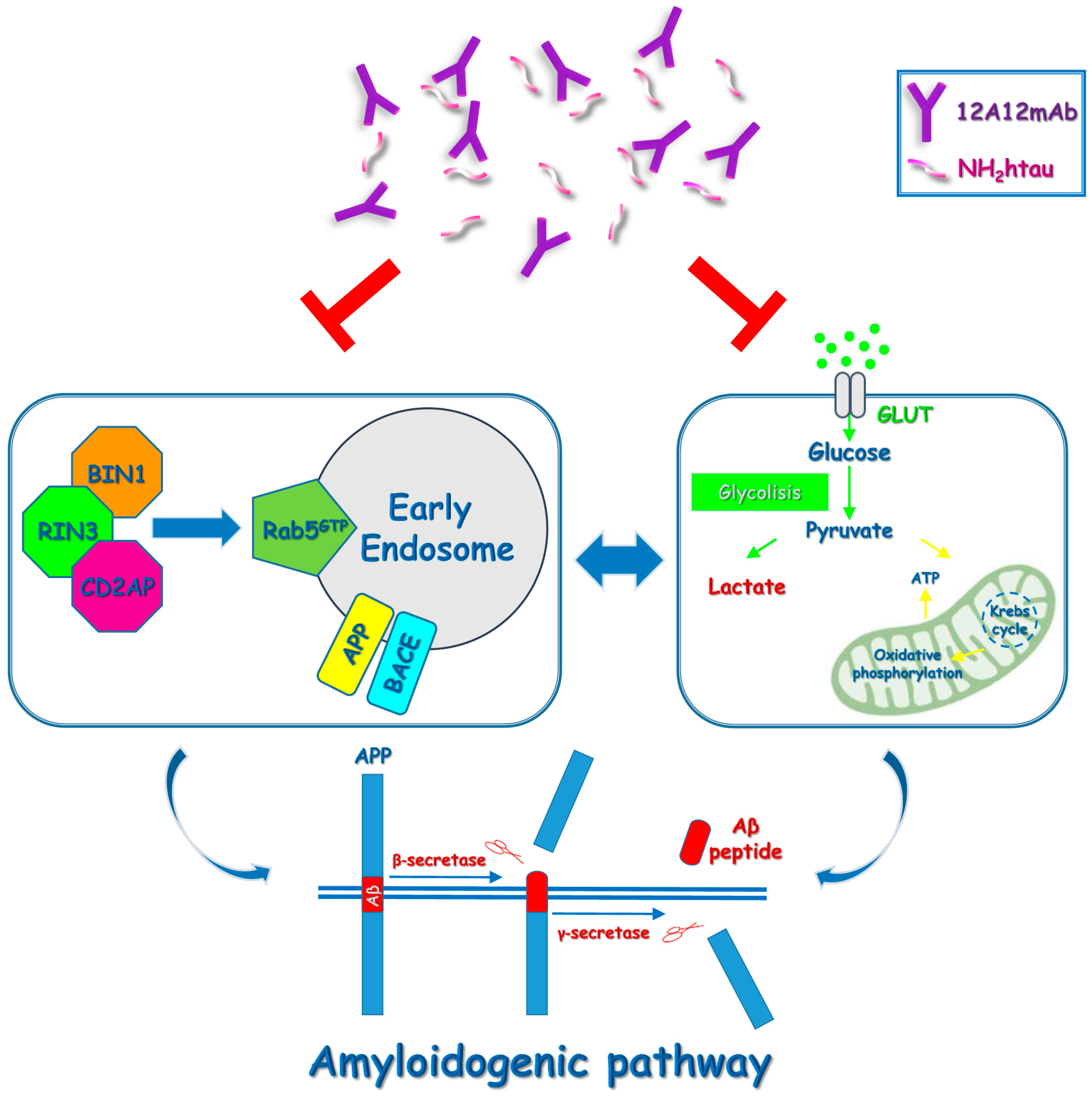
3. Discussion
4. Materials and Methods
4.1. Animals and Ethical Approval
4.2. Immunization Scheme
4.3. Tissue Collection, Harvesting and Preparation
4.4. Western Blot Analysis and Semi-Quantitative Densitometry
4.5. Hematoxylin and Eosin (H/E) Staining
4.6. Glucose Metabolism Analysis
4.6.1. Tissue Homogenate Preparation
4.6.2. Enzymatic Activity Measurements
4.6.3. L-Lactate Production Measurements
4.6.4. Determination of the Glycolytic Index
4.7. Data Management and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain: From eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, D.; Votruba, M. Can the retina be used to diagnose and plot the progression of Alzheimer’s disease? Acta Ophthalmol. 2017, 95, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.H.; Li, Q.X.; He, Z.; Vingrys, A.J.; Wong, V.H.Y.; Currier, N.; Mullen, J.; Bui, B.V.; Nguyen, C.T.O. The eye as a bi-omarker for Alzheimer’s disease. Front. Neurosci. 2016, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011, 54, S204–S217. [Google Scholar] [CrossRef]
- Chiquita, S.; Rodrigues-Neves, A.C.; Baptista, F.I.; Carecho, R.; Moreira, P.I.; Castelo-Branco, M.; Ambrósio, A.F. The Retina as a Window or Mirror of the Brain Changes Detected in Alzheimer’s Disease: Critical Aspects to Unravel. Mol. Neurobiol. 2019, 56, 5416–5435. [Google Scholar] [CrossRef]
- Romaus-Sanjurjo, D.R.; Regueiro, U.; López-Lopez, M.L.; Vázquez-Vazquez, L.V.; Ouro, A.; Lema, I.; Sobrino, T. Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 2486. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, L.; Shen, Y. The retina: A window in which to view the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101590. [Google Scholar] [CrossRef]
- Liao, C.; Xu, J.; Chen, Y.; Ip, N.Y. Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers. Biomolecules 2021, 11, 1215. [Google Scholar] [CrossRef]
- Reed, B.T.; Behar-Cohen, F.; Krantic, S. Seeing Early Signs of Alzheimer’s Disease Through the Lens of the Eye. Curr. Alzheimer Res. 2017, 14, 6–17. [Google Scholar] [CrossRef]
- Jindal, V. Interconnection between brain and retinal neurodegenerations. Mol. Neurobiol. 2015, 51, 885–892. [Google Scholar] [CrossRef]
- Hart, N.J.; Koronyo, Y.; Black, K.L.; Koronyo-Hamaoui, M. Ocular indicators of Alzheimer’s: Exploring disease in the retina. Acta Neuropathol. 2016, 132, 767–787. [Google Scholar] [CrossRef] [PubMed]
- Kusne, Y.; Wolf, A.B.; Townley, K.; Conway, M.; Peyman, G.A. Visual system manifestations of Alzheimer’s disease. Acta Ophthalmol. 2017, 95, e668–e676. [Google Scholar] [CrossRef] [PubMed]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, C.; Cerri, E.; Fabiani, C.; Capsoni, S.; Cattaneo, A.; Domenici, L. The retina as a window to early dysfunctions of Alzheimer’s disease following studies with a 5xFAD mouse model. Neurobiol. Aging 2018, 67, 181–188. [Google Scholar] [CrossRef]
- Latina, V.; Giacovazzo, G.; Cordella, F.; Balzamino, B.O.; Micera, A.; Varano, M.; Marchetti, C.; Malerba, F.; Florio, R.; Ercole Bruni, B.; et al. Systemic delivery of a specific antibody targeting the pathological N-terminal truncated tau peptide reduces retinal degeneration in a mouse model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 38. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Pogue, A.; Bhattacharjee, S.; Lukiw, W.J. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. Neuroreport 2011, 22, 623–627. [Google Scholar] [CrossRef]
- den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147. [Google Scholar] [CrossRef]
- Hadoux, X.; Hui, F.; Lim, J.K.H.; Masters, C.L.; Pébay, A.; Chevalier, S.; Ha, J.; Loi, S.; Fowler, C.J.; Rowe, C.; et al. Non-invasive in vivo hyperspectral imaging of the retina for potential biomarker use in Alzheimer’s disease. Nat. Commun. 2019, 10, 4227. [Google Scholar] [CrossRef]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, e93621. [Google Scholar] [CrossRef]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef]
- Schön, C.; Hoffmann, N.A.; Ochs, S.M.; Burgold, S.; Filser, S.; Steinbach, S.; Seeliger, M.W.; Arzberger, T.; Goedert, M.; Kretzschmar, H.A.; et al. Long-term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLoS ONE 2012, 7, e53547. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Byman, E.; Netherlands Brain Bank; Wennström, M. Levels of Retinal Amyloid-β Correlate with Levels of Retinal IAPP and Hippocampal Amyloid-β in Neuropathologically Evaluated Individuals. J. Alzheimers Dis. 2020, 73, 1201–1209. [Google Scholar] [CrossRef]
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Fuchs, D.T.; Sheyn, J.; Black, K.L.; Mirzaei, N.; Koronyo-Hamaoui, M. Retinal Vascu-lopathy in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 731614. [Google Scholar] [CrossRef]
