
The Brain at High Altitude: From Molecular Signaling to Cognitive Performance

 ,
,  , and
, and 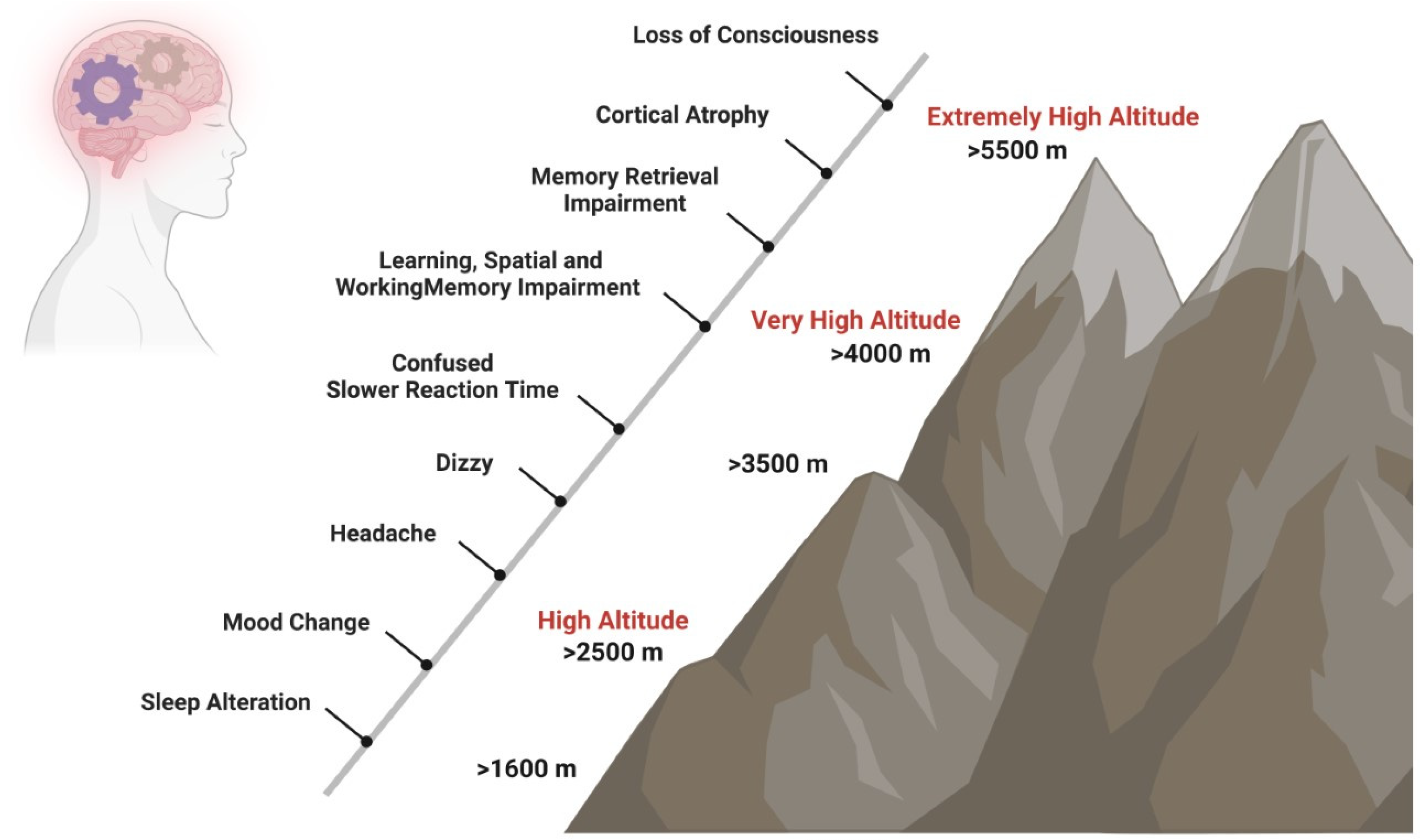{kind=link}
{kind=link}
Abstract
1. High Altitude and Cognition
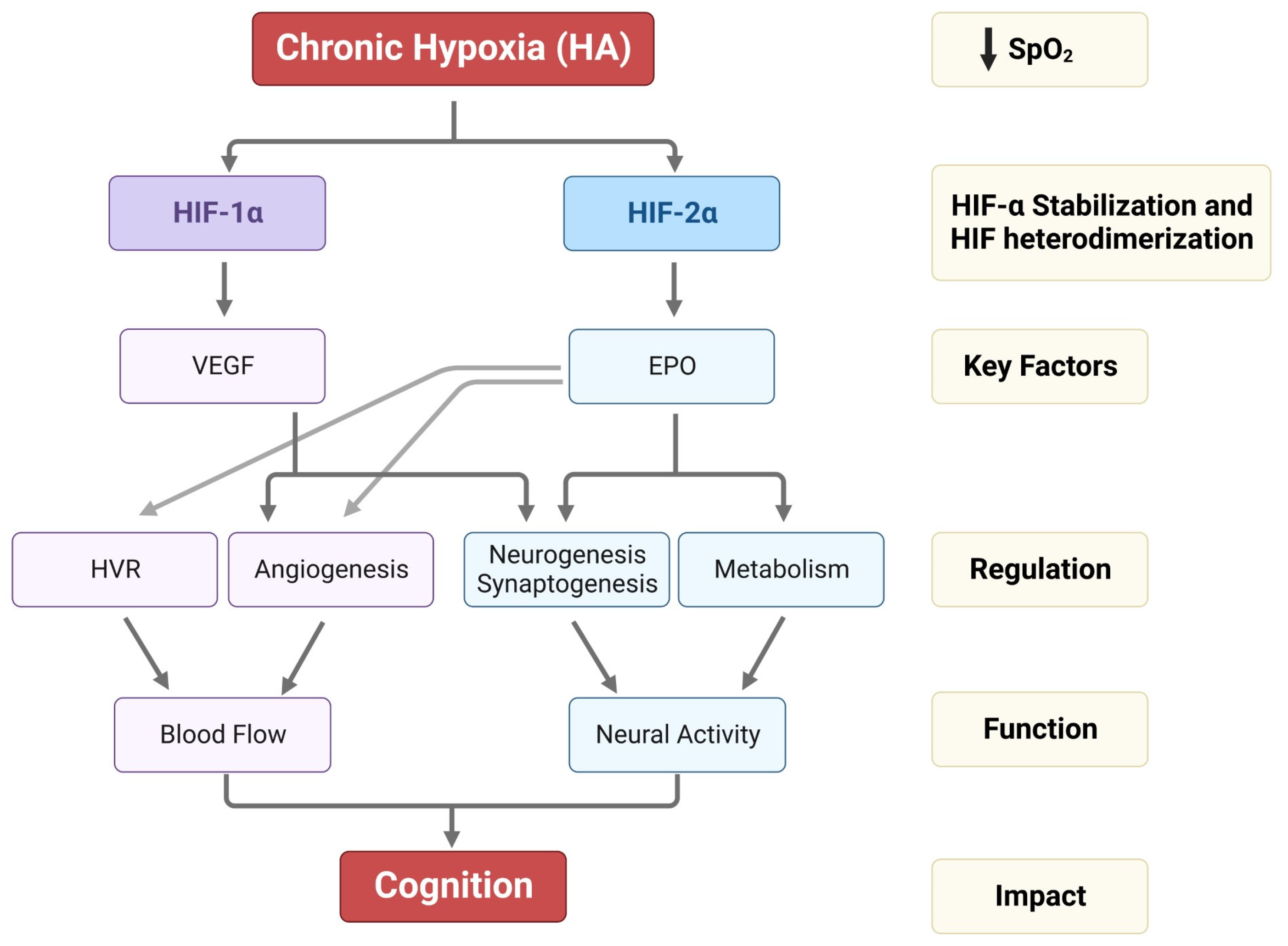
2. Hypoxia-Inducible Factors (HIFs) in the Brain Adaptation to HA
3. Signaling Pathways Interacting with HIFs
4. Central Respiratory Acclimatization to HA
5. The Neuronal Response to HA
5.1. Neurogenesis
5.2. Neuronal Survival
5.3. Neuronal Metabolism
5.4. Neurotransmitter Release
5.5. Neuronal Membrane Potential and Ion Exchange
6. HIF-2 and Synaptic Plasticity
7. Potential Therapeutic Implications of Targeting HIF-2-Mediated Modifications in HA-Related Illnesses
- Targeting HIF-2-mediated pathways of neural plasticity may offer novel approaches for enhancing cognitive function and memory formation in HA environments, which can be impaired by hypoxia-induced changes in brain function.
