
Identification and Isolation of α-Glucosidase Inhibitors from Siraitia grosvenorii Roots Using Bio-Affinity Ultrafiltration and Comprehensive Chromatography

Abstract
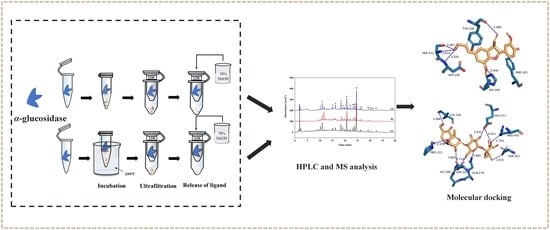
1. Introduction
2. Results and Discussion
2.1. Inhibition of α-Glucosidase by Crude S. grosvenorii Root Extract and the Eluted Fractions
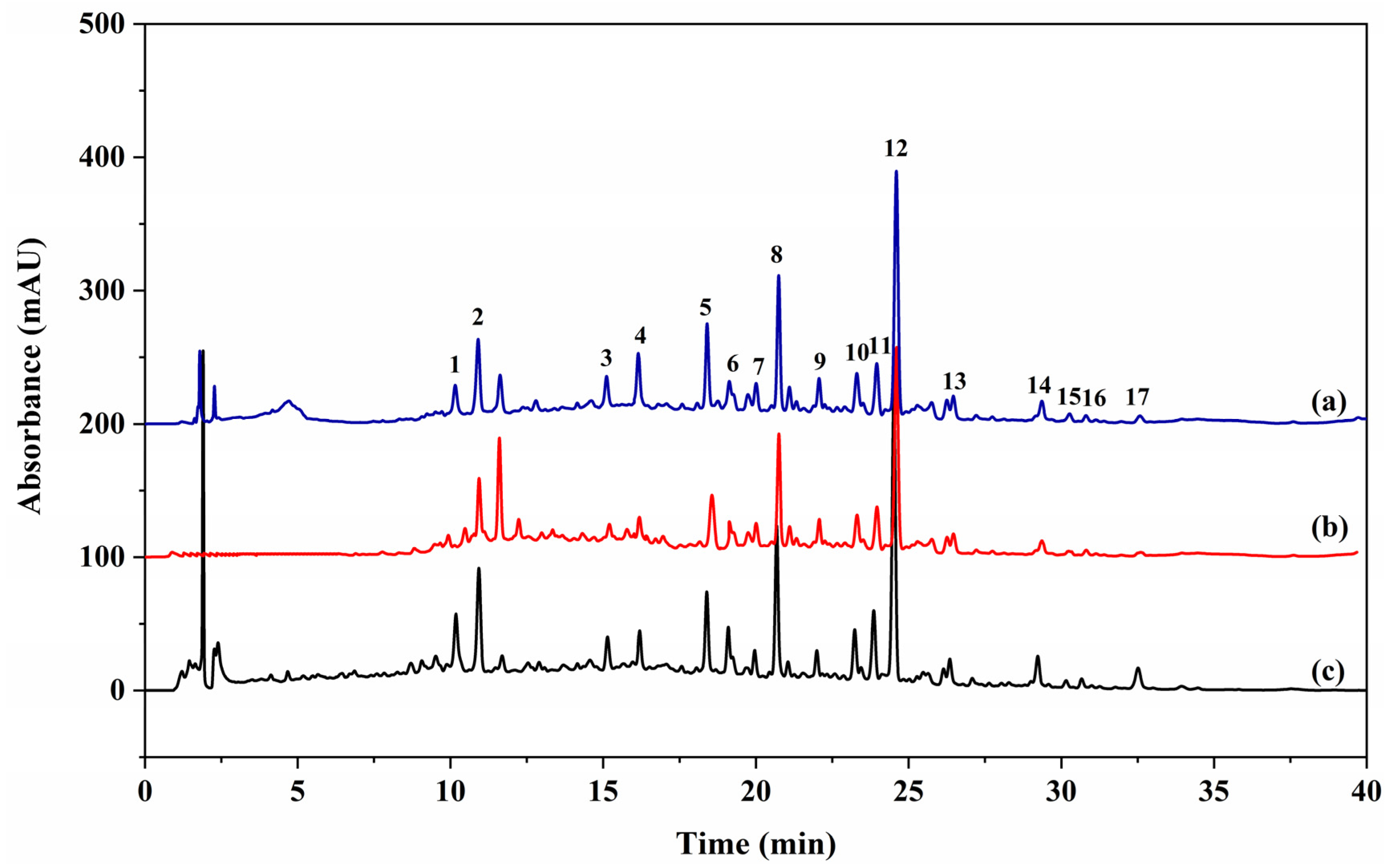
2.2. Screening for Potential α-Glucosidase Inhibitors in SGR2
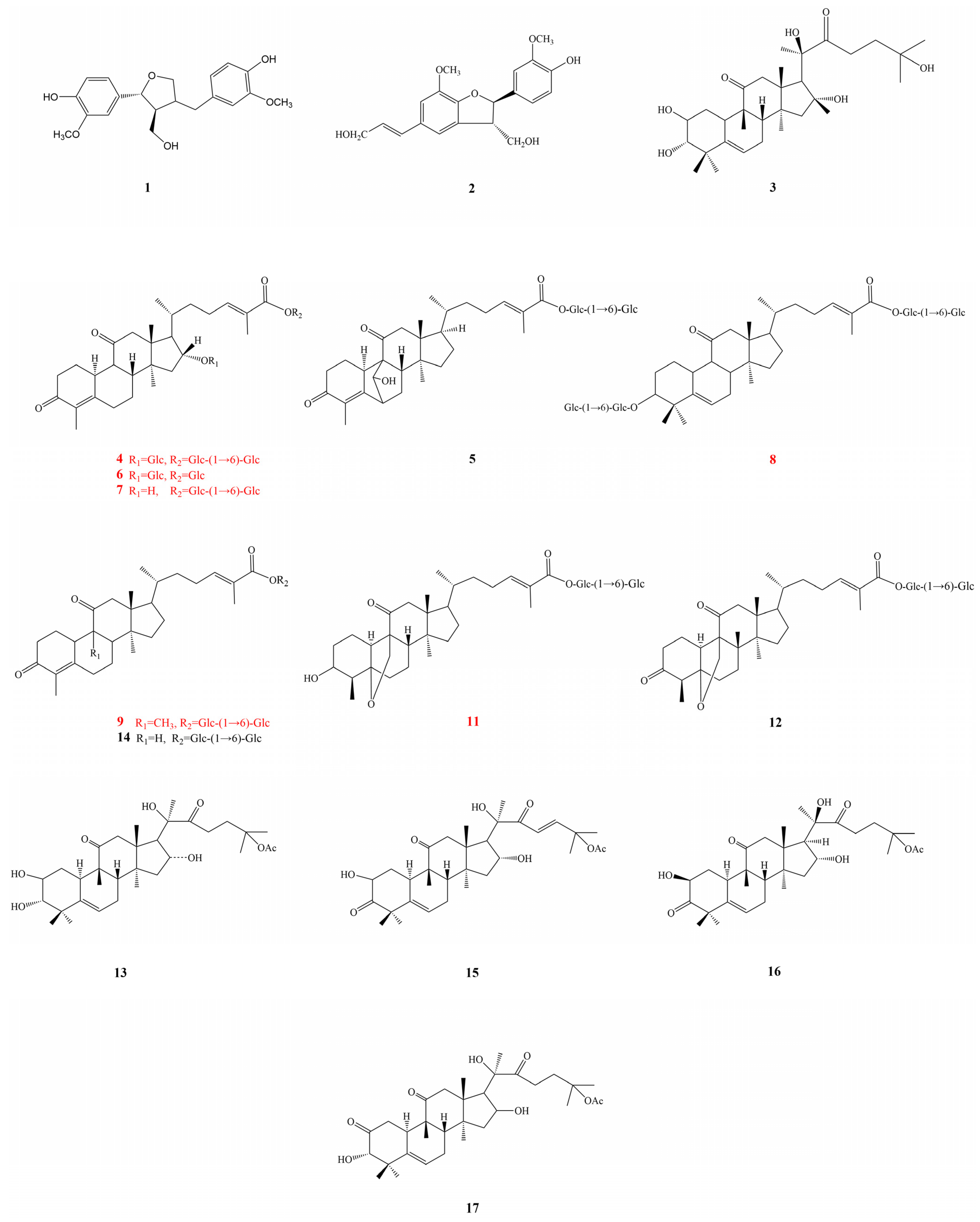
2.3. Isolation and Structural Identification
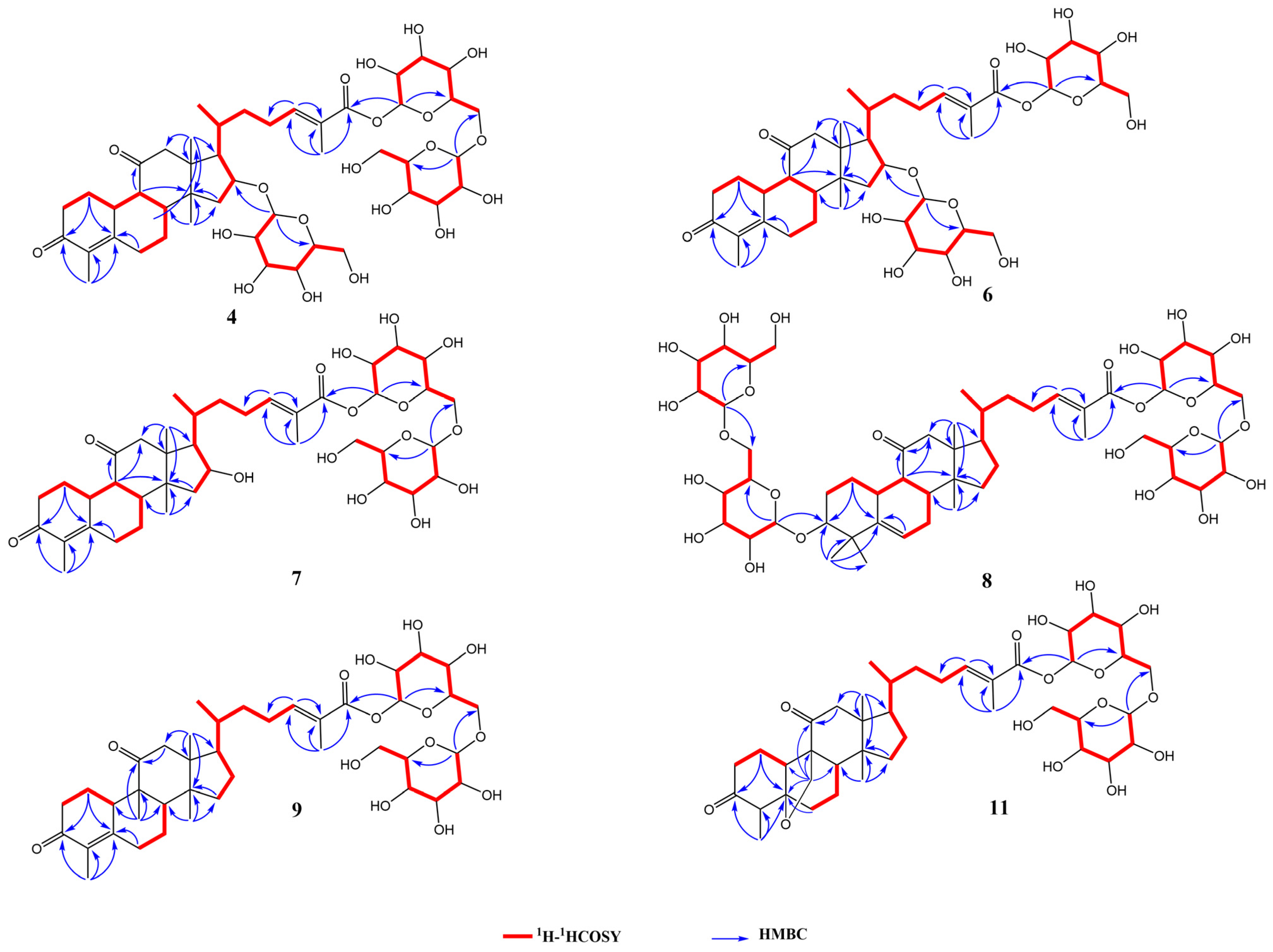
2.3.1. Elucidating the Structure of Novel Compounds
2.3.2. Identification of the Known Isolated Compounds
2.4. Inhibition of α-Glucosidase and Structural-Activity Relationship
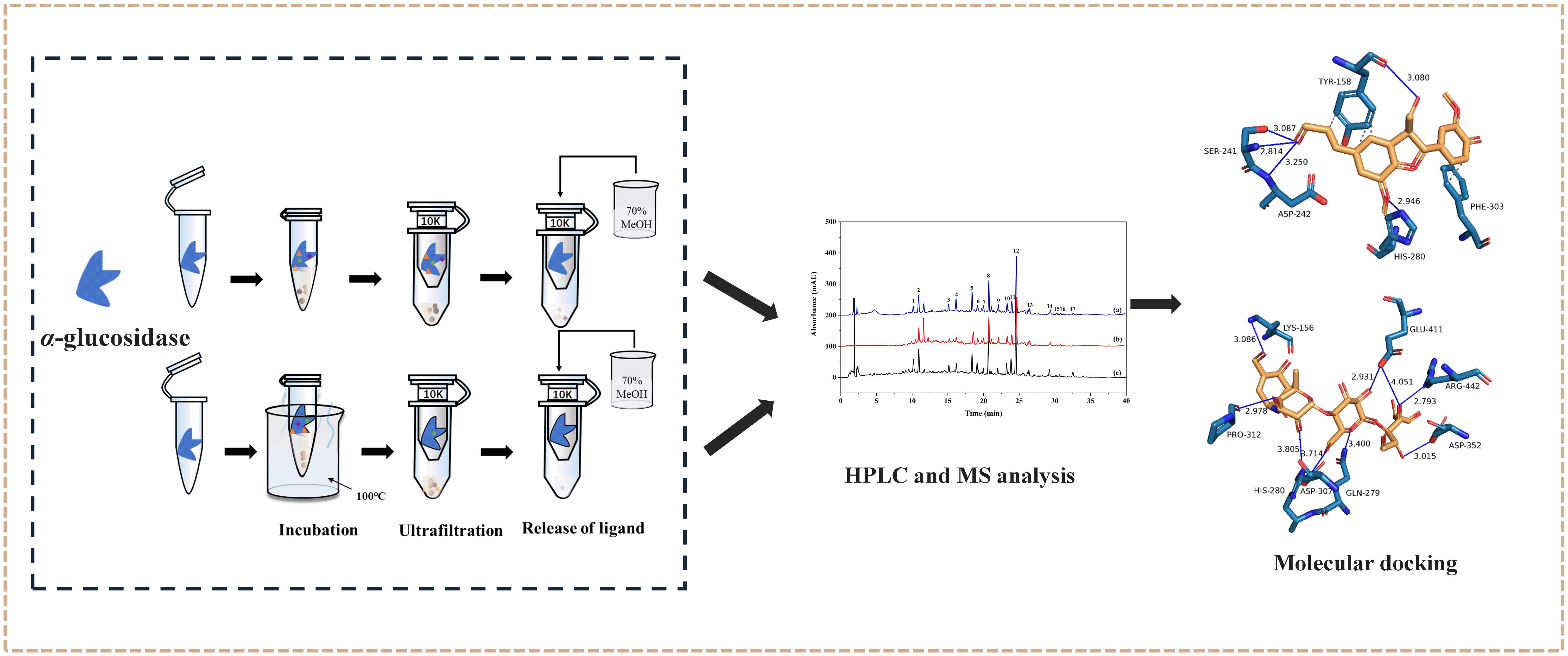
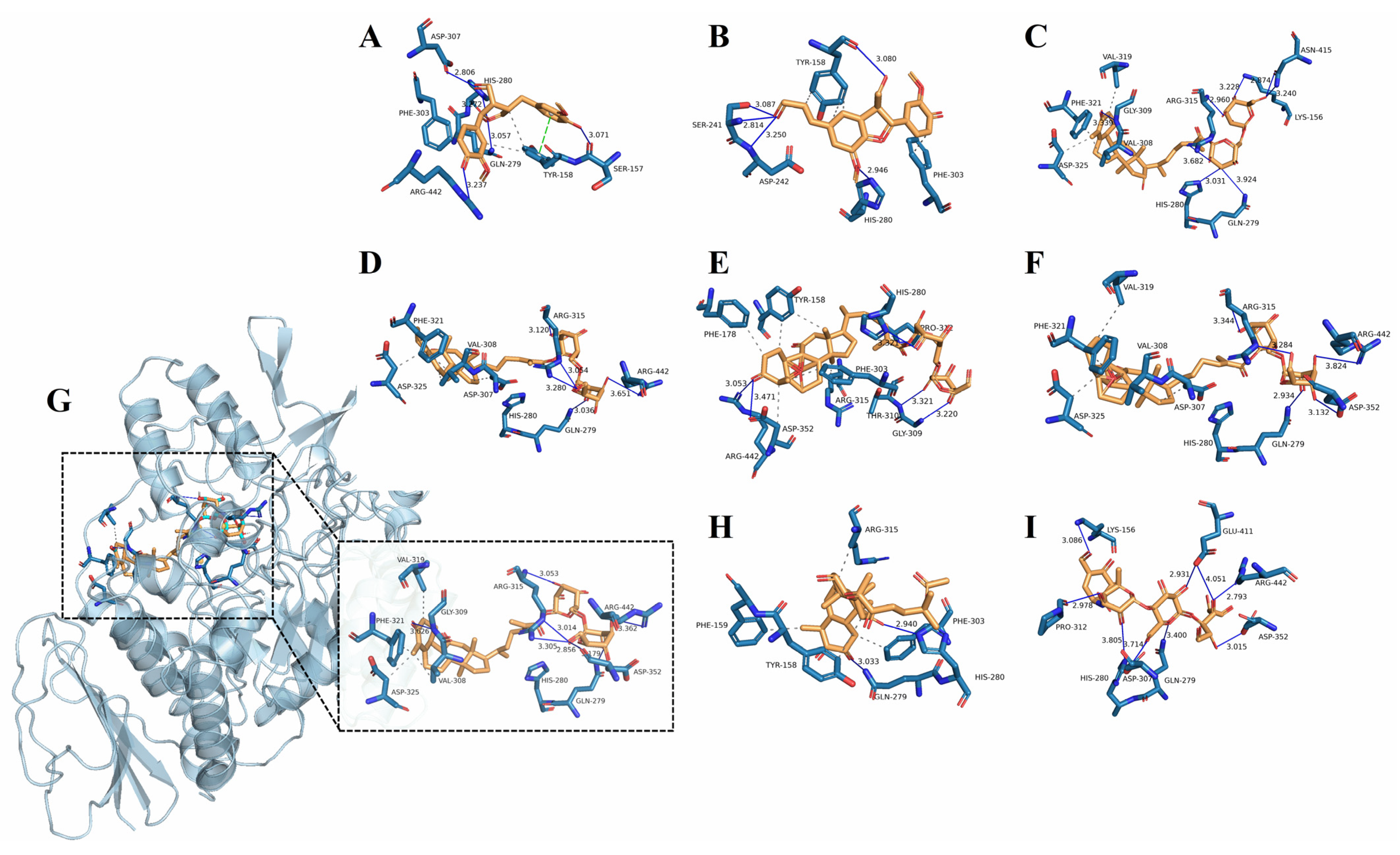
2.5. Molecular Docking Analysis
3. Materials and Methods
3.1. Materials and Reagents
3.2. Extraction and Preparation of Fractions from S. grosvenorii Root Extract
3.3. Assessment of α-Glucosidase Inhibition
3.4. Screening and Identification of α-Glucosidase Inhibitors
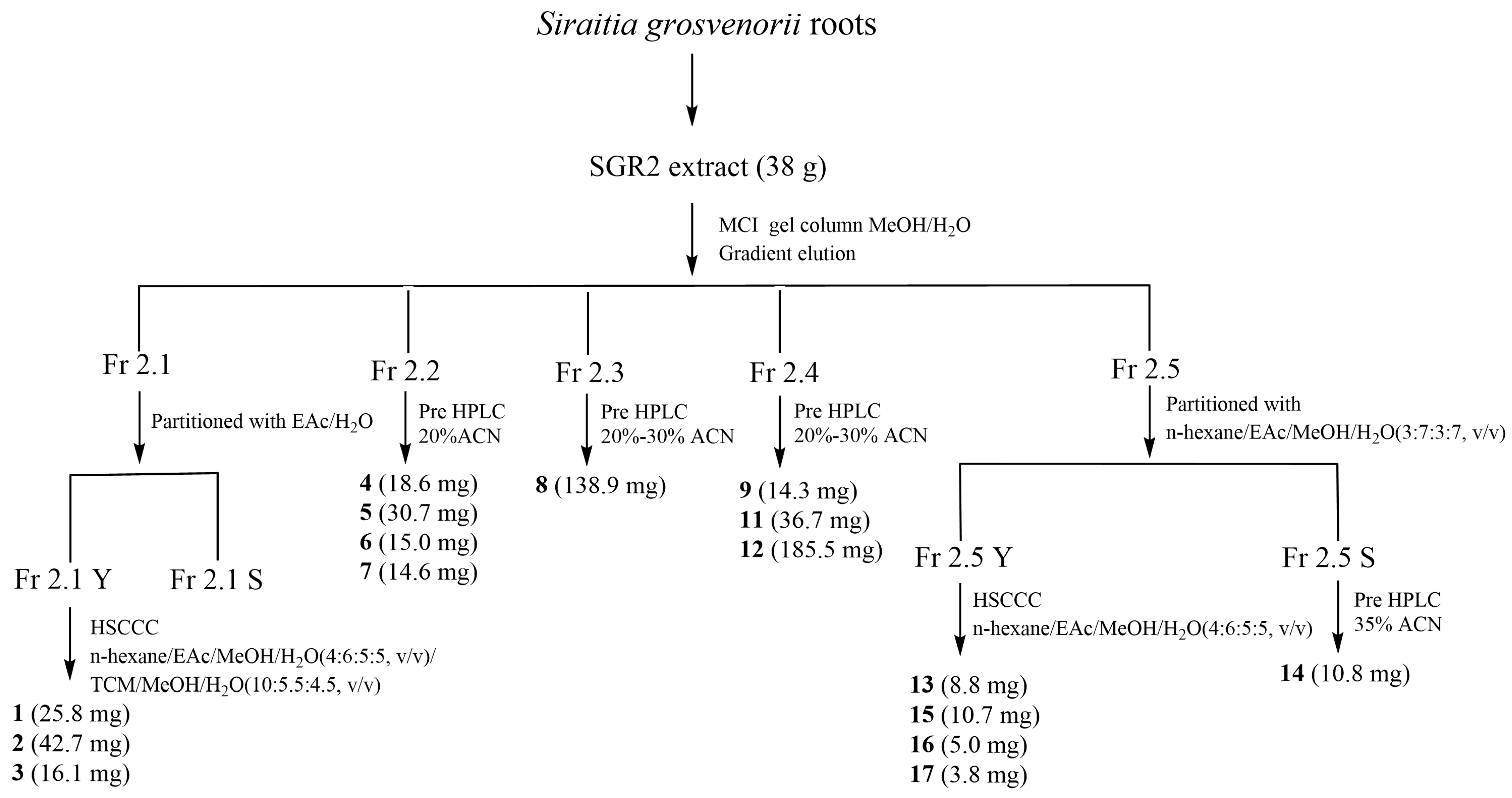
3.5. Extraction and Isolation
3.6. Spectroscopic Data
3.6.1. Siraitic Acid III E (4)
3.6.2. Siraitic Acid IIb E (6)
3.6.3. Siraitic Acid II E (7)
3.6.4. Siraitic Acid IV H (8)
3.6.5. Siraitic Acid II G (9)
3.6.6. Siraitic Acid II A (11)
3.7. HPLC Conditions
3.8. Determination of Sugar Configuration
3.9. Molecular Docking Analysis
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 31 January 2023).
- Morais, F.S.; Canuto, K.M.; Ribeiro, P.R.V.; Silva, A.B.; Pessoa, O.D.L.; Freitas, C.D.T.; Alencar, N.M.N.; Oliveira, A.C.; Ramos, M.V. Chemical profiling of secondary metabolites from Himatanthus drasticus (Mart.) Plumel latex with inhibitory action against the enzymes α-amylase and α-glucosidase: In vitro and in silico assays. J. Ethnopharmacol. 2020, 253, 112644. [Google Scholar] [CrossRef]
- Tan, K.; Tesar, C.; Wilton, R.; Jedrzejczak, R.P.; Joachimiak, A. Interaction of antidiabetic α-glucosidase inhibitors and gut bacteria α-glucosidase. Protein Sci. 2018, 27, 1498–1508. [Google Scholar] [CrossRef]
- Kim, J.H.; Ahn, J.H.; Kim, S.K.; Lee, D.H.; Kim, H.S.; Shon, H.S.; Jeon, H.J.; Kim, T.H.; Cho, Y.W.; Kim, J.T.; et al. Combined use of basal insulin analog and acarbose reduces postprandial glucose in patients with uncontrolled type 2 diabetes. J. Diabetes Investig. 2015, 6, 219–226. [Google Scholar] [CrossRef]
- Kumar, R.V.; Sinha, V.R. Newer insights into the drug delivery approaches of α-glucosidase inhibitors. Expert Opin. Drug Deliv. 2012, 9, 403–416. [Google Scholar] [CrossRef]
