
Experimental Insights into the Interplay between Histone Modifiers and p53 in Regulating Gene Expression


Abstract
1. Introduction
2. The Interplay between Histone Modifiers and p53
2.1. Histone Modifiers on p53

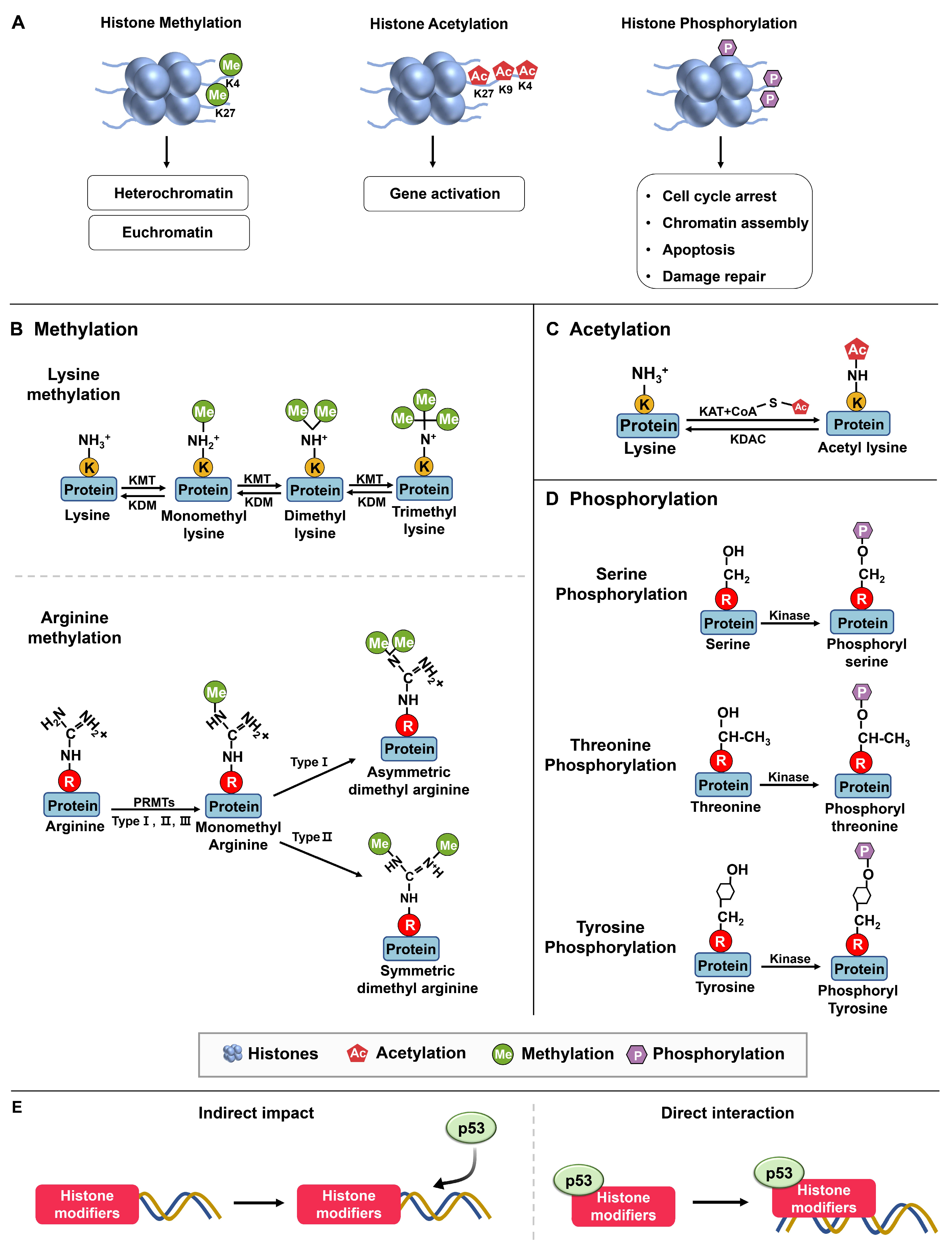{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromatin Modification | Associated Transcription State | References | |
| Acetylation of H3 | Euchromatin/Active | [27] | |
| Acetylation of H4 | Euchromatin/Active | [27] | |
| Di-/Trimethylation of H3K4 | Euchromatin/Active | [29,30] | |
| Di-/Trimethylation of H3K9 | Heterochromatin/Inactive | [29,30] | |
| Di-/Trimethylation of H3K27 | Heterochromatin/Inactive | [31,32] | |
| Enzyme Category | Enzyme Types | Description | Reference |
| Writers | DNA methyltransferases (DNMTs) | Enzymes responsible for adding DNA methylation | [28] |
| Histone lysine methyltransferases (KMTs) | Enzymes responsible for adding histone lysine methylation | ||
| Histone acetyltransferases (HATs) | Enzymes responsible for adding histone acetylation | ||
| Erasers | Histone lysine demethylases (KDMs) | Enzymes responsible for removing histone lysine methylation | |
| Histone deacetylases (HDACs) | Enzymes responsible for removing histone acetylation | ||
| Readers | Bromodomain proteins | Proteins that can “read” acetylated residues on histones | |
| Chromodomain proteins | Proteins that can “read” methylated residues on histones | ||
| Movers | Nucleosome remodeling complexes | Enzymes that can move nucleosomes, aiding gene transcription | |
| Histone Modifications | Amino Acid Residue | ||
| Acetylation | Lysine (K) | ||
| Methylation | Lysine (K), Arginine (R) | ||
| Phosphorylation | Serine (S), Threonine (T), Tyrosine (Y) | ||
| Ubiquitination | Lysine (K) | ||
| Sumoylation | Lysine (K) | ||
| ADP-ribosylation | Various (E, D, R, C, K, and more) |
2.1.1. Methylation
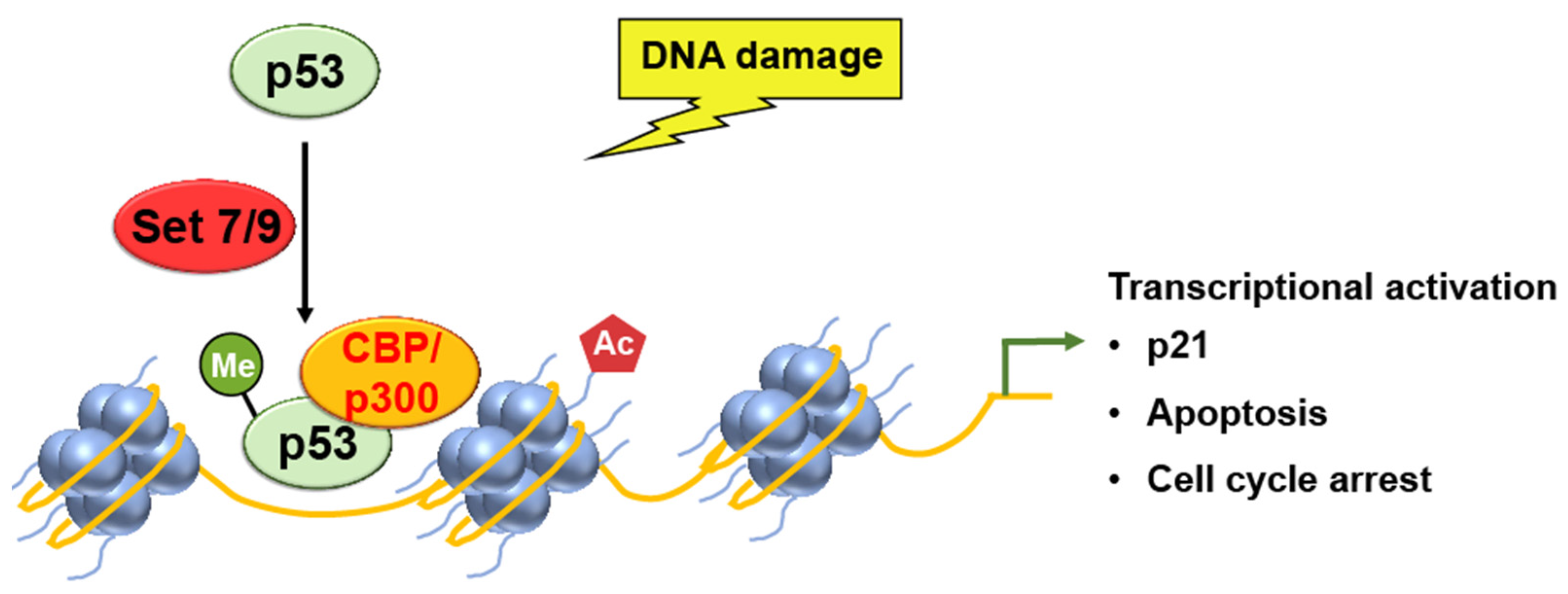
SET7/9
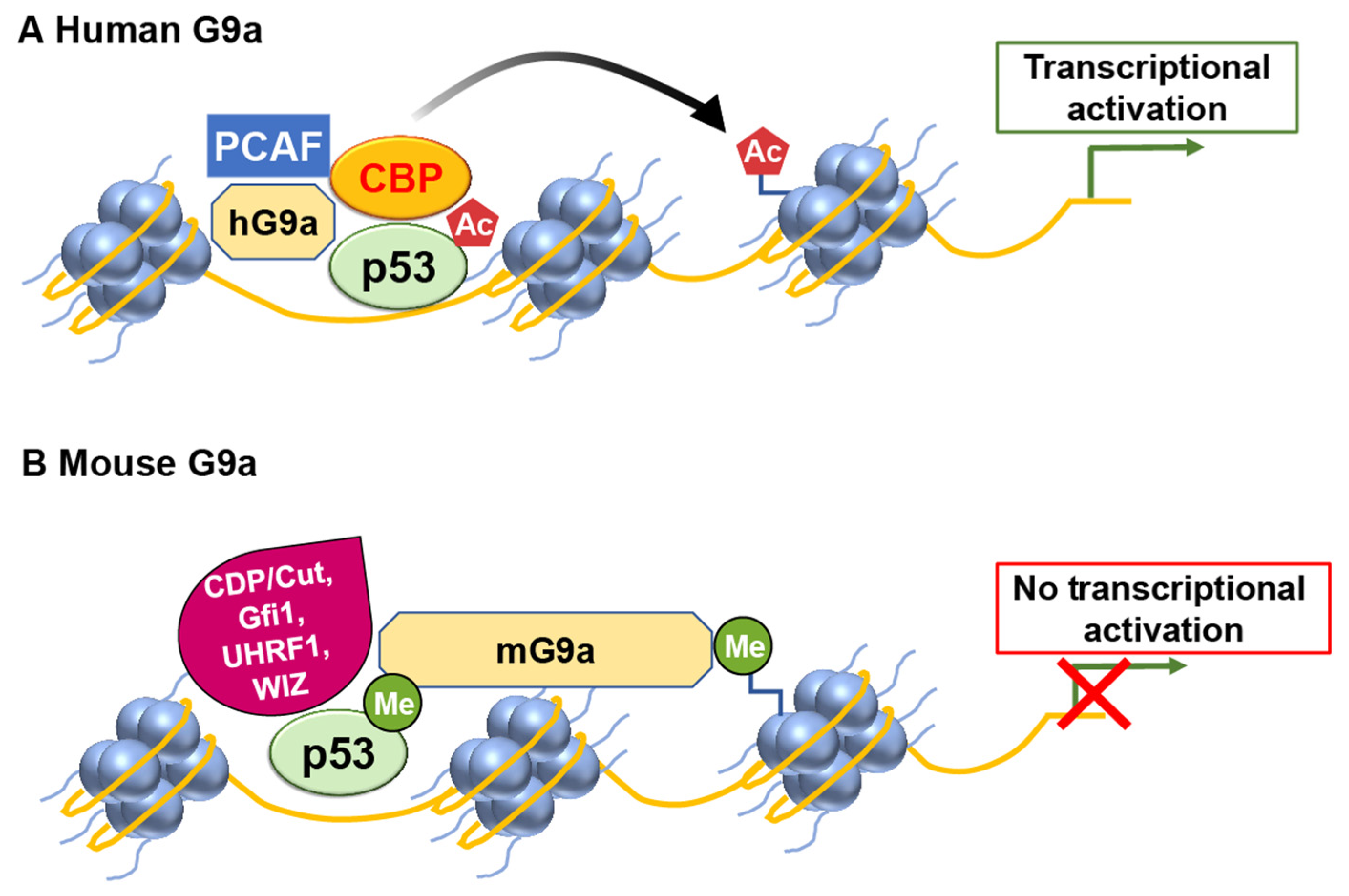
G9a
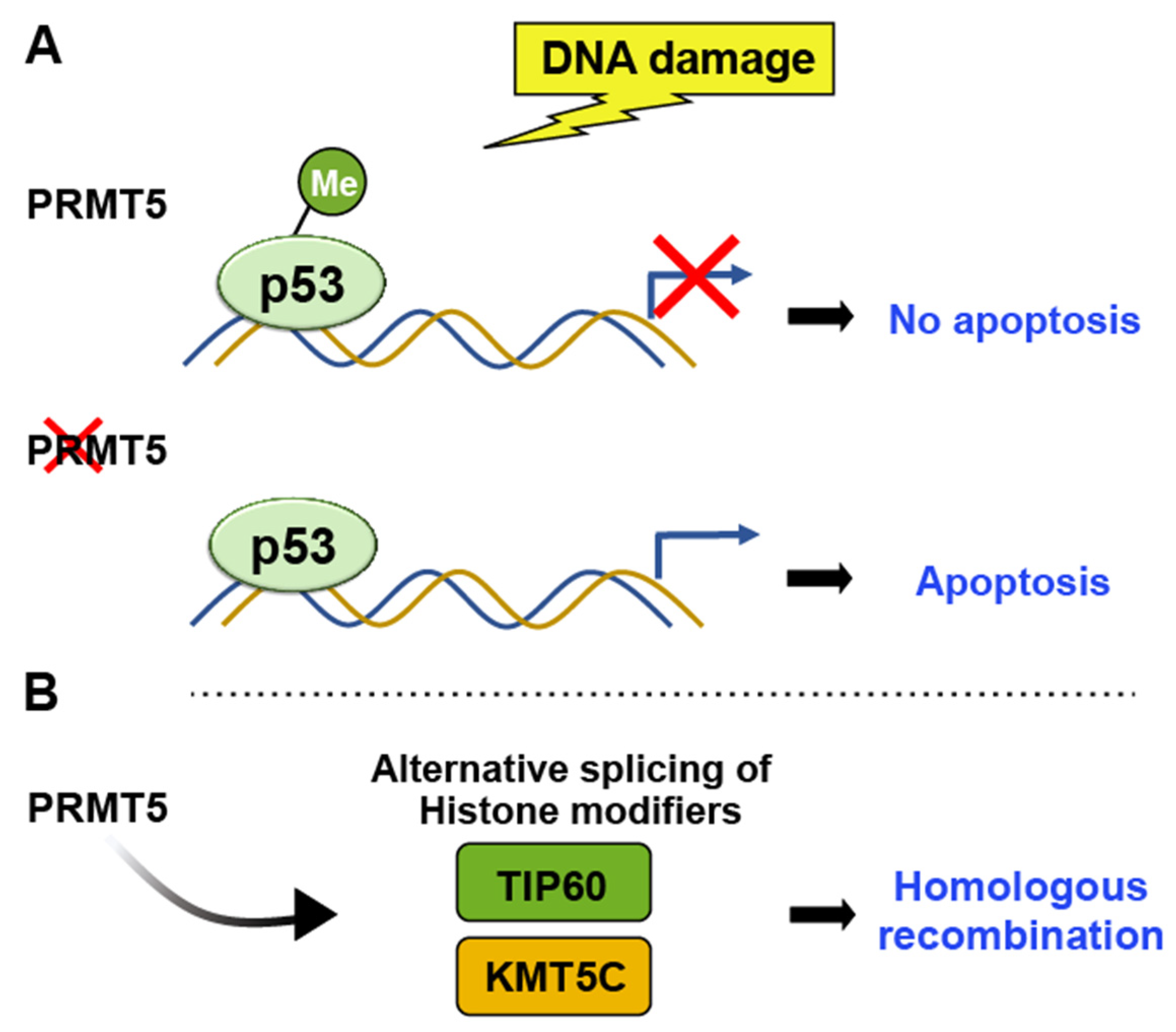
PRMTs (Protein Arginine Methyltransferases)
JMJD2
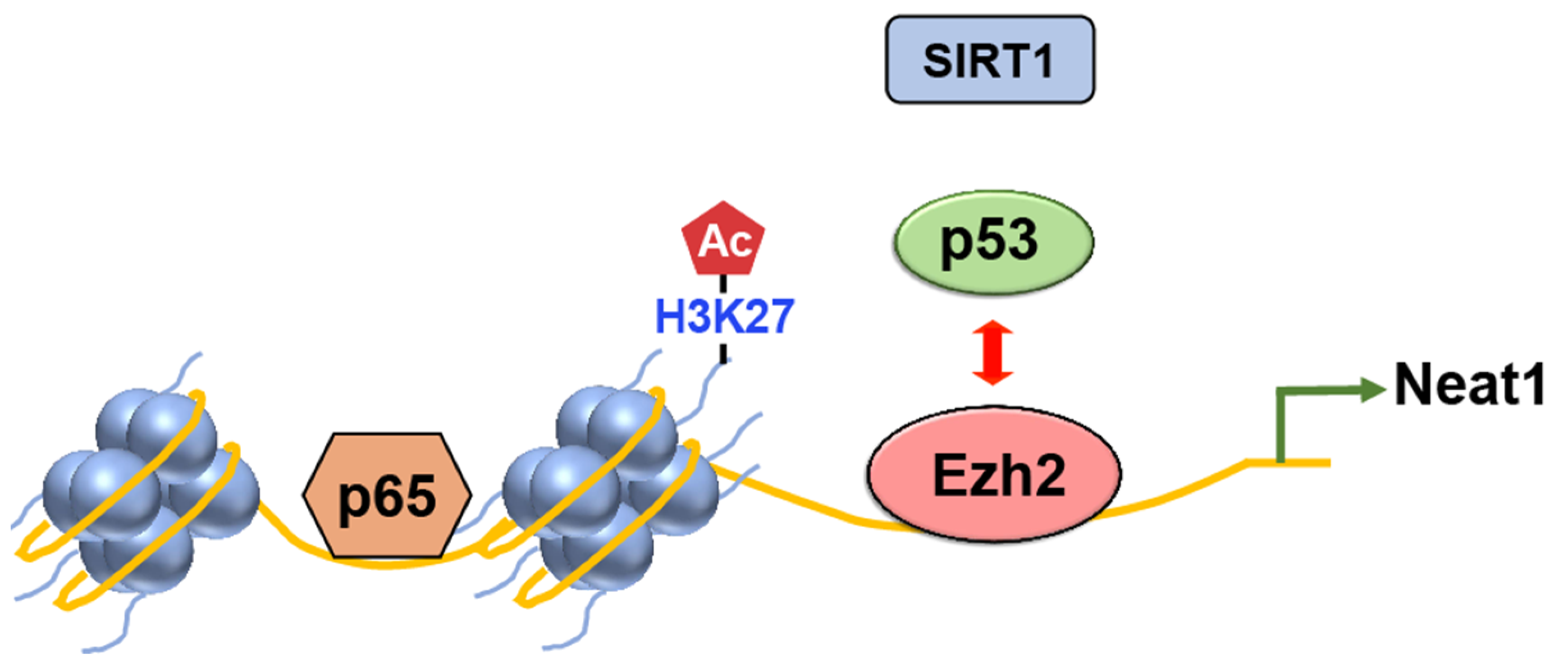
EZH2