- Gene therapy approaches targeting specific HIF-2-regulated genes or signaling pathways may provide a more targeted and long-lasting way to modulate the effects of hypoxia on the brain and improve HA-related illness outcomes.
- Developing personalized medicine approaches that consider an individual’s genetic and epigenetic profiles may help identify those at higher risk for HA-related illnesses and tailor interventions accordingly.
- Investigating the potential use of non-pharmacological interventions, such as cognitive or physical training, to enhance HIF-2-mediated modifications that promote neural plasticity and cognitive function in HA environments.
8. Other Factors than Hypoxia Might Affect the Brain at HA
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DelMastro, K.; Hellem, T.; Kim, N.; Kondo, D.; Sung, Y.H.; Renshaw, P.F. Incidence of major depressive episode correlates with elevation of substate region of residence. J. Affect. Disord. 2011, 129, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Kious, B.M.; Bakian, A.; Zhao, J.; Mickey, B.; Guille, C.; Renshaw, P.; Sen, S. Altitude and risk of depression and anxiety: Findings from the intern health study. Int. Rev. Psychiatry 2019, 31, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Koester-Hegmann, C.; Bengoetxea, H.; Kosenkov, D.; Thiersch, M.; Haider, T.; Gassmann, M.; Schneider Gasser, E.M. High-Altitude Cognitive Impairment Is Prevented by Enriched Environment Including Exercise via VEGF Signaling. Front. Cell. Neurosci. 2018, 12, 532. [Google Scholar] [CrossRef] [PubMed]
- Virues-Ortega, J.; Buela-Casal, G.; Garrido, E.; Alcazar, B. Neuropsychological functioning associated with high-altitude exposure. Neuropsychol. Rev. 2004, 14, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, H.; Fu, S.; Guo, S.; Yang, X.; Luo, P.; Han, B. Long-Term Exposure to High Altitude Affects Voluntary Spatial Attention at Early and Late Processing Stages. Sci. Rep. 2014, 4, 4443. [Google Scholar] [CrossRef]
- Bahrke, M.S.; Shukitt-Hale, B. Effects of altitude on mood, behaviour and cognitive functioning. A review. Sports Med. 1993, 16, 97–125. [Google Scholar] [CrossRef]
- Ma, H.; Zhang, D.; Li, X.; Ma, H.; Wang, N.; Wang, Y. Long-term exposure to high altitude attenuates verbal and spatial working memory: Evidence from an event-related potential study. Brain Behav. 2019, 9, e01256. [Google Scholar] [CrossRef]
- Virues-Ortega, J.; Bucks, R.; Kirkham, F.J.; Baldeweg, T.; Baya-Botti, A.; Hogan, A.M.; Bolivian Children Living at Altitude project. Changing patterns of neuropsychological functioning in children living at high altitude above and below 4000 m: A report from the Bolivian Children Living at Altitude (BoCLA) study. Dev. Sci. 2011, 14, 1185–1193. [Google Scholar] [CrossRef]
- Shukitt, B.L.; Banderet, L.E. Mood states at 1600 and 4300 meters terrestrial altitude. Aviat. Space Environ. Med. 1988, 59, 530–532. [Google Scholar]
- Shukitt-Hale, B.; Lieberman, H.R. The Effect of Altitude on Cognitive Performance and Mood States; National Academies Press (US), Institute of Medicine (US) Committee on Military Nutrition Research: Washington, DC, USA, 1996; Volume 22. [Google Scholar]