- Lianza, M.; Poli, F.; Nascimento, A.M.d.; Soares da Silva, A.; da Fonseca, T.S.; Toledo, M.V.; Simas, R.C.; Chaves, A.R.; Leitão, G.G.; Leitão, S.G. In vitro α-glucosidase inhibition by Brazilian medicinal plant extracts characterised by ultra-high performance liquid chromatography coupled to mass spectrometry. J. Enzym. Inhib. Med. Chem. 2022, 37, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Shafique, Z.; Amjad, S.T.; Bin Asad, M.H.H. Enzymes inhibitors from natural sources with antidiabetic activity: A review. Phytother. Res. 2019, 33, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, F.; Islam, M.; Saeed, H.; Ahmed, A.; Hashmi, F.K.; Khan, K.M.; Dar, U.I.; Ullah, K.; Rana, S.M.; Saleem, B.; et al. Prediction of α-Glucosidase Inhibitory Activity of LC-ESI-TQ-MS/MS-Identified Compounds from Tradescantia pallida Leaves. Pharmaceutics 2022, 14, 2578. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Singha, S.; Kar, A.; Chanda, J.; Banerjee, S.; Dasgupta, B.; Haldar, P.K.; Sharma, N. Therapeutic importance of Cucurbitaceae: A medicinally important family. J. Ethnopharmacol. 2022, 282, 114599. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zou, L.Y.; Li, T.; Lai, D.N.; Wu, Y.Y.; Qin, S. Mogroside V inhibits LPS-induced COX-2 expression/ROS production and overexpression of HO-1 by blocking phosphorylation of AKT1 in RAW264.7 cells. Acta Biochim. Biophys. Sin. 2019, 51, 365–374. [Google Scholar] [CrossRef]
- Li, L.H.; Zheng, W.F.; Wang, C.; Qi, J.M.; Li, H.B. Mogroside V Protects against Hepatic Steatosis in Mice on a High-Fat Diet and LO2 Cells Treated with Free Fatty Acids via AMPK Activation. J. Evid.-Based Complement. Altern. Med. 2020, 2020, 7826874. [Google Scholar] [CrossRef]
- Liu, C.; Dai, L.; Liu, Y.; Rong, L.; Dou, D.; Sun, Y.; Ma, L. Antiproliferative Activity of Triterpene Glycoside Nutrient from Monk Fruit in Colorectal Cancer and Throat Cancer. Nutrients 2016, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.S.; Wang, C.C.; Qi, X.Y.; Zou, J.; Sun, Z.D. Antiglycation and antioxidant activities of mogroside extract from Siraitia grosvenorii (Swingle) fruits. J. Food Sci. Technol. 2018, 55, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fu, X.Y.; Wang, T.T.; Xu, R.; Shayiranbieke, A.; Zheng, X.X.; Jia, X.N.; Xiao, C.N.; Zhao, X.F. Screening of bioactive flavour compounds targeting muscarinic-3 acetylcholine receptor from Siraitia grosvenorii and evaluation of their synergistic anti-asthmatic activity. Food Chem. 2022, 395, 133593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, G.; Peng, Y.; Wang, M.; Li, X. Anti-hyperglycemic and anti-hyperlipidemic effects of a special fraction of Luohanguo extract on obese T2DM rats. J. Ethnopharmacol. 2020, 247, 112273. [Google Scholar] [CrossRef]
- Li, D.P.; El-Aasr, M.; Ikeda, T.; Ogata, M.; Miyashita, H.; Yoshimitsu, H.; Nohara, T. Two new cucurbitane-type glycosides obtained from roots of Siraitia grosvenori SWINGLE. Chem. Pharm. Bull. 2009, 57, 870–872. [Google Scholar] [CrossRef]