- Tsai, Y.; Lu, B.; Ljubimov, A.V.; Girman, S.; Ross-Cisneros, F.N.; Sadun, A.A.; Svendsen, C.N.; Cohen, R.M.; Wang, S. Ocular changes in TgF344-AD rat model of Alzheimer’s disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.B.; Chitranshi, N.; den Haan, J.; Mirzaei, M.; You, Y.; Lim, J.K.; Basavarajappa, D.; Godinez, A.; Di Angelantonio, S.; Sachdev, P. Retinal changes in Alzheimer’s disease- integrated prospects of imaging, functional and molecular advances. Prog. Retin. Eye Res. 2021, 82, 100899. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, N.; Shi, H.; Oviatt, M.; Doustar, J.; Rentsendorj, A.; Fuchs, D.T.; Sheyn, J.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Alzheimer’s Retinopathy: Seeing Disease in the Eyes. Front. Neurosci. 2020, 14, 921. [Google Scholar] [CrossRef] [PubMed]
- Albers, M.W.; Gilmore, G.C.; Kaye, J.; Murphy, C.; Wingfield, A.; Bennett, D.A.; Boxer, A.L.; Buchman, A.S.; Cruickshanks, K.J.; Devanand, D.P.; et al. At the interface of sensory and motor dysfunctions and Alzheimer’s disease. Alzheimers Dement. 2015, 11, 70–98. [Google Scholar] [CrossRef]
- Glosser, G.; Gallo, J.; Duda, N.; de Vries, J.J.; Clark, C.M.; Grossman, M. Visual perceptual functions predict instrumental ac-tivities of daily living in patients with dementia. Neuropsychiatry Neuropsychol. Behav. Neurol. 2002, 15, 198–206. [Google Scholar]
- Rizzo, M.; Anderson, S.W.; Dawson, J.; Nawrot, M. Vision and cognition in Alzheimer’s disease. Neuropsychologia 2000, 38, 1157–1169. [Google Scholar] [CrossRef]
- Tippett, L.J.; Blackwood, K.; Farah, M.J. Visual object and face processing in mild-to-moderate Alzheimer’s disease: From segmentation to imagination. Neuropsychologia 2003, 41, 453–468. [Google Scholar] [CrossRef]
- Cui, J.G.; Hill, J.M.; Zhao, Y.; Lukiw, W.J. Expression of inflammatory genes in the primary visual cortex of late-stage Alzheimer’s disease. Neuroreport 2007, 18, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Dehabadi, M.H.; Davis, B.M.; Wong, T.K.; Cordeiro, M.F. Retinal manifestations of Alzheimer’s disease. Neurodegener. Dis. Manag. 2014, 4, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Tzekov, R.; Mullan, M. Vision function abnormalities in Alzheimer disease. Surv. Ophthalmol. 2014, 59, 414–433. [Google Scholar] [CrossRef]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.M.; Clement, C.; Sambamurti, K.; Dua, P.; Lukiw, W.J. Beta-amyloid precursor protein (βAPP) processing in Alzheimer’s disease (AD) and age-related macular megeneration (AMD). Mol. Neurobiol. 2015, 52, 533–544. [Google Scholar] [CrossRef]
- Hill, J.M.; Dua, P.; Clement, C.; Lukiw, W.J. An evaluation of progressive amyloidogenic and pro-inflammatory change in the primary visual cortex and retina in Alzheimer’s disease (AD). Front. Neurosci. 2014, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Marwick, B.; Yu, H.; Fadavi, H.; Tavakoli, M. Ophthalmic Biomarkers for Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2021, 13, 720167. [Google Scholar] [CrossRef]
- Ge, Y.J.; Xu, W.; Ou, Y.N.; Qu, Y.; Ma, Y.H.; Huang, Y.Y.; Shen, X.N.; Chen, S.D.; Tan, L.; Zhao, Q.H.; et al. Retinal biomarkers in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Ageing Res. Rev. 2021, 69, 101361. [Google Scholar] [CrossRef]
- Lemos, R.; Santana, I.; Caetano, G.; Bernardino, I.; Morais, R.; Farivar, R.; Castelo-Branco, M. Three-Dimensional Face Recog-nition in Mild Cognitive Impairment: A Psychophysical and Structural MR Study. J. Int. Neuropsychol. Soc. 2016, 22, 744–754. [Google Scholar] [CrossRef]
- Graewe, B.; Lemos, R.; Ferreira, C.; Santana, I.; Farivar, R.; De Weerd, P.; Castelo-Branco, M. Impaired processing of 3D mo-tion-defined faces in mild cognitive impairment and healthy aging: An fMRI study. Cereb. Cortex 2013, 23, 2489–2499. [Google Scholar] [CrossRef]
- Ngolab, J.; Honma, P.; Rissman, R.A. Reflections on the Utility of the Retina as a Biomarker for Alzheimer’s Disease: A Literature Review. Neurol. Ther. 2019, 8, 57–72. [Google Scholar] [CrossRef]
- Koronyo, Y.; Salumbides, B.C.; Black, K.L.; Koronyo-Hamaoui, M. Alzheimer’s disease in the retina: Imaging retinal aβ plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 2012, 10, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Blazes, M.; Lee, C.S. Understanding the Brain through Aging Eyes. Adv. Geriatr. Med. Res. 2021, 3, e210008. [Google Scholar] [CrossRef]
- Doustar, J.; Rentsendorj, A.; Torbati, T.; Regis, G.C.; Fuchs, D.T.; Sheyn, J.; Mirzaei, N.; Graham, S.L.; Shah, P.K.; Mastali, M. Parallels between retinal and brain pathology and response to immunotherapy in old, late-stage Alzheimer’s disease mouse models. Aging Cell 2020, 19, e13246. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Borreca, A.; Latina, V.; Giacovazzo, G.; Pignataro, A.; Krashia, P.; Natale, F.; Cocco, S.; Rinaudo, M.; Malerba, F.; et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun. 2020, 2, fcaa039. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diagnostic and therapeutic approach. Biochim. Biophys. Acta Mol. Basis. Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Latina, V.; Calissano, P. A long story for a short peptide: Therapeutic efficacy of a cleavage-specific tau antibody. Neural. Regen. Res. 2021, 16, 2417–2419. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Alldred, M.J.; Counts, S.E.; Cataldo, A.M.; Neve, R.L.; Jiang, Y.; Wuu, J.; Chao, M.V.; Mufson, E.J.; Nixon, R.A.; et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 2010, 68, 885–893. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef]