- Shukitt-Hale, B.; Banderet, L.E.; Lieberman, H.R. Relationships between symptoms, moods, performance, and acute mountain sickness at 4700 meters. Aviat. Space Environ. Med. 1991, 62, 865–869. [Google Scholar]
- Hernandez-Vasquez, A.; Vargas-Fernandez, R.; Rojas-Roque, C.; Gamboa-Unsihuay, J.E. Association between altitude and depression in Peru: An 8-year pooled analysis of population-based surveys. J. Affect. Disord. 2022, 299, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Pun, M.; Guadagni, V.; Bettauer, K.M.; Drogos, L.L.; Aitken, J.; Hartmann, S.E.; Furian, M.; Muralt, L.; Lichtblau, M.; Bader, P.R.; et al. Effects on Cognitive Functioning of Acute, Subacute and Repeated Exposures to High Altitude. Front. Physiol. 2018, 9, 1131. [Google Scholar] [CrossRef] [PubMed]
- Shukitt-Hale, B.; Stillman, M.J.; Welch, D.I.; Levy, A.; Devine, J.A.; Lieberman, H.R. Hypobaric hypoxia impairs spatial memory in an elevation-dependent fashion. Behav. Neural Biol. 1994, 62, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Drevets, W.C.; Gautier, C.; Price, J.C.; Kupfer, D.J.; Kinahan, P.E.; Grace, A.A.; Price, J.L.; Mathis, C.A. Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol. Psychiatry 2001, 49, 81–96. [Google Scholar] [CrossRef]
- Young, S.N. Elevated incidence of suicide in people living at altitude, smokers and patients with chronic obstructive pulmonary disease and asthma: Possible role of hypoxia causing decreased serotonin synthesis. J. Psychiatry Neurosci. 2013, 38, 423–426. [Google Scholar] [CrossRef]
- Barath, A.S.; Rusheen, A.E.; Rojas Cabrera, J.M.; Price, J.B.; Owen, R.L.; Shin, H.; Jang, D.P.; Blaha, C.D.; Lee, K.H.; Oh, Y. Hypoxia-Associated Changes in Striatal Tonic Dopamine Release: Real-Time in vivo Measurements with a Novel Voltammetry Technique. Front. Neurosci. 2020, 14, 869. [Google Scholar] [CrossRef]
- Huang, C.C.; Lajevardi, N.S.; Tammela, O.; Pastuszko, A.; Delivoria-Papadopoulos, M.; Wilson, D.F. Relationship of extracellular dopamine in striatum of newborn piglets to cortical oxygen pressure. Neurochem. Res. 1994, 19, 649–655. [Google Scholar] [CrossRef]
- Pitychoutis, P.M.; Dalla, C.; Sideris, A.C.; Tsonis, P.A.; Papadopoulou-Daifoti, Z. 5-HT(1A), 5-HT(2A), and 5-HT(2C) receptor mRNA modulation by antidepressant treatment in the chronic mild stress model of depression: Sex differences exposed. Neuroscience 2012, 210, 152–167. [Google Scholar] [CrossRef]
- Ma, S.; Mifflin, S.W.; Cunningham, J.T.; Morilak, D.A. Chronic intermittent hypoxia sensitizes acute hypothalamic-pituitary-adrenal stress reactivity and Fos induction in the rat locus coeruleus in response to subsequent immobilization stress. Neuroscience 2008, 154, 1639–1647. [Google Scholar] [CrossRef]
- Kondo, D.G.; Forrest, L.N.; Shi, X.; Sung, Y.H.; Hellem, T.L.; Huber, R.S.; Renshaw, P.F. Creatine target engagement with brain bioenergetics: A dose-ranging phosphorus-31 magnetic resonance spectroscopy study of adolescent females with SSRI-resistant depression. Amino Acids 2016, 48, 1941–1954. [Google Scholar] [CrossRef]
- Jain, K.; Prasad, D.; Singh, S.B.; Kohli, E. Hypobaric Hypoxia Imbalances Mitochondrial Dynamics in Rat Brain Hippocampus. Neurol. Res. Int. 2015, 2015, 742059. [Google Scholar] [CrossRef]