- Lu, F.L.; Sun, J.Y.; Jiang, X.H.; Yang, X.R.; Liu, H.W.; Yan, X.J.; Chen, Y.Y.; Li, D.P. The generally useful estimate of solvent systems method facilitates off-line two-dimensional countercurrent chromatography for isolating compositions from Siraitia grosvenorii roots. J. Sep. Sci. 2023, 46, e2200708. [Google Scholar] [CrossRef]
- Si, J.Y.; Chen, D.H.; Tu, G.Z. Siraitic acid F, a new nor-cucurbitacin with novel skeleton, from the roots of Siraitia grosvenorii. J. Asian Nat. Prod. Res. 2005, 7, 37–41. [Google Scholar] [CrossRef]
- Sun, J.Y.; Sun, J.Q.; Li, H.P.; Yan, X.J.; Li, D.P.; Lu, F.L. Preparation of cucurbitacin compounds in Siraitia grosvenorii roots by high speed countercurrent chromatography. Se Pu Chin. J. Chromatogr. 2022, 40, 364–371. [Google Scholar] [CrossRef]
- Si, J.Y.; Chen, D.H.; Shen, L.G.; Tu, G.Z. Studies on the chemical constituents from the root of Siraitia grosvenorii. Acta Pharm. Sin. 1999, 34, 918–920. [Google Scholar]
- Abudurexiti, A.; Zhang, R.; Zhong, Y.; Tan, H.; Yan, J.; Bake, S.; Ma, X. Identification of α-glucosidase inhibitors from Mulberry using UF-UPLC-QTOF-MS/MS and molecular docking. J. Funct. Foods 2023, 101, 105362. [Google Scholar] [CrossRef]
- Miranda de Souza Duarte-Filho, L.A.; Ortega de Oliveira, P.C.; Yanaguibashi Leal, C.E.; de Moraes, M.C.; Picot, L. Ligand fishing as a tool to screen natural products with anticancer potential. J. Sep. Sci. 2023, 2200964. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.Z.; Wu, L.F.; Lin, X.; Hu, X.P.; Wang, L. Phenolic profiles and screening of potential α-glucosidase inhibitors from Polygonum aviculare L. leaves using ultra-filtration combined with HPLC-ESI-qTOF-MS/MS and molecular docking analysis. Ind. Crops Prod. 2020, 154, 112673. [Google Scholar] [CrossRef]
- Muchiri, R.N.; van Breemen, R.B. Drug discovery from natural products using affinity selection-mass spectrometry. Drug Discov. Today Technol. 2021, 40, 59–63. [Google Scholar] [CrossRef]
- Choi, Y.; Jung, Y.; Kim, S.-N. Identification of Eupatilin from Artemisia argyi as a Selective PPARα Agonist Using Affinity Selection Ultrafiltration LC-MS. Molecules 2015, 20, 13753–13763. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.J.; Jiang, H.J.; Xu, C.; Tong, S.Q.; Yan, J.Z. Screening and identification of α-glucosidase inhibitors from Shenqi Jiangtang Granule by ultrafiltration liquid chromatography and mass spectrometry. J. Sep. Sci. 2018, 41, 797–805. [Google Scholar] [CrossRef]
- Abdulla, R.; Mansur, S.; Lai, H.; Ubul, A.; Sun, G.; Huang, G.; Aisa, H.A. Qualitative Analysis of Polyphenols in Macroporous Resin Pretreated Pomegranate Husk Extract by HPLC-QTOF-MS. Phytochem. Anal. 2017, 28, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.K. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry 1992, 31, 3307–3330. [Google Scholar] [CrossRef]