- Nixon, R.A. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 2005, 26, 373–382. [Google Scholar] [CrossRef]
- Choi, J.H.; Kaur, G.; Mazzella, M.J.; Morales-Corraliza, J.; Levy, E.; Mathews, P.M. Early endosomal abnormalities and cholinergic neurondegeneration in amyloid-beta protein precursor transgenic mice. J. Alzheimers Dis. 2013, 34, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Fang, F.; Ding, J.; Wu, C. Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease. Traffic 2018, 19, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Perdigão, C.; Barata, M.A.; Araújo, M.N.; Mirfakhar, F.S.; Castanheira, J.; Guimas Almeida, C. Intracellular Trafficking Mech-anisms of Synaptic Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Houben, S.; Homa, M.; de Fisenne, M.A.; Potier, M.C.; Erneux, C.; Brion, J.P.; Leroy, K. Alzheimer’s Disease: Tau Pathology and Dysfunction of Endocytosis. Front. Mol. Neurosci. 2021, 13, 583755. [Google Scholar] [CrossRef]
- Thomas, R.S.; Lelos, M.J.; Good, M.A.; Kidd, E.J. Clathrin-mediated endocytic proteins are upregulated in the cortex of the Tg2576 mouse model of Alzheimer’s disease-like amyloid pathology. Biochem. Biophys. Res. Commun. 2011, 415, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Tacal, O. Trafficking and proteolytic processing of amyloid precursor protein and secretases in Alzheimer’s disease development: An up-to-date review. Eur. J. Pharmacol. 2019, 856, 172415. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef]
- .Mañucat-Tan, N.B.; Saadipour, K.; Wang, Y.J.; Bobrovskaya, L.; Zhou, X.F. Cellular Trafficking of Amyloid Precursor Protein in Amyloidogenesis Physiological and Pathological Significance. Mol. Neurobiol. 2019, 56, 812–830. [Google Scholar] [CrossRef]
- Tan, J.Z.A.; Gleeson, P.A. The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimer’s disease. Biochim. Biophys. Acta Biomembr. 2019, 1861, 697–712. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Roy, S. The physical approximation of APP and BACE-1: A key event in alzheimer’s disease pathogenesis. Dev. Neurobiol. 2018, 78, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Chia, P.Z.; Gleeson, P.A. Intracellular trafficking of the β-secretase and processing of amyloid precursor protein. IUBMB Life 2011, 63, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Szabo, M.P.; Mishra, S.; Knupp, A.; Young, J.E. The role of Alzheimer’s disease risk genes in endolysosomal pathways. Neurobiol. Dis. 2022, 162, 105576. [Google Scholar] [CrossRef]
- Tan, L.; Wang, X.; Ni, Z.F.; Zhu, X.; Wu, W.; Zhu, L.Q.; Liu, D. A systematic analysis of genomic changes in Tg2576 mice. Mol. Neurobiol. 2013, 47, 883–891. [Google Scholar] [CrossRef]
- Miyagawa, T.; Ebinuma, I.; Morohashi, Y.; Hori, Y.; Chang, M.Y.; Hattori, H.; Maehara, T.; Yokoshima, S.; Fukuyama, T.; Tsuji, S.; et al. BIN1 regulates BACE1 intracellular trafficking and amyloid-β production. Hum. Mol. Genet. 2016, 25, 2948–2958. [Google Scholar] [CrossRef]
- Ubelmann, F.; Burrinha, T.; Salavessa, L.; Gomes, R.; Ferreira, C.; Moreno, N.; Guimas Almeida, C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017, 18, 102–122. [Google Scholar] [CrossRef]
- Gao, P.; Ye, L.; Cheng, H.; Li, H. The Mechanistic Role of Bridging Integrator 1 (BIN1) in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2021, 41, 1431–1440. [Google Scholar] [CrossRef]
- Lasorsa, A.; Malki, I.; Cantrelle, F.X.; Merzougui, H.; Boll, E.; Lambert, J.C.; Landrieu, I. Structural Basis of Tau Interaction With BIN1 and Regulation by Tau Phosphorylation. Front. Mol. Neurosci. 2018, 11, 421. [Google Scholar] [CrossRef]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Glennon, E.B.; Lau, D.H.W.; Gabriele, R.M.C.; Taylor, M.F.; Troakes, C.; Opie-Martin, S.; Elliott, C.; Killick, R.; Hanger, D.P.; Perez-Nievas, B.G.; et al. Bridging Integrator-1 protein loss in Alzheimer’s disease promotes synaptic tau accumulation and disrupts tau release. Brain Commun. 2020, 2, fcaa011. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Simmons, D.; Sorkin, A. Differential subcellular distribution of endosomal compartments and the dopamine trans-porter in dopaminergic neurons. Mol. Cell. Neurosci. 2011, 46, 148–158. [Google Scholar] [CrossRef]
- Del Prete, D.; Lombino, F.; Liu, X.; D’Adamio, L. APP Is Cleaved by Bace1 in Pre-Synaptic Vesicles and Establishes a Pre-Synaptic Interactome, via Its Intracellular Domain, with Molecular Complexes that Regulate Pre-Synaptic Vesicles Functions. PLoS ONE 2014, 9, e108576. [Google Scholar] [CrossRef]
- Zhou, Y.; Hayashi, I.; Wong, J.; Tugusheva, K.; Renger, J.J.; Zerbinatti, C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLoS ONE 2014, 9, e103187. [Google Scholar] [CrossRef]
- Taga, M.; Petyuk, V.A.; White, C.; Marsh, G.; Ma, Y.; Klein, H.U.; Connor, S.M.; Kroshilina, A.; Yung, C.J.; Khairallah, A.; et al. BIN1 protein isoforms are differentially expressed in astrocytes, neurons, and microglia: Neuronal and astrocyte BIN1 are implicated in tau pathology. Mol. Neurodegener. 2020, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Barral, S.; Bird, T.; Goate, A.; Farlow, M.R.; Diaz-Arrastia, R.; Bennett, D.A.; Graff-Radford, N.; Boeve, B.F.; Sweet, R.A.; Stern, Y.; et al. Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology 2012, 78, 1464–1471. [Google Scholar] [CrossRef]