2.1.2. Phosphorylation
MAP Kinase Cascade
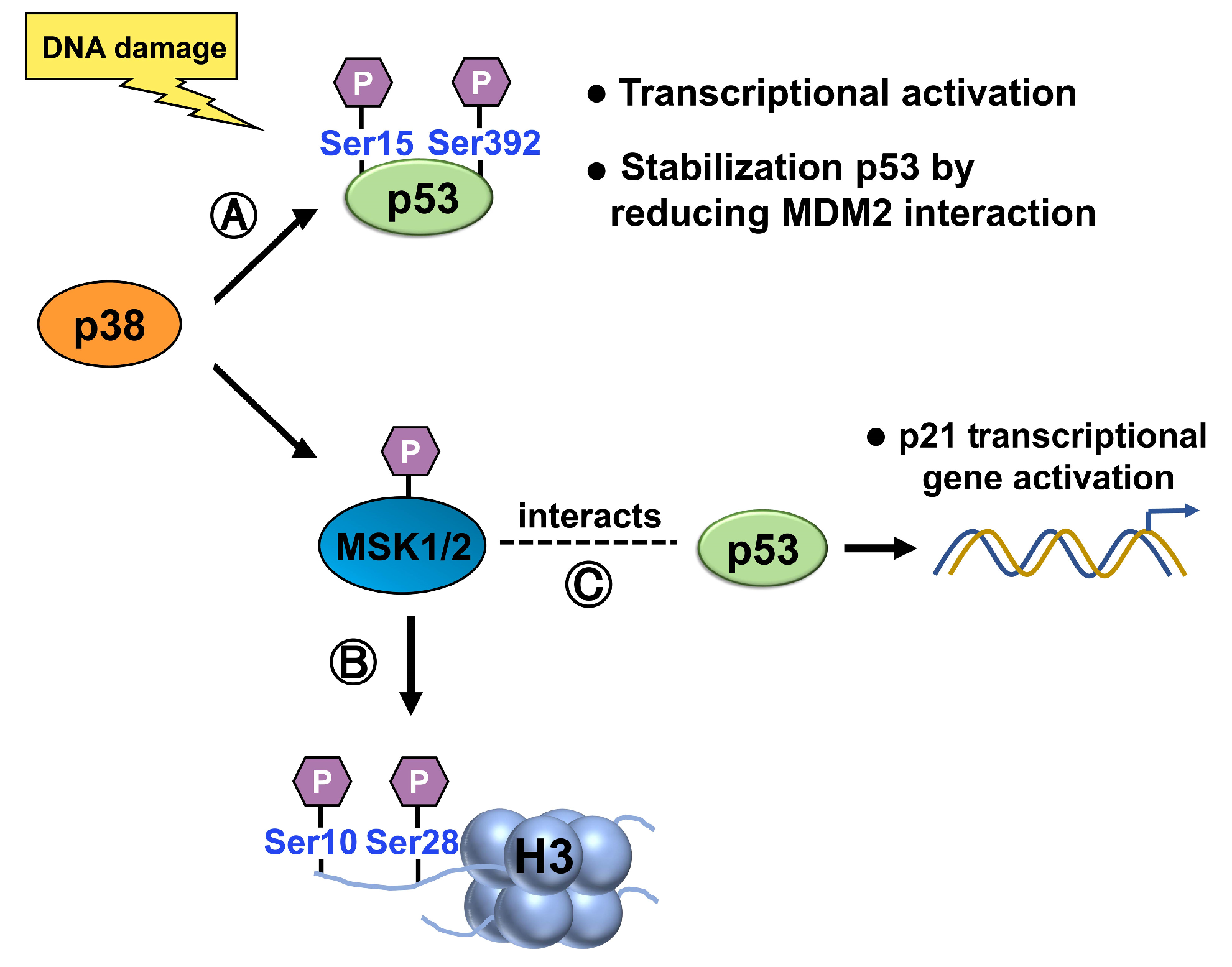
p38 MAP Kinase
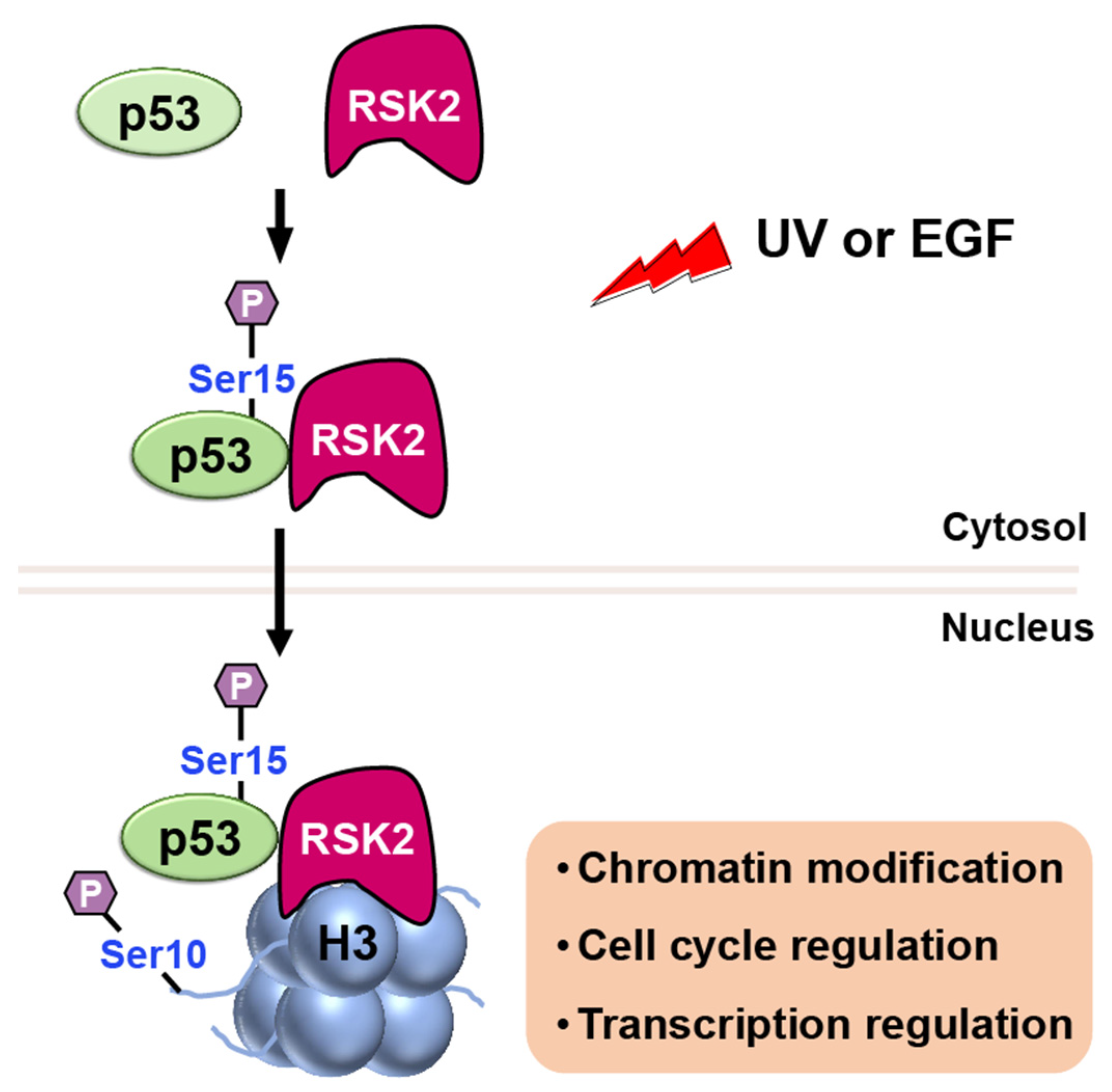
RSK2
2.1.3. Acetylation
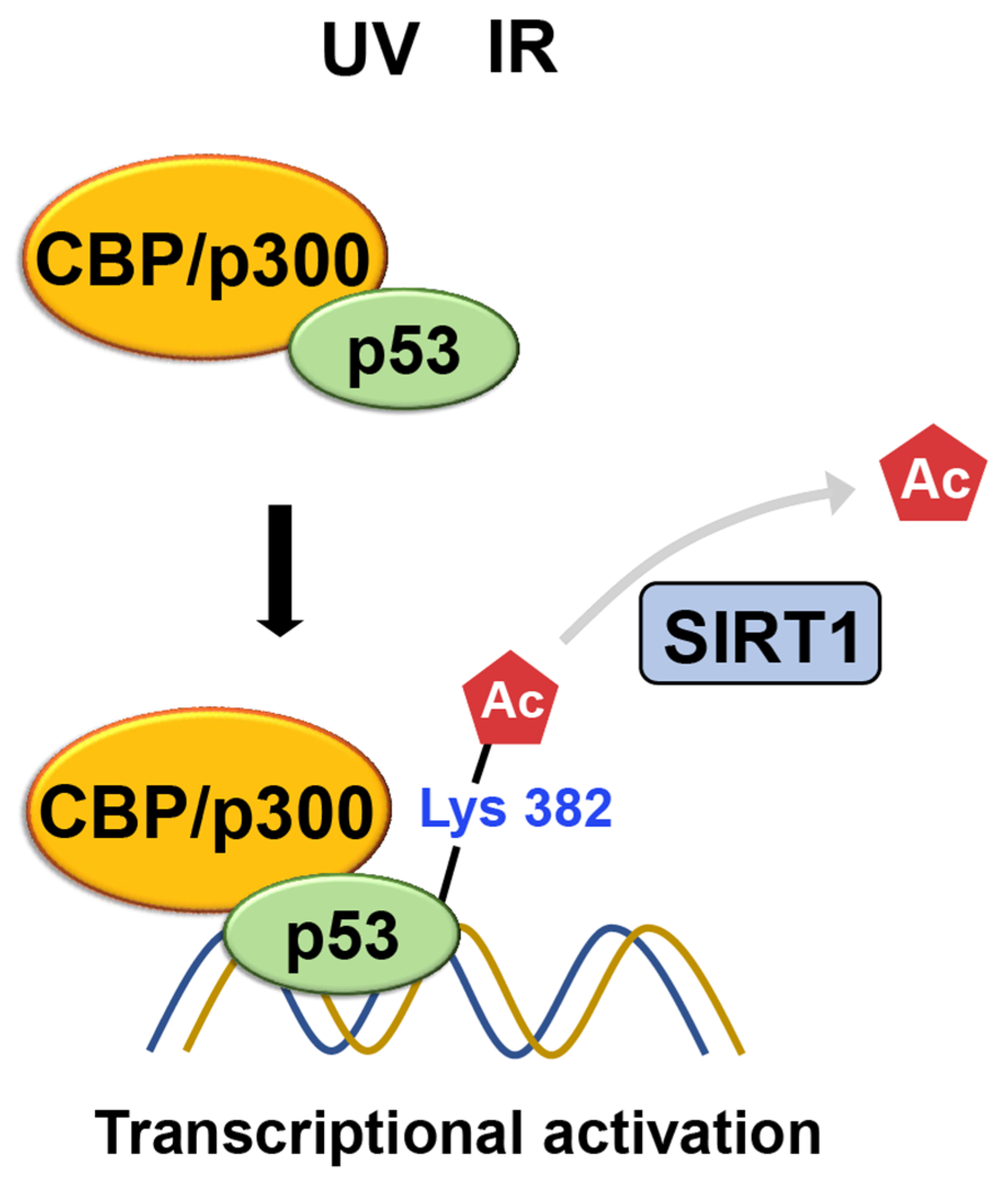
CBP/p300
SIRT1
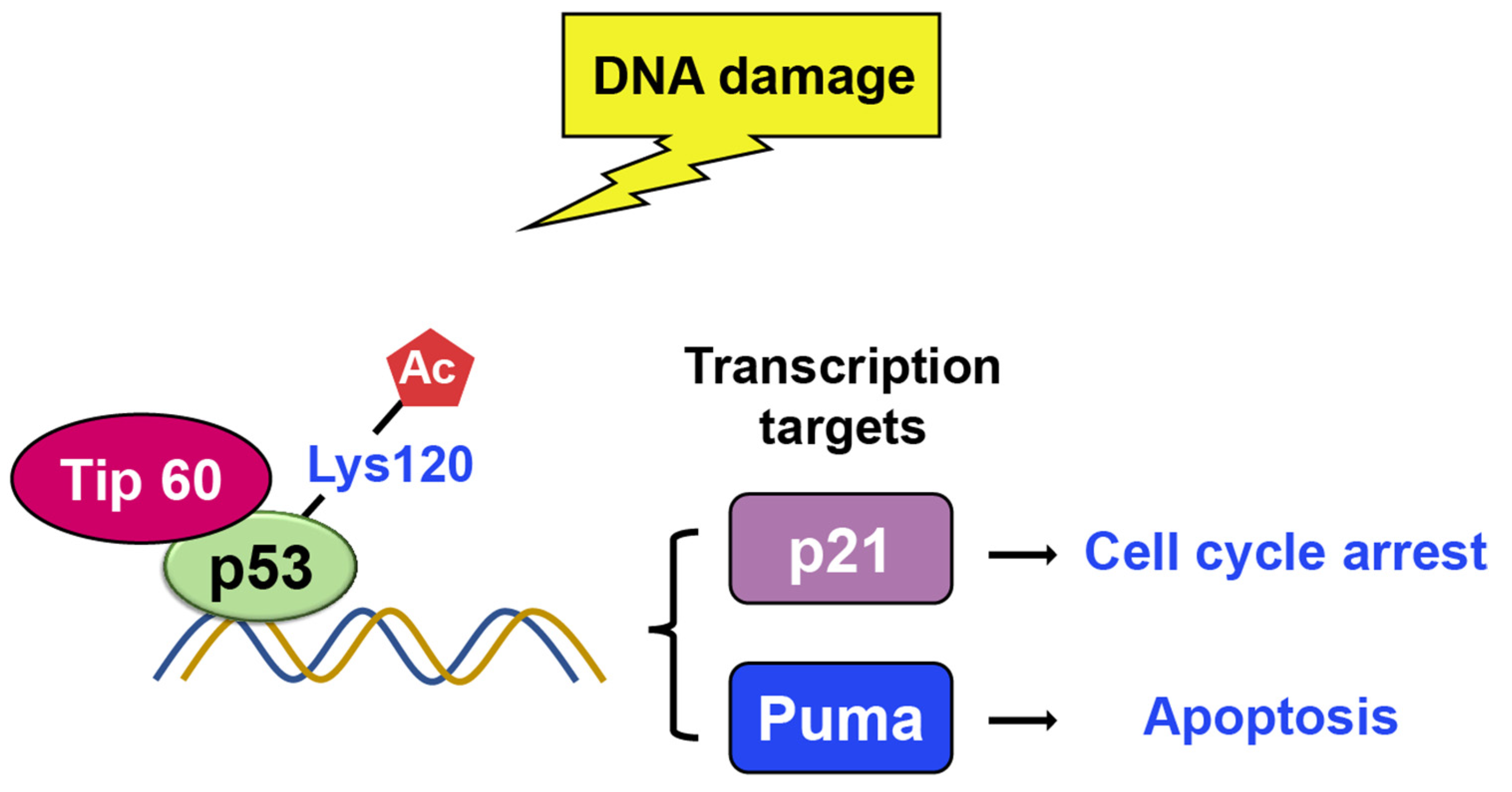
TIP60
3. Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Reviews. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Sarkar, S.; Tan, D. Histone variants and chromatin structure, update of advances. Comput. Struct. Biotechnol. J. 2023, 21, 299–311. [Google Scholar] [CrossRef]
- Zaib, S.; Rana, N.; Khan, I. Histone Modifications and their Role in Epigenetics of Cancer. Curr. Med. Chem. 2022, 29, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Et Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef]
- Ren, X.; Tian, S.; Meng, Q.; Kim, H.M. Histone Demethylase AMX-1 Regulates Fertility in a p53/CEP-1 Dependent Manner. Front. Genet. 2022, 13, 929716. [Google Scholar] [CrossRef] [PubMed]
- Kukita, A.; Sone, K.; Kaneko, S.; Kawakami, E.; Oki, S.; Kojima, M.; Wada, M.; Toyohara, Y.; Takahashi, Y.; Inoue, F.; et al. The Histone Methyltransferase SETD8 Regulates the Expression of Tumor Suppressor Genes via H4K20 Methylation and the p53 Signaling Pathway in Endometrial Cancer Cells. Cancers 2022, 14, 5367. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Perez, R.E.; Lai, X.; Chen, L.; Maki, C.G. The histone demethylase JMJD2B is critical for p53-mediated autophagy and survival in Nutlin-treated cancer cells. J. Biol. Chem. 2019, 294, 9186–9197. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Diehl, J.A. PRMT5-dependent p53 escape in tumorigenesis. Oncoscience 2015, 2, 700–702. [Google Scholar] [CrossRef]
- DeHart, C.J.; Chahal, J.S.; Flint, S.J.; Perlman, D.H. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol. Cell Proteom. 2014, 13, 1–17. [Google Scholar] [CrossRef]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef]
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F., 3rd; Reinberg, D.; Barlev, N.A. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell Biol. 2007, 27, 6756–6769. [Google Scholar] [CrossRef]