- Shi, X.F.; Carlson, P.J.; Kim, T.S.; Sung, Y.H.; Hellem, T.L.; Fiedler, K.K.; Kim, S.E.; Glaeser, B.; Wang, K.; Zuo, C.S.; et al. Effect of altitude on brain intracellular pH and inorganic phosphate levels. Psychiatry Res. 2014, 222, 149–156. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Penaloza, D.; Arias-Stella, J. The heart and pulmonary circulation at high altitudes: Healthy highlanders and chronic mountain sickness. Circulation 2007, 115, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Camayo, J.; Mejia, C.R.; Callacondo, D.; Dawson, J.A.; Posso, M.; Galvan, C.A.; Davila-Arango, N.; Bravo, E.A.; Loescher, V.Y.; Padilla-Deza, M.M.; et al. Reference values for oxygen saturation from sea level to the highest human habitation in the Andes in acclimatised persons. Thorax 2018, 73, 776–778. [Google Scholar] [CrossRef]
- Soliz, J.; Joseph, V.; Soulage, C.; Becskei, C.; Vogel, J.; Pequignot, J.M.; Ogunshola, O.; Gassmann, M. Erythropoietin regulates hypoxic ventilation in mice by interacting with brainstem and carotid bodies. J. Physiol. 2005, 568, 559–571. [Google Scholar] [CrossRef]
- Lombardi, C.; Meriggi, P.; Agostoni, P.; Faini, A.; Bilo, G.; Revera, M.; Caldara, G.; Di Rienzo, M.; Castiglioni, P.; Maurizio, B.; et al. High-altitude hypoxia and periodic breathing during sleep: Gender-related differences. J. Sleep. Res. 2013, 22, 322–330. [Google Scholar] [CrossRef]
- Julian, C.G.; Vargas, E.; Gonzales, M.; Davila, R.D.; Ladenburger, A.; Reardon, L.; Schoo, C.; Powers, R.W.; Lee-Chiong, T.; Moore, L.G. Sleep-disordered breathing and oxidative stress in preclinical chronic mountain sickness (excessive erythrocytosis). Respir. Physiol. Neurobiol. 2013, 186, 188–196. [Google Scholar] [CrossRef]
- Beall, C.M. Oxygen saturation increases during childhood and decreases during adulthood among high altitude native Tibetians residing at 3800–4200 m. High. Alt. Med. Biol. 2000, 1, 25–32. [Google Scholar] [CrossRef]
- Hill, C.M.; Baya, A.; Gavlak, J.; Carroll, A.; Heathcote, K.; Dimitriou, D.; L’Esperance, V.; Webster, R.; Holloway, J.; Virues-Ortega, J.; et al. Adaptation to Life in the High Andes: Nocturnal Oxyhemoglobin Saturation in Early Development. Sleep 2016, 39, 1001–1008. [Google Scholar] [CrossRef]
- Ucros, S.; Granados, C.M.; Castro-Rodriguez, J.A.; Hill, C.M. Oxygen Saturation in Childhood at High Altitude: A Systematic Review. High. Alt. Med. Biol. 2020, 21, 114–125. [Google Scholar] [CrossRef]
- Ucros, S.; Castro-Guevara, J.A.; Hill, C.M.; Castro-Rodriguez, J.A. Breathing Patterns and Oxygenation Saturation during Sleep in Children Habitually Living at High Altitude in the Andes: A Systematic Review. Front. Pediatr. 2021, 9, 798310. [Google Scholar] [CrossRef] [PubMed]
- Ucros, S.; Granados, C.; Hill, C.; Castro-Rodriguez, J.A.; Ospina, J.C. Normal values for respiratory sleep polygraphy in children aged 4 to 9 years at 2560 m above sea level. J. Sleep. Res. 2021, 30, e13341. [Google Scholar] [CrossRef] [PubMed]