- Prakash, I.; Chaturvedula, V.S. Additional new minor cucurbitane glycosides from Siraitia grosvenorii. Molecules 2014, 19, 3669–3680. [Google Scholar] [CrossRef]
- Wang, X.; Lu, W.; Chen, J.; Gong, M.; Li, Y.; Lu, D.; Lv, Y.; Zheng, Q. Studies on the chemical constituents of root of Luohanguo (Siraitia grosvenori). Chin. Herb. Med. 1996, 27, 515–518. [Google Scholar]
- Yang, L.; Wang, H.; Yan, H.; Wang, K.; Wu, S.; Li, Y. (−)-Lariciresinol Isolated from the Roots of Isatis indigotica Fortune ex Lindl. Inhibits Hepatitis B Virus by Regulating Viral Transcription. Molecules 2022, 27, 3223. [Google Scholar] [CrossRef]
- Yu, D.; Yang, J. Nuclear magnetic resonance spectroscopy. In Analytical Chemistry Manual, 2nd ed.; Chemical Industry Press: Beijing, China, 1999; Volume VII, p. 873. [Google Scholar]
- Torres-Moreno, H.; Marcotullio, M.C.; Velázquez, C.; Ianni, F.; Robles-Zepeda, R.E. Cucurbitacin IIb, a steroidal triterpene from Ibervillea sonorae induces antiproliferative and apoptotic effects on cervical and lung cancer cells. Steroids 2020, 157, 108597. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.L.; Morita, T.; Kasai, R.; Zhou, J.; Wu, C.Y.; Tanaka, O. Saponins from Chinese medicinal plants. (I). Isolation and structures of hemslosides. Planta Med. 1984, 50, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, J.Y.; Cheon, C.; Ko, S.-G. Development and Validation of a New Analytical HPLC-PDA Method for Simultaneous Determination of Cucurbitacins B and D from the Roots of Trichosanthes kirilowii. J. Chem. 2022, 2022, 2109502. [Google Scholar] [CrossRef]
- Li, H.H.; Li, J.; Zhang, X.J.; Li, J.M.; Xi, C.; Wang, W.Q.; Lu, Y.L.; Xuan, L.J. 23,24-Dihydrocucurbitacin B promotes lipid clearance by dual transcriptional regulation of LDLR and PCSK9. Acta Pharmacol. Sin. 2020, 41, 327–335. [Google Scholar] [CrossRef]
- Li, Z.-R.; Qiu, M.-H.; Xu, X.-P.; Tian, J.; Nie, R.-L.; Duan, Z.-H.; Lei, Z.-M. Triterpenoid Saponins and a Cucurbitcin from Thladiatha cordifolia. Plant Divers. 1998, 20, 1–3. [Google Scholar]
- Kaushik, U.; Aeri, V.; Mir, S.R. Cucurbitacins—An insight into medicinal leads from nature. Pharmacogn. Rev. 2015, 9, 12–18. [Google Scholar]
- Kim, K.H.; Lee, I.S.; Park, J.Y.; Kim, Y.; An, E.J.; Jang, H.J. Cucurbitacin B Induces Hypoglycemic Effect in Diabetic Mice by Regulation of AMP-Activated Protein Kinase Alpha and Glucagon-Like Peptide-1 via Bitter Taste Receptor Signaling. Front. Pharmacol. 2018, 9, 1071. [Google Scholar] [CrossRef]
- Proença, C.; Ribeiro, D.; Freitas, M.; Fernandes, E. Flavonoids as potential agents in the management of type 2 diabetes through the modulation of α-amylase and α-glucosidase activity: A review. Crit. Rev. Food Sci. Nutr. 2022, 62, 3137–3207. [Google Scholar] [CrossRef]