- Karch, C.M.; Jeng, A.T.; Nowotny, P.; Cady, J.; Cruchaga, C.; Goate, A.M. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE 2012, 7, e50976. [Google Scholar] [CrossRef]
- Marques-Coelho, D.; Iohan, L.D.C.C.; Melo de Farias, A.R.; Flaig, A.; Brainbank Neuro–CEB Neuropathology Network; Lambert, J.C.; Costa, M.R. Differential transcript usage unravels gene expression alterations in Alzheimer’s disease human brains. NPJ Aging Mech. Dis. 2021, 7, 2. [Google Scholar] [CrossRef]
- Glennon, E.B.; Whitehouse, I.J.; Miners, J.S.; Kehoe, P.G.; Love, S.; Kellett, K.A.; Hooper, N.M. BIN1 is decreased in sporadic but not familial Alzheimer’s disease or in aging. PLoS ONE 2013, 8, e78806. [Google Scholar] [CrossRef]
- Crotti, A.; Sait, H.R.; McAvoy, K.M.; Estrada, K.; Ergun, A.; Szak, S.; Marsh, G.; Jandreski, L.; Peterson, M.; Reynolds, T.L.; et al. BIN1 favors the spreading of Tau via extracellular vesicles. Sci. Rep. 2019, 9, 9477. [Google Scholar] [CrossRef] [PubMed]
- McAvoy, K.M.; Rajamohamed Sait, H.; Marsh, G.; Peterson, M.; Reynolds, T.L.; Gagnon, J.; Geisler, S.; Leach, P.; Roberts, C.; Cahir-McFarland, E.; et al. Cell-autonomous and non-cell autonomous effects of neuronal BIN1 loss in vivo. PLoS ONE 2019, 14, e0220125. [Google Scholar] [CrossRef]
- Borreca, A.; Gironi, K.; Amadoro, G.; Ammassari-Teule, M. Opposite Dysregulation of Fragile-X Mental Retardation Protein and Heteronuclear Ribonucleoprotein C Protein Associates with Enhanced APP Translation in Alzheimer Disease. Mol. Neurobiol. 2016, 53, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Zohar, O.; Pick, C.G.; Cavallaro, S.; Chapman, J.; Katzav, A.; Milman, A.; Alkon, D.L. Age-dependent differential expression of BACE splice variants in brain regions of tg2576 mice. Neurobiol. Aging 2005, 26, 1167–1175. [Google Scholar] [CrossRef]
- Velliquette, R.A.; O’Connor, T.; Vassar, R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005, 25, 10874–10883. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef]
- Holsinger, R.M.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased expression of the amyloid precursor be-ta-secretase in Alzheimer’s disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar] [CrossRef]
- Tyler, S.J.; Dawbarn, D.; Wilcock, G.K.; Allen, S.J. alpha- and beta-secretase: Profound changes in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 299, 373–376. [Google Scholar] [CrossRef]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated be-ta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef]
- Shen, R.; Murphy, C.J.; Xu, X.; Hu, M.; Ding, J.; Wu, C. Ras and Rab Interactor 3: From Cellular Mechanisms to Human Diseases. Front. Cell. Dev. Biol. 2022, 10, 824961. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, K.; Takasugi, T.; Chiu, Y.W.; Hori, Y.; Tomita, T.; Fukuda, M.; Hisanaga, S.I. CD2-associated protein (CD2AP) overexpression accelerates amyloid precursor protein (APP) transfer from early endosomes to the lysosomal degradation pathway. J. Biol. Chem. 2019, 294, 10886–10899. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Jiang, H.; Srivatsan, S.; Xiao, Q.; Lefton, K.B.; Yamada, K.; Mahan, T.E.; Lee, J.M.; Shaw, A.S.; Holtzman, D.M. Effects of CD2-associated protein deficiency on amyloid-β in neuroblastoma cells and in an APP transgenic mouse model. Mol. Neurodegener. 2015, 10, 12. [Google Scholar] [CrossRef]
- Xue, Y.Y.; Chen, Y.H.; Lin, R.R.; Huang, H.F.; Wu, Z.Y.; Tao, Q.Q. Alzheimer’s disease susceptibility locus in CD2AP is associated with increased cerebrospinal fluid tau levels in mild cognitive impairment. Alzheimer’s Disease Neuroimaging Initiative. Neurosci. Lett. 2022, 771, 136419. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Chen, K.; Keenan, B.T.; Chibnik, L.B.; Fleisher, A.; Thiyyagura, P.; Roontiva, A.; McCabe, C.; Patsopoulos, N.A.; Corneveaux, J.J.; et al. Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol. 2013, 70, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Zhao, X.; He, L.; Ding, Y.; Xu, W.; Lin, S.; Fang, S.; Yang, W.; Sung, K.; Spencer, B.; et al. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Kajiho, H.; Sakurai, K.; Minoda, T.; Yoshikawa, M.; Nakagawa, S.; Fukushima, S.; Kontani, K.; Katada, T. Characterization of RIN3 as a guanine nucleotide exchange factor for the Rab5 subfamily GTPase Rab31. J. Biol. Chem. 2011, 286, 24364–24373. [Google Scholar] [CrossRef]
- Tan, M.S.; Yang, Y.X.; Xu, W.; Wang, H.F.; Tan, L.; Zuo, C.T.; Dong, Q.; Tan, L.; Suckling, J.; Yu, J.T.; et al. Associations of Alzheimer’s disease risk variants with gene expression, amyloidosis, tauopathy, and neurodegeneration. Alzheimers Res. Ther. 2021, 13, 15. [Google Scholar] [CrossRef]
- Li, G. Rab GTPases, membrane trafficking and diseases. Curr. Drug Targets 2011, 12, 1188–1193. [Google Scholar] [CrossRef]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell. Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef]