- de Aquino Lemos, V.; Antunes, H.K.; dos Santos, R.V.; Lira, F.S.; Tufik, S.; de Mello, M.T. High altitude exposure impairs sleep patterns, mood, and cognitive functions. Psychophysiology 2012, 49, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Bondi, M.W.; Gayles, E.; Delis, D.C. Mechanisms of Memory Dysfunction during High Altitude Hypoxia Training in Military Aircrew. J. Int. Neuropsychol. Soc. 2017, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pavlicek, V.; Schirlo, C.; Nebel, A.; Regard, M.; Koller, E.A.; Brugger, P. Cognitive and emotional processing at high altitude. Aviat. Space Environ. Med. 2005, 76, 28–33. [Google Scholar]
- Zhang, D.; Ma, H.; Huang, J.; Zhang, X.; Ma, H.; Liu, M. Exploring the impact of chronic high-altitude exposure on visual spatial attention using the ERP approach. Brain Behav. 2018, 8, e00944. [Google Scholar] [CrossRef]
- Fayed, N.; Modrego, P.J.; Morales, H. Evidence of brain damage after high-altitude climbing by means of magnetic resonance imaging. Am. J. Med. 2006, 119, 168.e1–168.e6. [Google Scholar] [CrossRef]
- Fan, C.; Zhao, Y.; Yu, Q.; Yin, W.; Liu, H.; Lin, J.; Yang, T.; Fan, M.; Gesang, L.; Zhang, J. Reversible Brain Abnormalities in People without Signs of Mountain Sickness during High-Altitude Exposure. Sci. Rep. 2016, 6, 33596. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, J.; Sun, Y.; Huang, Y.; Ye, H.; Wang, X.; Yang, T.; Jiang, X.; Zhang, J. Compromised white matter microstructural integrity after mountain climbing: Evidence from diffusion tensor imaging. High. Alt. Med. Biol. 2012, 13, 118–125. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.; Han, Q.; Lin, J.; Yang, T.; Chen, Z.; Zhang, J. Long-term acclimatization to high-altitude hypoxia modifies interhemispheric functional and structural connectivity in the adult brain. Brain Behav. 2016, 6, e00512. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J. The human brain in a high altitude natural environment: A review. Front. Hum. Neurosci. 2022, 16, 915995. [Google Scholar] [CrossRef]
- Richardson, C.; Hogan, A.M.; Bucks, R.S.; Baya, A.; Virues-Ortega, J.; Holloway, J.W.; Rose-Zerilli, M.; Palmer, L.J.; Webster, R.J.; Kirkham, F.J.; et al. Neurophysiological evidence for cognitive and brain functional adaptation in adolescents living at high altitude. Clin. Neurophysiol. 2011, 122, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Wehby, G.L. Living on higher ground reduces child neurodevelopment-evidence from South America. J. Pediatr. 2013, 162, 606–611.e1. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.S.; Kim, T.S.; Kim, N.; Kuykendall, M.D.; Sherwood, S.N.; Renshaw, P.F.; Kondo, D.G. Association between Altitude and Regional Variation of ADHD in Youth. J. Atten. Disord. 2018, 22, 1299–1306. [Google Scholar] [CrossRef]
- Mallet, R.T.; Burtscher, J.; Pialoux, V.; Pasha, Q.; Ahmad, Y.; Millet, G.P.; Burtscher, M. Molecular Mechanisms of High-Altitude Acclimatization. Int. J. Mol. Sci. 2023, 24, 1968. [Google Scholar] [CrossRef]
- Gassmann, N.N.; van Elteren, H.A.; Goos, T.G.; Morales, C.R.; Rivera-Ch, M.; Martin, D.S.; Peralta, P.C.; Passano Del Carpio, A.; Aranibar Machaca, S.; Huicho, L.; et al. Pregnancy at high altitude in the Andes leads to increased total vessel density in healthy newborns. J. Appl. Physiol. 2016, 121, 709–715. [Google Scholar] [CrossRef]