- Oh, S.T.; Kim, K.B.; Chae, Y.C.; Kang, J.Y.; Hahn, Y.; Seo, S.B. H3K9 histone methyltransferase G9a-mediated transcriptional activation of p21. FEBS Lett. 2014, 588, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; Macip, S.; Melino, G.; Barlev, N.A. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2017, 36, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Oh, S.; Ro, D.H.; Yoo, H.; Kwon, Y.W. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419–434. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef]
- Keck, F.; Ataey, P.; Amaya, M.; Bailey, C.; Narayanan, A. Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses 2015, 7, 5257–5273. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Wang, Z.; Schones, D.E.; Zhao, K.; DeSalle, R.; Zhang, M.Q. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genom. 2009, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.D.; Tempst, P.; Reinberg, D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Daks, A.; Vasileva, E.; Fedorova, O.; Shuvalov, O.; Barlev, N.A. The Role of Lysine Methyltransferase SET7/9 in Proliferation and Cell Stress Response. Life 2022, 12, 362. [Google Scholar] [CrossRef]
- Kurash, J.K.; Lei, H.; Shen, Q.; Marston, W.L.; Granda, B.W.; Fan, H.; Wall, D.; Li, E.; Gaudet, F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 2008, 29, 392–400. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef]
- Karkhanis, V.; Hu, Y.J.; Baiocchi, R.A.; Imbalzano, A.N.; Sif, S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633–641. [Google Scholar] [CrossRef]
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef]
- Custodio, G.; Parise, G.A.; Kiesel Filho, N.; Komechen, H.; Sabbaga, C.C.; Rosati, R.; Grisa, L.; Parise, I.Z.; Pianovski, M.A.; Fiori, C.M.; et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J. Clin. Oncol. 2013, 31, 2619–2626. [Google Scholar] [CrossRef]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell. Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Tang, X.; Milyavsky, M.; Shats, I.; Erez, N.; Goldfinger, N.; Rotter, V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 2004, 23, 5759–5769. [Google Scholar] [CrossRef]