- Ibrahim, R.M.; Elmasry, G.F.; Refaey, R.H.; El-Shiekh, R.A. Lepidium meyenii (Maca) Roots: UPLC-HRMS, Molecular Docking, and Molecular Dynamics. ACS Omega 2022, 7, 17339–17357. [Google Scholar] [CrossRef]
- Shahzad, D.; Saeed, A.; Larik, F.A.; Channar, P.A.; Abbas, Q.; Alajmi, M.F.; Arshad, M.I.; Erben, M.F.; Hassan, M.; Raza, H.; et al. Novel C-2 Symmetric Molecules as α-Glucosidase and α-Amylase Inhibitors: Design, Synthesis, Kinetic Evaluation, Molecular Docking and Pharmacokinetics. Molecules 2019, 24, 1511. [Google Scholar] [CrossRef]
- Liu, B.R.; Zheng, H.R.; Jiang, X.J.; Zhang, P.Z.; Wei, G.Z. Serratene triterpenoids from Lycopodium cernuum L. as α-glucosidase inhibitors: Identification, structure-activity relationship and molecular docking studies. Phytochemistry 2022, 195, 113056. [Google Scholar] [CrossRef] [PubMed]
- Flores-Bocanegra, L.; González-Andrade, M.; Bye, R.; Linares, E.; Mata, R. α-Glucosidase Inhibitors from Salvia circinata. J. Nat. Prod. 2017, 80, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.L.; Liu, M.L.; Li, S.Y.; Song, W.Q.; Ouyang, H.; Xiao, Z.P.; Zhu, H.L. Identification, potency evaluation, and mechanism clarification of α-glucosidase inhibitors from tender leaves of Lithocarpus polystachyus Rehd. Food Chem. 2022, 371, 131128. [Google Scholar] [CrossRef]
- Tanaka, T.; Tomii, K.; Ueda, T.; Kouno, I.; Nakashima, T. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Inhibition (%) 1 | IC50 (μg/mL) 2 |
|---|---|---|
| crude extract | 9.81 ± 1.21 | 4850.0 ± 217.6 |
| SGR1 | 48.09 ± 0.83 | 990.7 ± 21.6 |
| SGR2 | 59.07 ± 2.73 | 631.1 ± 50.2 |
| Acarbose | 55.15 ± 1.55 | 860.3 ± 31.3 3 |
| Peak | Retention Time (min) | Compound Names | Specific Binding Factors Mean ± SD (n = 3) | Inhibition (%) 1 | IC50 (μM) 2 |
|---|---|---|---|---|---|
| 1 | 10.16 | (-)-lariciresinol | 4.97 ± 0.15 | 78.18 ± 1.54 | 1832.87 ± 31.33 |
| 2 | 10.91 | 3,4′-dimethoxy-4,9,9′-trihydroxy-benzofuranolignan-7′-ene | 1.87 ± 0.77 | 63.43 ± 0.23 | 2275.47 ± 13.80 |
| 3 | 15.12 | 23,24-dihydrocucurbitacin F | 3.76 ± 0.33 | 38.27 ± 0.70 | n.d. |
| 4 | 16.19 | Siraitic acid III E | 2.93 ± 0.72 | 36.74 ± 0.36 | n.d. |
| 5 | 18.40 | Siraitic glycoside II F | 4.39 ± 0.42 | 42.73 ± 1.79 | n.d. |
| 6 | 19.13 | Siraitic acid IIb E | 3.91 ± 0.17 | 42.44 ± 0.81 | n.d. |
| 7 | 20.00 | Siraitic acid II E | 3.46 ± 1.03 | 58.48 ± 1.19 | 1206.84 ± 5.49 |
| 8 | 20.74 | Siraitic acid IV H | 0.27 ± 0.18 | 23.38 ± 1.05 | n.d. |
| 9 | 22.07 | Siraitic acid II G | 5.33 ± 2.11 | 53.12 ± 0.99 | 1580.96 ± 12.54 |
| 10 | 23.30 | Unknown | 6.27 ± 1.08 | - | - |
| 11 | 23.96 | Siraitic acid II A | 6.59 ± 0.91 | 50.60 ± 1.23 | 1239.78 ± 20.49 |
| 12 | 24.60 | Siraitic acid II B | 6.65 ± 1.18 | 51.34 ± 0.83 | 1034.53 ± 36.95 |
| 13 | 26.46 | 23,24-dihydrocucurbitacin F-25-acetate | 1.31 ± 0.06 | 21.67 ± 0.58 | n.d. |