- Iwasaki, K.; Zhang, R.; Zuckerman, J.H.; Ogawa, Y.; Hansen, L.H.; Levine, B.D. Impaired dynamic cerebral autoregulation at extreme high altitude even after acclimatization. J. Cereb. Blood Flow. Metab. 2011, 31, 283–292. [Google Scholar] [CrossRef]
- Merz, T.M.; Treyer, V.; Hefti, U.; Spengler, C.M.; Schwarz, U.; Buck, A.; Maggiorini, M. Changes in cerebral glucose metabolism after an expedition to high altitudes. High. Alt. Med. Biol. 2006, 7, 28–38. [Google Scholar] [CrossRef]
- Hogan, A.M.; Virues-Ortega, J.; Botti, A.B.; Bucks, R.; Holloway, J.W.; Rose-Zerilli, M.J.; Palmer, L.J.; Webster, R.J.; Baldeweg, T.; Kirkham, F.J. Development of aptitude at altitude. Dev. Sci. 2010, 13, 533–544. [Google Scholar] [CrossRef]
- Yan, X. Cognitive impairments at high altitudes and adaptation. High Alt. Med. Biol. 2014, 15, 141–145. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Wang, J.; Xin, Z.; Zhang, Q.; Zhang, W.; Xi, Y.; Zhu, Y.; Li, C.; Li, J.; et al. Altered resting-state networks may explain the executive impairment in young health immigrants into high-altitude area. Brain Imaging Behav. 2021, 15, 147–156. [Google Scholar] [CrossRef]
- Ledford, H.; Callaway, E. Biologists who decoded how cells sense oxygen win medicine Nobel. Nature 2019, 574, 161–162. [Google Scholar] [CrossRef]
- Bracken, C.P.; Fedele, A.O.; Linke, S.; Balrak, W.; Lisy, K.; Whitelaw, M.L.; Peet, D.J. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 2006, 281, 22575–22585. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Xiao, J.; Liu, J.; Fan, X.; Fan, F.; Lei, H. Genetic changes in the EPAS1 gene between Tibetan and Han ethnic groups and adaptation to the plateau hypoxic environment. PeerJ 2019, 7, e7943. [Google Scholar] [CrossRef] [PubMed]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.; Klaus, S.J.; Bachmann, S. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010, 77, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Percy, M.J. The HIF pathway and erythrocytosis. Annu. Rev. Pathol. 2011, 6, 165–192. [Google Scholar] [CrossRef] [PubMed]
- Kleszka, K.; Leu, T.; Quinting, T.; Jastrow, H.; Pechlivanis, S.; Fandrey, J.; Schreiber, T. Hypoxia-inducible factor-2alpha is crucial for proper brain development. Sci. Rep. 2020, 10, 19146. [Google Scholar] [CrossRef]
- Jacobson, L.O.; Goldwasser, E.; Fried, W.; Plzak, L.F. Studies on erythropoiesis. VII. The role of the kidney in the production of erythropoietin. Trans. Assoc. Am. Physicians 1957, 70, 305–317. [Google Scholar]
- Kimakova, P.; Solar, P.; Solarova, Z.; Komel, R.; Debeljak, N. Erythropoietin and Its Angiogenic Activity. Int. J. Mol. Sci. 2017, 18, 1519. [Google Scholar] [CrossRef]
- Khalid, K.; Frei, J.; Aboouf, M.A.; Koester-Hegmann, C.; Gassmann, M.; Fritschy, J.M.; Schneider Gasser, E.M. Erythropoietin Stimulates GABAergic Maturation in the Mouse Hippocampus. eNeuro 2021, 8, 0006–0021. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Garcia-Agudo, L.F.; Steixner-Kumar, A.A.; Wilke, J.B.H.; Butt, U.J. Introducing the brain erythropoietin circle to explain adaptive brain hardware upgrade and improved performance. Mol. Psychiatry 2022, 27, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Aboouf, M.A.; Koester-Hegmann, C.; Muttathukunnel, P.; Laouafa, S.; Arias-Reyes, C.; Thiersch, M.; Soliz, J.; Gassmann, M.; Schneider Gasser, E.M. Erythropoietin promotes hippocampal mitochondrial function and enhances cognition in mice. Commun. Biol. 2021, 4, 938. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis 2008, 11, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar] [CrossRef]