- Yuan, J.; Zhu, Q.; Zhang, X.; Wen, Z.; Zhang, G.; Li, N.; Pei, Y.; Wang, Y.; Pei, S.; Xu, J.; et al. Ezh2 competes with p53 to license lncRNA Neat1 transcription for inflammasome activation. Cell Death Differ. 2022, 29, 2009–2023. [Google Scholar] [CrossRef]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1495. [Google Scholar] [CrossRef] [PubMed]
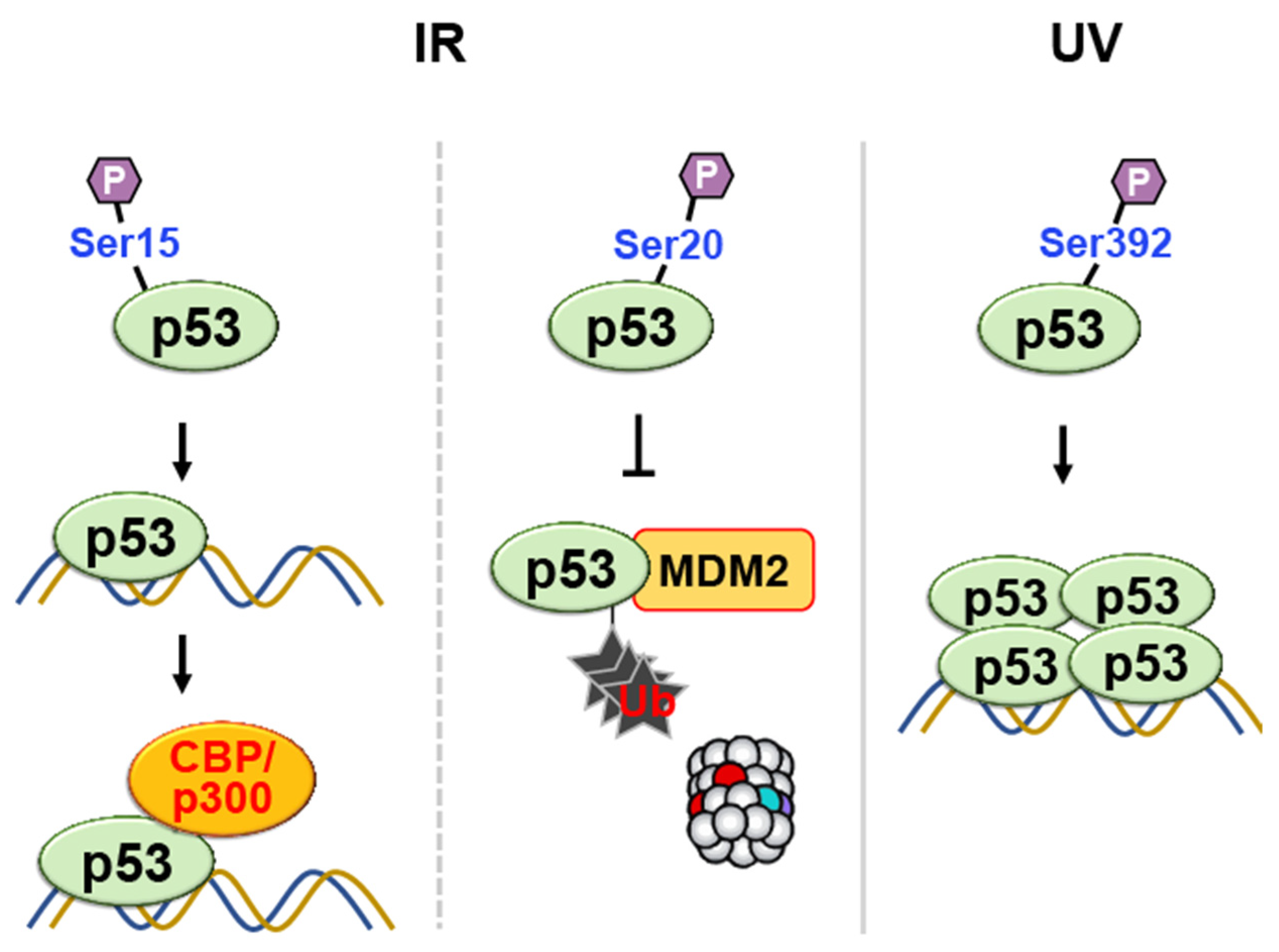
- Lambert, P.F.; Kashanchi, F.; Radonovich, M.F.; Shiekhattar, R.; Brady, J.N. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 1998, 273, 33048–33053. [Google Scholar] [CrossRef]
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef]
- Chehab, N.H.; Malikzay, A.; Stavridi, E.S.; Halazonetis, T.D. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13777–13782. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Taya, Y.; Prives, C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999, 18, 1815–1823. [Google Scholar] [CrossRef]
- Unger, T.; Juven-Gershon, T.; Moallem, E.; Berger, M.; Vogt Sionov, R.; Lozano, G.; Oren, M.; Haupt, Y. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 1999, 18, 1805–1814. [Google Scholar] [CrossRef]
- Kapoor, M.; Lozano, G. Functional activation of p53 via phosphorylation following DNA damage by UV but not gamma radiation. Proc. Natl. Acad. Sci. USA 1998, 95, 2834–2837. [Google Scholar] [CrossRef]
- Lu, H.; Taya, Y.; Ikeda, M.; Levine, A.J. Ultraviolet radiation, but not gamma radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc. Natl. Acad. Sci. USA 1998, 95, 6399–6402. [Google Scholar] [CrossRef] [PubMed]
- Hupp, T.R.; Meek, D.W.; Midgley, C.A.; Lane, D.P. Regulation of the specific DNA binding function of p53. Cell 1992, 71, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Sakamoto, H.; Lewis, M.S.; Anderson, C.W.; Erickson, J.W.; Appella, E.; Xie, D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997, 36, 10117–10124. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, N.; Premkumar Reddy, E. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar] [CrossRef]