- Caudano, F.; Montalto, G.; Passalacqua, M.; Pronzato, M.A.; Fedele, E.; Ricciarelli, R. cGMP favors the interaction between APP and BACE1 by inhibiting Rab5 GTPase activity. Sci. Rep. 2020, 10, 1358. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Teves, C.A.F.; Long, A.; Hofert, M.; Tanzi, R.E. The neuronal-specific isoform of BIN1 regulates β-secretase cleavage of APP and Aβ generation in a RIN3-dependent manner. Sci. Rep. 2022, 12, 3486. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, P.; Nomura, T.; Andrew, R.J.; Masse, N.Y.; Sampathkumar, V.; Musial, T.F.; Sudwarts, A.; Recupero, A.J.; Le Metayer, T.; Hansen, M.T.; et al. Neuronal BIN1 Regulates Presynaptic Neurotransmitter Release and Memory Consolidation. Cell Rep. 2020, 30, 3520–3535.e7. [Google Scholar] [CrossRef] [PubMed]
- Pensalfini, A.; Jiang, Y.; Kim, S.; Nixon, R.A. Assessing Rab5 Activation in Health and Disease. Methods Mol. Biol. 2021, 2293, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Viegas, F.O.; Neuhauss, S.C.F. A Metabolic Landscape for Maintaining Retina Integrity and Function. Front. Mol. Neurosci. 2021, 14, 656000. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Aldana, B.I.; Nissen, J.D.; Tanila, H.; Waagepetersen, H.S. Alterations in Cerebral Cortical Glucose and Glutamine Metabolism Precedes Amyloid Plaques in the APPswe/PSEN1dE9 Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2017, 42, 1589–1598. [Google Scholar] [CrossRef]
- Zilberter, Y.; Zilberter, M. The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J. Neurosci. Res. 2017, 95, 2217–2235. [Google Scholar] [CrossRef]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef]
- Macdonald, I.R.; DeBay, D.R.; Reid, G.A.; O’Leary, T.P.; Jollymore, C.T.; Mawko, G.; Darvesh, S. Early detection of cerebral glucose uptake changes in the 5XFAD mouse. Curr. Alzheimer Res. 2014, 11, 450–460. [Google Scholar] [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef]
- Bouter, C.; Bouter, Y. 18F-FDG-PET in Mouse Models of Alzheimer’s Disease. Front. Med. 2019, 6, 71. [Google Scholar] [CrossRef]
- Ross, J.M.; Öberg, J.; Brené, S.; Coppotelli, G.; Terzioglu, M.; Pernold, K.; Goiny, M.; Sitnikov, R.; Kehr, J.; Trifunovic, A.; et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. USA 2010, 107, 20087–20092. [Google Scholar] [CrossRef]
- Bero, A.W.; Yan, P.; Roh, J.H.; Cirrito, J.R.; Stewart, F.R.; Raichle, M.E.; Lee, J.M.; Holtzman, D.M. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 2011, 14, 750–756. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef]
- Macauley, S.L.; Stanley, M.; Caesar, E.E.; Yamada, S.A.; Raichle, M.E.; Perez, R.; Mahan, T.E.; Sutphen, C.L.; Holtzman, D.M. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Investig. 2015, 125, 2463–2467. [Google Scholar] [CrossRef]
- Harris, R.A.; Tindale, L.; Lone, A.; Singh, O.; Macauley, S.L.; Stanley, M.; Holtzman, D.M.; Bartha, R.; Cumming, R.C. Aerobic glycolysis in the frontal cortex correlates with memory performance in wild-type mice but not the APP/PS1 mouse model of cerebral amyloidosis. J. Neurosci. 2016, 36, 1871–1878. [Google Scholar] [CrossRef]
- Traxler, L.; Herdy, J.R.; Stefanoni, D.; Eichhorner, S.; Pelucchi, S.; Szücs, A.; Santagostino, A.; Kim, Y.; Agarwal, R.K.; Schlachetzki, J.C.M.; et al. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell. Metab. 2022, 34, 1248–1263.e6. [Google Scholar] [CrossRef]
- Weaver, K.E.; Richards, T.L.; Logsdon, R.G.; McGough, E.L.; Minoshima, S.; Aylward, E.H.; Kleinhans, N.M.; Grabowski, T.J.; McCurry, S.M.; Teri, L. Posterior cingulate lactate as a metabolic biomarker in amnestic mild cognitive impairment. Biomed. Res. Int. 2015, 2015, 610605. [Google Scholar] [CrossRef]
- Liguori, C.; Stefani, A.; Sancesario, G.; Sancesario, G.M.; Marciani, M.G.; Pierantozzi, M. CSF lactate levels, τ proteins, cognitive decline: A dynamic relationship in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, 655–659. [Google Scholar] [CrossRef]
- Liguori, C.; Chiaravalloti, A.; Sancesario, G.; Stefani, A.; Sancesario, G.M.; Mercuri, N.B.; Schillaci, O.; Pierantozzi, M. Cere-brospinal fluid lactate levels and brain [18F]FDG PET hypometabolism within the default mode network in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Redjems-Bennani, N.; Jeandel, C.; Lefebvre, E.; Blain, H.; Vidailhet, M.; Guéant, J.L. Abnormal substrate levels that depend upon mitochondrial function in cerebrospinal fluid from Alzheimer patients. Gerontology 1998, 44, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.K.; Wood, J.P.M.; Chidlow, G.; Han, G.; Kittipassorn, T.; Peet, D.J.; Casson, R.J. Cancer-like metabolism of the mammalian retina. Clin. Exp. Ophthalmol. 2015, 43, 367–376. [Google Scholar] [CrossRef]
- Haydinger, C.D.; Kittipassorn, T.; Peet, D.J. Power to see-Drivers of aerobic glycolysis in the mammalian retina: A review. Clin. Exp. Ophthalmol. 2020, 48, 1057–1071. [Google Scholar] [CrossRef]
- Rajala, R.V.S. Aerobic Glycolysis in the Retina: Functional Roles of Pyruvate Kinase Isoforms. Front. Cell. Dev. Biol. 2020, 8, 266. [Google Scholar] [CrossRef]
- Ardanaz, C.G.; Ramírez, M.J.; Solas, M. Brain Metabolic Alterations in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3785. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Wang, L.; Ganguly, A.; Saikia, M.; Wagner, S.L.; Koo, E.H.; Roy, S. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–64. [Google Scholar] [CrossRef]