| 14 | 29.36 | Siraitic acid II C | 17.26 ± 3.14 | 72.01 ± 2.18 | 430.13 ± 13.33 |
| 15 | 30.27 | Cucurbitacin B | 7.98 ± 1.11 | 50.59 ± 0.98 | 1505.41 ± 57.02 |
| 16 | 30.81 | 23,24-dihydrocucurbitacin B | 4.70 ± 0.76 | 27.54 ± 3.80 | n.d. |
| 17 | 32.58 | Dihydroisocucurbitacin B-25-acetate | 2.28 ± 0.44 | 23.74 ± 0.33 | n.d. |
| Position | 4 | 6 | 7 | 8 | 9 | 11 |
|---|---|---|---|---|---|---|
| aglycone | ||||||
| 1 | 2.60, 1.42, m | 2.59, 1.41, m | 2.65, 1.51, m | 1.85, m | 2.02, 1.87, m | 1.11, 2.05, m |
| 2 | 2.47, 2.38, m | 2.47, 2.38, m | 2.53, 2.44, m | 2.58, 2.16, m | 1.54, 1.91, m | |
| 3 | 3.73, m | 3.86, d (7.5) | ||||
| 4 | 1.58, dd (7.1, 3.0) | |||||
| 6 | 2.79, 1.80, m | 2.80, 1.78, m | 2.84, 1.86, m | 5.51, m | 2.45, m | 1.84, 1.98, m |
| 7 | 1.51, 1.24, m | 1.48, 1.23, m | 1.58, 1.33, m | 1.59, 1.19, m | 1.68, m | 1.25, 1.96, m |
| 8 | 1.82, m | 1.82, m | 1.86, m | 1.96, m | 1.90, m | 2.04, br t (7.2) |
| 9 | 2.27, m | 2.28, t, (11.1) | 2.37, t (11.4) | 2.18, m | ||
| 10 | 2.72, m | 2.71, m | 2.75, m | 3.14, m | 2.91, m | 2.40, dd (11.8, 5.8) |
| 11 | ||||||
| 12 | 2.80, 2.47, m | 2.80, 2.47, m | 2.93, 2.56, d (12.4) | 2.50, m | 2.82, 2.61, d (16.3) | |
| 15 | 1.64, 1.95, m | 1.93, 1.62, m | 1.95, 1.68, m | 1.17, m | 1.29, m | |
| 16 | 4.52, m | 4.52, t (7.0) | 4.30, m | 1.89, m | 1.94, m | |
| 17 | 2.21, m | 2.13, m | 2.12, m | 1.70, m | ||
| 18 | 0.75, s | 0.74, s | 0.83, s | 0.70, s | 0.76, s | 0.68, s |
| 19 | 1.11, s | 3.67, d (8.4), 4.73, d (8.4) | ||||
| 20 | 1.60, m | 1.61, m | 1.61, m | 1.30, m | 1.34, m | |
| 21 | 0.86, d (6.0) | 0.87, d (6.6) | 0.93, d (6.2) | 0.82, d (6.4) | 0.84, d (6.5) | 0.85, d (6.4) |
| 22 | 2.11, 1.72, m | 2.07, 1.79, m | 1.98, m | 1.37, 0.98, m | 1.44, m | |
| 23 | 2.36, 2.08, m | 2.44, 2,10, m | 2.47, 2.04, m | 2.15, 1.94, m | 2.19, m | |
| 24 | 7.36, t (7.7) | 7.40, t (7.1) | 7.38, t (6.7) | 7.06, t (6.9) | 7.09, t (7.5) | |
| 26 | 1.93, s | 1.95, s | 1.93, s | 1.91, s | 1.92, s | 1.93, s |
| 28 | 1.91, s | 1.91, s | 1.89, s | 1.11, s | 1.90, s | 1.35, d (7.0) |
| 29 | 1.52, s | |||||
| 30 | 1.50, s | 1.51, s | 1.48, s | 0.90, s | 1.16, s | 1.19, s |
| sugar | ||||||
| GI1 | 4.87, d (7.7) | 4.88, d (7.7) | 6.42, d (8.2) | 4.81, m | 6.45, d (8.0) | 6.47, d (8.0) |
| GI2 | 4.01, m | 3.95, t (8.5) | 4.16, m | 3.90, m | 4.25, m | 4.01, m |
| GI3 | 4.24, m | 4.23, m | 4.15, m | 4.20, m | 3.89, m | 3.90, m |
| GI4 | 4.16, m | 4.17, m | 4.41, t (9.7) | 3.97, m | 4.44, m | 4.21, m |
| GI5 | 3.92, m | 4.01, m | 4.28, m | 4.11, m | 4.22, m | 4.18, m |
| GI6 | 4.34, 4.46, m | 4.44, 4.37, m | 4.77, 4.37, m | 4.83, 4.30, m | 4.79, d (11.9) 4.38, m | 4.78, 4.38, m |
| GII1 | 6.37, d (7.6) | 6.50, d (7.6) | 6.47, d (7.6) | 5.15, m | 5.02, m | 4.93, d (8.3) |
| GII2 | 4.25, m | 4.34, m | 4.01, t (8.2) | 4.04, m | 4.05, t (8.2) | 4.01, m |