- She, Q.B.; Chen, N.; Dong, Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 2000, 275, 20444–20449. [Google Scholar] [CrossRef]
- Cox, M.L.; Meek, D.W. Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell Signal 2010, 22, 564–571. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Sattarifard, H.; Safaei, A.; Khazeeva, E.; Rastegar, M.; Davie, J.R. Mitogen- and stress-activated protein kinase (MSK1/2) regulated gene expression in normal and disease states. Biochem. Cell Biol. 2023, 101. [Google Scholar] [CrossRef] [PubMed]
- Crosio, C.; Heitz, E.; Allis, C.D.; Borrelli, E.; Sassone-Corsi, P. Chromatin remodeling and neuronal response: Multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J. Cell Sci. 2003, 116 Pt 24, 4905–4914. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.L.; Rose, S.; Barratt, M.J.; Mahadevan, L.C. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000, 19, 3714–3726. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, J.G.; Chin, C.; In, S.; Yang, A.; Park, H.S.; Kim, J.; Park, J.H. MSK1 functions as a transcriptional coactivator of p53 in the regulation of p21 gene expression. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ginty, D.D.; Greenberg, M.E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 1996, 273, 959–963. [Google Scholar] [CrossRef]
- Cho, Y.Y.; He, Z.; Zhang, Y.; Choi, H.S.; Zhu, F.; Choi, B.Y.; Kang, B.S.; Ma, W.Y.; Bode, A.M.; Dong, Z. The p53 protein is a novel substrate of ribosomal S6 kinase 2 and a critical intermediary for ribosomal S6 kinase 2 and histone H3 interaction. Cancer Res. 2005, 65, 3596–3603. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of p53 by Histone Acetylation and Acetyltransferases. Biochem. Soc. Trans. 2011, 39, 349–355. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef]
- Gong, F.; Chiu, L.Y.; Miller, K.M. Acetylation Reader Proteins: Linking Acetylation Signaling to Genome Maintenance and Cancer. PLoS Genet. 2016, 12, e1006272. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Timmermann, S.; Lehrmann, H.; Polesskaya, A.; Harel-Bellan, A. Histone acetylation and disease. Cell Mol. Life Sci. 2001, 58, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Tang, Z.; Chen, W.Y.; Shimada, M.; Nguyen, U.T.T.; Kim, J.; Sun, X.J.; Sengoku, T.; McGinty, R.K.; Fernandez, J.P.; Muir, T.W.; et al. SET1 and p300 Act Synergistically, through Coupled Histone Modifications, in Transcriptional Activation by p53. Cell 2023, 186, 2280. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Shimizu, H.; Perkins, N.D.; Hupp, T.R. DNA-dependent acetylation of p53 by the transcription coactivator p300. J. Biol. Chem. 2003, 278, 13431–13441. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Tsay, Y.G.; Tan, B.C.; Lo, W.Y.; Lee, S.C. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem. 2003, 278, 25568–25576. [Google Scholar] [CrossRef]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Sasca, D.; Hähnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.H.; Leng, W.; Jung, S.Y.; Zhang, H.; Deng, M.; Evans, D.; Li, Y.; Luo, K.; Qin, B.; et al. A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Mol. Cell 2014, 56, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, H.; Xu, Z.; Tang, H.; Geng, A.; Cai, B.; Su, T.; Shi, J.; Jiang, C.; Tian, X.; et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. 2019, 47, 8563–8580. [Google Scholar] [CrossRef] [PubMed]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar] [CrossRef]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef]
- Tyteca, S.; Vandromme, M.; Legube, G.; Chevillard-Briet, M.; Trouche, D. Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. EMBO J. 2006, 25, 1680–1689. [Google Scholar] [CrossRef]
- Wang, X.; Lupton, C.; Lauth, A.; Wan, T.C.; Foster, P.; Patterson, M.; Auchampach, J.A.; Lough, J.W. Evidence that the acetyltransferase Tip60 induces the DNA damage response and cell-cycle arrest in neonatal cardiomyocytes. J. Mol. Cell Cardiol. 2021, 155, 88–98. [Google Scholar] [CrossRef]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef]
- Robert, F.; Hardy, S.; Nagy, Z.; Baldeyron, C.; Murr, R.; Dery, U.; Masson, J.Y.; Papadopoulo, D.; Herceg, Z.; Tora, L. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol. Cell Biol. 2006, 26, 402–412. [Google Scholar] [CrossRef]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Lopez-Banuelos, L.; Vega, L. Inhibition of acetylation, is it enough to fight cancer? Crit. Rev. Oncol. Hematol. 2022, 176, 103752. [Google Scholar] [CrossRef]