- Saadipour, K.; Mañucat-Tan, N.B.; Lim, Y.; Keating, D.J.; Smith, K.S.; Zhong, J.H.; Liao, H.; Bobrovskaya, L.; Wang, Y.J.; Chao, M.V. p75 neurotrophin receptor interacts with and promotes BACE1 localization in endosomes aggravating amyloidogenesis. J. Neurochem. 2018, 144, 302–317. [Google Scholar] [CrossRef]
- Sannerud, R.; Declerck, I.; Peric, A.; Raemaekers, T.; Menendez, G.; Zhou, L.; Veerle, B.; Coen, K.; Munck, S.; De Strooper, B.; et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl. Acad. Sci. USA 2011, 108, E559–E568. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Schneider, A.; Schlechtingen, G.; Weidlich, S.; Ries, J.; Braxmeier, T.; Schwille, P.; Schulz, J.B.; Schroeder, C.; Simons, M.; et al. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 2008, 320, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Sambamurti, K.; Efthimiopoulos, S.; Pappolla, M.A.; Robakis, N.K. Evidence that secretase cleavage of cell surface Alzheimer amyloid precursor occurs after normal endocytic internalization. J. Neurosci. Res. 1995, 40, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. Cell. Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Grbovic, O.M.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Dinakar, R.; Summers-Terio, N.B.; Ceresa, B.P.; Nixon, R.A.; Anne, M.; Cataldo, A.M. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J. Biol. Chem. 2003, 278, 31261–31268. [Google Scholar] [CrossRef]
- Carey, R.M.; Balcz, B.A.; Lopez-Coviella, I.; Slack, B.E. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC Cell Biol. 2005, 6, 30. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Kang, J.E.; Lee, J.; Stewart, F.R.; Verges, D.K.; Silverio, L.M.; Bu, G.; Mennerick, S.; Holtzman, D.M. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 2008, 58, 42–51. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Kaether, C.; Schmitt, S.; Willem, M.; Haass, C. Amyloid precursor protein and Notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic 2006, 7, 408–415. [Google Scholar] [CrossRef]
- Bhalla, A.; Vetanovetz, C.P.; Morel, E.; Chamoun, Z.; Di Paolo, G.; Small, S.A. The location and trafficking routes of the neu-ronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis. 2012, 47, 126–134. [Google Scholar] [CrossRef]
- Agostinho, P.; Pliássova, A.; Oliveira, C.R.; Cunha, R.A. Localization and Trafficking of Amyloid-β Protein Precursor and Secretases: Impact on Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 329–347. [Google Scholar] [CrossRef]
- Lai, S.S.M.; Ng, K.Y.; Koh, R.Y.; Chok, K.C.; Chye, S.M. Endosomal-lysosomal dysfunctions in Alzheimer’s disease: Pathogenesis and therapeutic interventions. Metab. Brain Dis. 2021, 36, 1087–1100. [Google Scholar] [CrossRef]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- .Lambert, E.; Saha, O.; Soares Landeira, B.; Melo de Farias, A.R.; Hermant, X.; Carrier, A.; Pelletier, A.; Gadaut, J.; Davoine, L.; Dupont, C.; et al. The Alzheimer susceptibility gene BIN1 induces isoform-dependent neurotoxicity through early endosome defects. Acta Neuropathol. Commun. 2022, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.; Mirfakhar, S.F.; Perdigão, C.; Burrinha, T. Impact of late-onset Alzheimer’s genetic risk factors on beta-amyloid endocytic production. Cell. Mol. Life Sci. 2018, 75, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.J.; De Rossi, P.; Nguyen, P.; Kowalski, H.R.; Recupero, A.J.; Guerbette, T.; Krause, S.V.; Rice, R.C.; Laury-Kleintop, L.; Wagner, S.L.; et al. Reduction of the expression of the late-onset Alzheimer’s disease (AD) risk-factor BIN1 does not affect amyloid pathology in an AD mouse model. J. Biol. Chem. 2019, 294, 4477–4487. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, P.; Andrew, R.J.; Musial, T.F.; Buggia-Prevot, V.; Xu, G.; Ponnusamy, M.; Ly, H.; Krause, S.V.; Rice, R.C.; de l’Estoile, V.; et al. Aberrant accrual of BIN1 near Alzheimer’s disease amyloid deposits in transgenic models. Brain Pathol. 2019, 29, 485–501. [Google Scholar] [CrossRef]
- De Rossi, P.; Buggia-Prévot, V.; Clayton, B.L.L.; Vasquez, J.B.; van Sanford, C.; Andrew, R.J.; Lesnick, R.; Botté, A.; Deyts, C.; Salem, S.; et al. Predominant expression of Alzheimer’s disease-associated BIN1 in mature oligodendrocytes and localization to white matter tracts. Mol. Neurodegener. 2016, 11, 59. [Google Scholar] [CrossRef]
- Holler, C.J.; Davis, P.R.; Beckett, T.L.; Platt, T.L.; Webb, R.L.; Head, E.; Murphy, M.P. Bridging integrator 1 (BIN1) protein ex-pression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers Dis. 2014, 42, 1221–1227. [Google Scholar] [CrossRef]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef]
- Malki, I.; Cantrelle, F.X.; Sottejeau, Y.; Lippens, G.; Lambert, J.C.; Landrieu, I. Regulation of the interaction between the neu-ronal BIN1 isoform 1 and Tau proteins—Role of the SH3 domain. FEBS J. 2017, 284, 3218–3229. [Google Scholar] [CrossRef]