| GII3 | 4.19, m | 4.31, m | 3.90, m | 4.20, m | 4.31, m | 4.01, m |
| GII4 | 4.20, m | 4.36, m | 4.21, m | 4.11, m | 4.24, m | 4.18, m |
| GII5 | 4.24, m | 4.02, m | 4.18, m | 4.20, m | 4.22, m | 3.85, m |
| GII6 | 4.73, 4.34, m | 4.66, 4.42, m | 4.50, 4.36, m | 4.53, 4.37, m | 4.49, d (11.9) 4.37, m | 4.63, 4.40, m |
| GIII1 | 4.99, d (7.9) | 6.43, d (7.6) | ||||
| GIII2 | 4.01, m | 4.22, m | ||||
| GIII3 | 4.01, m | 4.23, m | ||||
| GIII4 | 4.20, m | 4.41, m | ||||
| GIII5 | 3.87, m | 4.20, m | ||||
| GIII6 | 4.65, 4.42, m | 4.77, 4.37, m | ||||
| GIV1 | 5.02, d (7.8) | |||||
| GIV2 | 4.01, m | |||||
| GIV3 | 3.88, m | |||||
| GIV4 | 4.23, m | |||||
| GIV5 | 4.20, m | |||||
| GIV6 | 4.47, 4.35, m |
| Position | 4 | 6 | 7 | 8 | 9 | 11 | Position | 4 | 6 | 7 | 8 | 9 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aglycone | sugar | ||||||||||||
| 1 | 28.8 | 28.8 | 28.9 | 26.2 | 24.9 | 18.5 | GI1 | 102.8 | 102.8 | 96.6 | 107.5 | 96.6 | 96.7 |
| 2 | 37.6 | 37.6 | 37.7 | 30.2 | 37.8 | 20.2 | GI2 | 75.4 | 75.5 | 74.5 | 75.7 | 74.5 | 75.6 |
| 3 | 198.9 | 198.8 | 198.9 | 88.2 | 197.9 | 71.0 | GI3 | 79.0 | 78.7 | 78.2 | 79.0 | 78.9 | 78.3 |
| 4 | 130.3 | 130.3 | 130.3 | 43.0 | 132.9 | 40.8 | GI4 | 72.6 | 72.8 | 71.1 | 72.1 | 71.2 | 71.3 |
| 5 | 158.2 | 158.1 | 158.2 | 144.8 | 157.3 | 86.4 | GI5 | 78.7 | 79.0 | 78.9 | 77.8 | 78.3 | 79.0 |
| 6 | 31.2 | 31.2 | 31.3 | 118.9 | 28.5 | 26.2 | GI6 | 62.9 | 62.4 | 69.8 | 70.9 | 69.9 | 70.0 |
| 7 | 27.9 | 27.9 | 27.9 | 28.7 | 21.7 | 28.5 | GII1 | 96.5 | 96.5 | 105.8 | 106.0 | 105.7 | 105.8 |
| 8 | 45.3 | 45.3 | 45.4 | 36.5 | 45.8 | 46.0 | GII2 | 74.4 | 74.7 | 75.5 | 75.6 | 75.5 | 74.6 |
| 9 | 55.0 | 55.0 | 55.1 | 50.7 | 50.0 | 60.6 | GII3 | 78.6 | 79.0 | 78.7 | 79.0 | 78.8 | 78.9 |
| 10 | 38.1 | 38.1 | 38.1 | 35.4 | 41.1 | 45.4 | GII4 | 71.8 | 71.3 | 71.8 | 72.1 | 71.8 | 71.9 |
| 11 | 210.9 | 210.8 | 211.0 | 212.5 | 214.8 | 210.3 | GII5 | 78.8 | 79.7 | 78.8 | 78.9 | 78.8 | 78.8 |
| 12 | 52.5 | 52.6 | 52.8 | 52.3 | 51.5 | 51.0 | GII6 | 69.8 | 64.2 | 62.9 | 63.2 | 63.0 | 63.0 |
| 13 | 47.7 | 47.8 | 47.9 | 47.0 | 47.4 | 49.3 | GIII1 | 105.5 | 96.7 | ||||
| 14 | 51.1 | 51.1 | 51.5 | 47.8 | 49.7 | 49.6 | GIII2 | 75.4 | 74.6 | ||||
| 15 | 40.0 | 40.0 | 45.4 | 33.4 | 34.9 | 34.2 | GIII3 | 78.8 | 78.9 | ||||
| 16 | 83.0 | 82.8 | 77.0 | 57.8 | 28.3 | 32.6 | GIII4 | 73.5 | 71.2 | ||||
| 17 | 56.3 | 56.3 | 58.7 | 50.5 | 50.8 | 49.9 | GIII5 | 78.8 | 78.4 | ||||
| 18 | 17.9 | 17.9 | 17.9 | 17.1 | 17.9 | 17.0 | GIII6 | 64.0 | 70.0 | ||||
| 19 | 23.2 | 74.9 | GIV1 | 105.8 | |||||||||
| 20 | 34.9 | 34.8 | 35.2 | 36.7 | 36.5 | 36.4 | GIV2 | 75.6 | |||||
| 21 | 19.2 | 19.2 | 19.1 | 18.8 | 18.6 | 18.7 | GIV3 | 78.8 | |||||
| 22 | 33.8 | 33.9 | 34.9 | 35.5 | 35.2 | 35.3 | GIV4 | 71.8 | |||||
| 23 | 26.2 | 26.0 | 26.2 | 26.3 | 26.2 | 26.2 | GIV5 | 78.7 | |||||