- Schnell, A.P.; Kohrt, S.; Aristodemou, A.; Taylor, G.P.; Bangham, C.R.M.; Thoma-Kress, A.K. HDAC inhibitors Panobinostat and Romidepsin enhance tax transcription in HTLV-1-infected cell lines and freshly isolated patients’ T-cells. Front. Immunol. 2022, 13, 978800. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Kiga, K.; Fukuda-Yuzawa, Y.; Tanabe, M.; Tsuji, S.; Sasakawa, C.; Fukao, T. Comprehensive silencing of target-sharing microRNAs is a mechanism for SIRT1 overexpression in cancer. RNA Biol. 2014, 11, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, H.; Lirussi, F.; Garrido, C.; Ye, X.Y.; Xie, T. Dual inhibitors of histone deacetylases and other cancer-related targets: A pharmacological perspective. Biochem. Pharm. 2020, 182, 114224. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.H. Dual Inhibitors Against Topoisomerases and Histone Deacetylases. J. Cancer Prev. 2015, 20, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Christman, J.K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Buocikova, V.; Tyciakova, S.; Pilalis, E.; Mastrokalou, C.; Urbanova, M.; Matuskova, M.; Demkova, L.; Medova, V.; Longhin, E.M.; Rundén-Pran, E.; et al. Decitabine-induced DNA methylation-mediated transcriptomic reprogramming in human breast cancer cell lines; the impact of DCK overexpression. Front. Pharmacol. 2022, 13, 991751. [Google Scholar] [CrossRef]
- Shankar, S.; Davis, R.; Singh, K.P.; Kurzrock, R.; Ross, D.D.; Srivastava, R.K. Suberoylanilide hydroxamic acid (Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells orthotopically implanted in BALB/c nude mice. Mol. Cancer Ther. 2009, 8, 1596–1605. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef]
- Tuncer, Z.; Duran, T.; Gunes, C.E.; Kurar, E. Apoptotic effect of Belinostat (PXD101) on MCF-7 cancer cells. Ann. Med. Res. 2021, 28, 941–945. [Google Scholar] [CrossRef]