- Sartori, M.; Mendes, T.; Desai, S.; Lasorsa, A.; Herledan, A.; Malmanche, N.; Mäkinen, P.; Marttinen, M.; Malki, I.; Chapuis, J.; et al. BIN1 recovers tauopathy-induced long-term memory deficits in mice and interacts with Tau through Thr348 phosphor-ylation. Acta Neuropathol. 2019, 138, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, M.; Wang, S.; Yuksel, M.; Hansen, M.T.; Blazier, D.M.; McMillan, J.D.; Zhang, X.; Dammer, E.B.; Collier, L.; Thi-nakaran, G. Loss of forebrain BIN1 attenuates hippocampal pathology and neuroinflammation in a tauopathy model. Brain 2023, 146, 1561–1579. [Google Scholar] [CrossRef] [PubMed]
- Stefanoska, K.; Volkerling, A.; Bertz, J.; Poljak, A.; Ke, J.D.; Ittner, L.M.; Ittner, A. An N-terminal motif unique to primate tau enables differential protein-protein interactions. J. Biol. Chem. 2018, 293, 3710–3719. [Google Scholar] [CrossRef]
- Zhou, H.; Costaguta, G.; Payne, G.S. Clathrin Adaptor Complex-interacting Protein Irc6 Functions through the Conserved C-Terminal Domain. Sci. Rep. 2019, 9, 4436. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Arikan, M.C.; Andreadis, A. Modulation of the membrane-binding domain of tau protein: Splicing regulation of exon 2. Brain Res. Mol Brain Res. 2003, 116, 94–105. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Pollack, S.J.; Dakkak, D.; Guo, T.; Gómez-Suaga, P.; Noble, W.; Hanger, D.P. Autophagy and lysosomal defects in cells ex-pressing disease-associated tau. Alzheimers Dement. 2021, 17, e058299. [Google Scholar] [CrossRef]
- Mahendran, T.S.; Suresh, S.N.; Garimella, L.; Manjithaya, R. Soluble 4R0N Tau Abrogates Endocytic Vesicular Dynamics. Front. Aging Neurosci. 2020, 12, 537712. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Mufson, E.J.; Alldred, M.J.; Counts, S.E.; Wuu, J.; Nixon, R.A.; Che, S. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 2011, 42, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.I.; Rebelo, S.; Esselmann, H.; Wiltfang, J.; Lah, J.; Lane, R.; Small, S.A.; Gandy, S.; da Cruz e Silva, E.F.; da Cruz e Silva, O.A. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol. Neurodegener. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Praticò, D. The retromer complex system in a transgenic mouse model of AD: Influence of age. Neurobiol. Aging 2017, 52, 32–38. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Xu, G.; Weigel-Van Aken, K.A. Lactic acid induces aberrant amyloid precursor protein processing by promoting its interaction with endoplasmic reticulum chaperone proteins. PLoS ONE 2010, 5, e13820. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M.; Butterworth, J.; Tennant, M.C.; Gordon, A. Enzyme activities in relation to pH and lactate in postmortem brain in Alzheimer-type and other dementias. J. Neurochem. 1990, 55, 1624–1630. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Pankiewicz, J.; Scholtzova, H.; Ji, Y.; Quartermain, D.; Jensen, C.H.; Duff, K.; Nixon, R.A.; Gruen, R.J.; Wisniewski, T. Amyloid-beta deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice Comparative Study. J. Neuropathol. Exp. Neurol. 2004, 63, 418–428. [Google Scholar] [CrossRef]
- Oh, H.; Madison, C.; Baker, S.; Rabinovici, G.; Jagust, W. Dynamic relationships between age, amyloid-β deposition, and glucose metabolism link to the regional vulnerability to Alzheimer’s disease. Brain 2016, 139, 2275–2289. [Google Scholar] [CrossRef]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.; Morris, J.C.; Raichle, M.E. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360.e3. [Google Scholar] [CrossRef]
- Goyal, M.S.; Gordon, B.A.; Couture, L.E.; Flores, S.; Xiong, C.; Morris, J.C.; Raichle, M.E.; Benzinger, T.-L.; Vlassenko, A.G. Spatiotemporal relationship between subthreshold amyloid accumulation and aerobic glycolysis in the human brain. Neurobiol. Aging 2020, 96, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Raichle, M.E. Brain aerobic glycolysis functions and Alzheimer’s disease. Clin. Transl. Imaging 2015, 3, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, S.N.; Vlassenko, A.G.; Rundle, M.M.; Snyder, A.Z.; Mintun, M.A.; Raichle, M.E. Regional aerobic glycolysis in thehuman brain. Proc. Natl. Acad. Sci. USA 2010, 107, 17757–17762. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, L.H.; Witter, M.P.; Sonnewald, U. Neuronal and astrocyticmetabolism in a transgenic rat model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2014, 34, 906–914. [Google Scholar] [CrossRef]
- Lin, A.P.; Shic, F.; Enriquez, C.; Ross, B.D. Reduced glutamate neurotransmission in patients with Alzheimer’s disease—An in vivo (13)C magnetic resonance spectroscopy study. MAGMA 2003, 16, 29–42. [Google Scholar] [CrossRef]
- Mullins, R.; Reiter, D.; Kapogiannis, D. Magnetic resonance spectroscopy reveals abnormalities of glucose metabolism in the Alzheimer’s brain. Ann. Clin. Transl. Neurol. 2018, 5, 262–272. [Google Scholar] [CrossRef]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar] [CrossRef]
- Gatta, L.B.; Vitali, M.; Verardi, R.; Arosio, P.; Finazzi, D. Inhibition of heme synthesis alters amyloid precursor protein pro-cessing. J. Neural Transm. 2009, 116, 79–88. [Google Scholar] [CrossRef]