| 24 | 146.4 | 146.3 | 145.5 | 145.2 | 145.0 | 144.9 | GIV6 | 63.0 | |||||
| 25 | 127.2 | 127.3 | 127.8 | 128.0 | 127.9 | 127.9 | |||||||
| 26 | 13.0 | 13.1 | 12.9 | 13.0 | 12.8 | 12.9 | |||||||
| 27 | 167.6 | 167.6 | 167.5 | 167.6 | 167.5 | 167.6 | |||||||
| 28 | 11.5 | 11.6 | 11.6 | 27.3 | 11.6 | 13.5 | |||||||
| 29 | 25.6 | ||||||||||||
| 30 | 17.7 | 17.7 | 17.9 | 17.2 | 19.7 | 20.4 |
| Main Compound | Binding Affinity (kcal/mol) | Number of Binding to Residues | Residues Involved in H-Bond Formation | Hydrophobic Interaction |
|---|---|---|---|---|
| 1 | −8.5 | 5 | HIS 280, GLN 279, SER 157, ASP 307, ARG 442 | SER 157, GLN 279, HIS 280, ASP 307, ARG 442 π-π stacking: TYR158 |
| 2 | −8.5 | 5 | SER 241, ASP 242, TYR 158, HIS 280 | TYR 158, SER 241, ASP 242, HIS 280 |
| 7 | −9.9 | 8 | LYS 156, HIS 280, GLN 279, ARG 315, GLY 309, ASN 415 | VAL 319, VAL 308, ASP325, PHE 321 |
| 9 | −9.9 | 5 | ARG 315, ARG 442, GLN 279 | VAL 308, PHE 321, ASP 325, ASP 307 |
| 11 | −9.8 | 5 | HIS 280, THR 310, GLY 309, ASP 352, ARG 442 | ASP 352, PHE 303, ARG 315, TYR 158, PRO 312 |
| 12 | −10.0 | 5 | GLN 279, ARG 315, ASP 352, ARG442 | VAL 319, PHE 321, ASP325, ASP 307 |
| 14 | −10.0 | 7 | ARG 315, ASP 352, GLN 279, GLY 309, ARG442 | VAL 319, PHE 321, ASP 325, VAL 308 |
| 15 | −8.4 | 2 | GLN 279, HIS 280 | PHE159, PHE 303, ARG 315, TYR 158 |
| Acarbose | −8.4 | 9 | GLN 279, ASP352, ARG 442, GLU 411, PRO 312, HIS 280, LYS 156 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, F.; Sun, J.; Jiang, X.; Song, J.; Yan, X.; Teng, Q.; Li, D. Identification and Isolation of α-Glucosidase Inhibitors from Siraitia grosvenorii Roots Using Bio-Affinity Ultrafiltration and Comprehensive Chromatography. Int. J. Mol. Sci. 2023, 24, 10178. https://doi.org/10.3390/ijms241210178
Lu F, Sun J, Jiang X, Song J, Yan X, Teng Q, Li D. Identification and Isolation of α-Glucosidase Inhibitors from Siraitia grosvenorii Roots Using Bio-Affinity Ultrafiltration and Comprehensive Chromatography. International Journal of Molecular Sciences. 2023; 24(12):10178. https://doi.org/10.3390/ijms241210178
Chicago/Turabian StyleLu, Fenglai, Jiayi Sun, Xiaohua Jiang, Jingru Song, Xiaojie Yan, Qinghu Teng, and Dianpeng Li. 2023. "Identification and Isolation of α-Glucosidase Inhibitors from Siraitia grosvenorii Roots Using Bio-Affinity Ultrafiltration and Comprehensive Chromatography" International Journal of Molecular Sciences 24, no. 12: 10178. https://doi.org/10.3390/ijms241210178
APA StyleLu, F., Sun, J., Jiang, X., Song, J., Yan, X., Teng, Q., & Li, D. (2023). Identification and Isolation of α-Glucosidase Inhibitors from Siraitia grosvenorii Roots Using Bio-Affinity Ultrafiltration and Comprehensive Chromatography. International Journal of Molecular Sciences, 24(12), 10178. https://doi.org/10.3390/ijms241210178