- Meng, W.; Wang, B.; Mao, W.; Wang, J.; Zhao, Y.; Li, Q.; Zhang, C.; Ma, J. Enhanced efficacy of histone deacetylase inhibitor panobinostat combined with dual PI3K/mTOR inhibitor BEZ235 against glioblastoma. Nagoya J. Med. Sci. 2019, 81, 93–102. [Google Scholar]
- Bekric, D.; Neureiter, D.; Ablinger, C.; Dobias, H.; Beyreis, M.; Ritter, M.; Jakab, M.; Bischof, J.; Koller, U.; Kiesslich, T.; et al. Evaluation of Tazemetostat as a Therapeutically Relevant Substance in Biliary Tract Cancer. Cancers 2023, 15, 1569. [Google Scholar] [CrossRef]
- Khalil, A.F.; El-Moselhy, T.F.; El-Bastawissy, E.A.; Abdelhady, R.; Younis, N.S.; El-Hamamsy, M.H. Discovery of novel enasidenib analogues targeting inhibition of mutant isocitrate dehydrogenase 2 as antileukaemic agents. J. Enzyme Inhib. Med. Chem. 2023, 38, 2157411. [Google Scholar] [CrossRef] [PubMed]
- Reinbold, R.; Hvinden, I.C.; Rabe, P.; Herold, R.A.; Finch, A.; Wood, J.; Morgan, M.; Staudt, M.; Clifton, I.J.; Armstrong, F.A.; et al. Resistance to the isocitrate dehydrogenase 1 mutant inhibitor ivosidenib can be overcome by alternative dimer-interface binding inhibitors. Nat. Commun. 2022, 13, 4785. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-M.; Zheng, X.; Lee, E. Experimental Insights into the Interplay between Histone Modifiers and p53 in Regulating Gene Expression. Int. J. Mol. Sci. 2023, 24, 11032. https://doi.org/10.3390/ijms241311032
Kim H-M, Zheng X, Lee E. Experimental Insights into the Interplay between Histone Modifiers and p53 in Regulating Gene Expression. International Journal of Molecular Sciences. 2023; 24(13):11032. https://doi.org/10.3390/ijms241311032
Chicago/Turabian StyleKim, Hyun-Min, Xiaoyu Zheng, and Ethan Lee. 2023. "Experimental Insights into the Interplay between Histone Modifiers and p53 in Regulating Gene Expression" International Journal of Molecular Sciences 24, no. 13: 11032. https://doi.org/10.3390/ijms241311032
APA StyleKim, H.-M., Zheng, X., & Lee, E. (2023). Experimental Insights into the Interplay between Histone Modifiers and p53 in Regulating Gene Expression. International Journal of Molecular Sciences, 24(13), 11032. https://doi.org/10.3390/ijms241311032