- Fu, W.; Shi, D.; Westaway, D.; Jhamandas, J.H. Bioenergetic mechanisms in astrocytes may contribute to amyloid plaque deposition and toxicity. J. Biol. Chem. 2015, 290, 12504–12513. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Resende, R.; Ferreiro, E.; Rego, A.C.; Pereira, C.F. Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets 2010, 11, 1193–1206. [Google Scholar] [CrossRef]
- Blonz, E.R. Alzheimer’s Disease as the Product of a Progressive Energy Deficiency Syndrome in the Central Nervous System: The Neuroenergetic Hypothesis. J. Alzheimers Dis. 2017, 60, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Camblor-Perujo, S.; Barca, E.C.; Negrete-Hurtado, A.; Racho, J.; De Bruyckere, E.; Wittich, C.; Ellrich, N.; Martins, S.; Adjaye, J.; et al. AP-2 reduces amyloidogenesis by promoting BACE1 trafficking and degradation in neurons. EMBO Rep. 2020, 21, e47954. [Google Scholar] [CrossRef] [PubMed]
- Kurkinen, K.M.; Marttinen, M.; Turner, L.; Natunen, T.; Mäkinen, P.; Haapalinna, F.; Sarajärvi, T.; Gabbouj, S.; Kurki, M.; Paananen, J.; et al. SEPT8 modulates β-amyloidogenic processing of APP by affecting the sorting and accumulation of BACE1. J. Cell Sci. 2016, 129, 2224–2238. [Google Scholar] [CrossRef]
- Ye, X.; Feng, T.; Tammineni, P.; Chang, Q.; Jeong, Y.Y.; Margolis, D.J.; Cai, H.; Kusnecov, A.; Cai, Q. Regulation of Synaptic Amyloid-β Generation through BACE1 Retrograde Transport in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 2639–2655. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Sengupta, U.; Hernandez, C.; Barrett, A.D.; Dineley, K.; Kayed, R. Tau immu-notherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J. Neurosci. 2015, 35, 4857–4868. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; De Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011, 14, 69–76. [Google Scholar] [CrossRef]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Incontro, S.; Li, Y.; Nicoll, R.A.; Roche, K.W. The STEP61 interactome reveals subunit-specific AMPA receptor binding and synaptic regulation. Proc. Natl. Acad. Sci. USA 2019, 116, 8028–8037. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; von Jagow, G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.J.; Hill, C. A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med. 1956, 48, 311–314. [Google Scholar]
- Bobba, A.; Amadoro, G.; La Piana, G.; Calissano, P.; Atlante, A. Glycolytic enzyme upregulation and numbness of mitochon-drial activity characterize the early phase of apoptosis in cerebellar granule cells. Apoptosis 2015, 20, 10–28. [Google Scholar] [CrossRef]
- Favia, M.; de Bari, L.; Lassandro, R.; Atlante, A. Modulation of glucose-related metabolic pathways controls glucose level in airway surface liquid and fight oxidative stress in cystic fibrosis cells. J. Bioenerg. Biomembr. 2019, 51, 203–218. [Google Scholar] [CrossRef]
- Bernt, E.; Bergmeyer, H.U. Lactate dehydrogenase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Academic Press: London, UK, 1963; pp. 736–741. [Google Scholar]
- Niklas, J.; Melnyk, A.; Yuan, Y.; Heinzle, E. Selective permeabilization for the high-throughput measurement of compartmented enzyme activities in mammalian cells. Anal. Biochem. 2011, 416, 218–227. [Google Scholar] [CrossRef]
- Bahnemann, J.; Kayo, S.; Wahrheit, J.; Heinzle, E.; Pörtner, R.; Zeng, A.-P. In search of an effective cell disruption method to isolate intact mitochondria from Chinese hamster ovary cells. Eng. Life Sci. 2014, 14, 161–169. [Google Scholar] [CrossRef]
- Atlante, A.; Gagliardi, S.; Marra, E.; Calissano, P. Neuronal apoptosis in rats is accompanied by rapid impairment of cellular respiration and is prevented by scavengers of reactive oxygen species. Neurosci. Lett. 1998, 245, 127–130. [Google Scholar] [CrossRef]
- Chemnitius, J.M.; Häfner, P.; Kreuzer, H.; Zech, R. Latent and free citrate synthase activity as enzymatic indicators for respiratory potential of isolated porcine heart mitochondria. J. Appl. Cardiol. 1988, 3, 301–310. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latina, V.; Atlante, A.; Malerba, F.; La Regina, F.; Balzamino, B.O.; Micera, A.; Pignataro, A.; Stigliano, E.; Cavallaro, S.; Calissano, P.; et al. The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2023, 24, 9683. https://doi.org/10.3390/ijms24119683
Latina V, Atlante A, Malerba F, La Regina F, Balzamino BO, Micera A, Pignataro A, Stigliano E, Cavallaro S, Calissano P, et al. The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences. 2023; 24(11):9683. https://doi.org/10.3390/ijms24119683
Chicago/Turabian StyleLatina, Valentina, Anna Atlante, Francesca Malerba, Federico La Regina, Bijorn Omar Balzamino, Alessandra Micera, Annabella Pignataro, Egidio Stigliano, Sebastiano Cavallaro, Pietro Calissano, and et al. 2023. "The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer’s Disease Mouse Model" International Journal of Molecular Sciences 24, no. 11: 9683. https://doi.org/10.3390/ijms24119683
APA StyleLatina, V., Atlante, A., Malerba, F., La Regina, F., Balzamino, B. O., Micera, A., Pignataro, A., Stigliano, E., Cavallaro, S., Calissano, P., & Amadoro, G. (2023). The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences, 24(11), 9683. https://doi.org/10.3390/ijms24119683