- Droma, Y.; Hanaoka, M.; Kinjo, T.; Kobayashi, N.; Yasuo, M.; Kitaguchi, Y.; Ota, M. The blunted vascular endothelial growth factor-A (VEGF-A) response to high-altitude hypoxia and genetic variants in the promoter region of the VEGFA gene in Sherpa highlanders. PeerJ 2022, 10, e13893. [Google Scholar] [CrossRef]
- Espinoza, J.R.; Alvarez, G.; Leon-Velarde, F.; Preciado, H.F.; Macarlupu, J.L.; Rivera-Ch, M.; Rodriguez, J.; Favier, J.; Gimenez-Roqueplo, A.P.; Richalet, J.P. Vascular endothelial growth factor-A is associated with chronic mountain sickness in the Andean population. High Alt. Med. Biol. 2014, 15, 146–154. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Nieto-Estevez, V.; Defterali, C.; Vicario-Abejon, C. IGF-I: A Key Growth Factor that Regulates Neurogenesis and Synaptogenesis from Embryonic to Adult Stages of the Brain. Front. Neurosci. 2016, 10, 52. [Google Scholar] [CrossRef]
- Balkowiec, A.; Katz, D.M. Brain-derived neurotrophic factor is required for normal development of the central respiratory rhythm in mice. J. Physiol. 1998, 510, 527–533. [Google Scholar] [CrossRef]
- Erickson, J.T.; Conover, J.C.; Borday, V.; Champagnat, J.; Barbacid, M.; Yancopoulos, G.; Katz, D.M. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J. Neurosci. 1996, 16, 5361–5371. [Google Scholar] [CrossRef]
- Caravagna, C.; Soliz, J.; Seaborn, T. Brain-derived neurotrophic factor interacts with astrocytes and neurons to control respiration. Eur. J. Neurosci. 2013, 38, 3261–3269. [Google Scholar] [CrossRef] [PubMed]
- Enette, L.; Vogel, T.; Fanon, J.L.; Lang, P.O. Effect of Interval and Continuous Aerobic Training on Basal Serum and Plasma Brain-Derived Neurotrophic Factor Values in Seniors: A Systematic Review of Intervention Studies. Rejuvenation Res. 2017, 20, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.; Muller, P.; Dordevic, M.; Lessmann, V.; Brigadski, T.; Muller, N.G. Daily Intermittent Normobaric Hypoxia Over 2 Weeks Reduces BDNF Plasma Levels in Young Adults—A Randomized Controlled Feasibility Study. Front. Physiol. 2018, 9, 1337. [Google Scholar] [CrossRef]
- Richalet, J.P.; Letournel, M.; Souberbielle, J.C. Effects of high-altitude hypoxia on the hormonal response to hypothalamic factors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1685–R1692. [Google Scholar] [CrossRef]
- Roitbak, T.; Surviladze, Z.; Cunningham, L.A. Continuous expression of HIF-1alpha in neural stem/progenitor cells. Cell. Mol. Neurobiol. 2011, 31, 119–133. [Google Scholar] [CrossRef]
- Tomita, S.; Ueno, M.; Sakamoto, M.; Kitahama, Y.; Ueki, M.; Maekawa, N.; Sakamoto, H.; Gassmann, M.; Kageyama, R.; Ueda, N.; et al. Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol. Cell. Biol. 2003, 23, 6739–6749. [Google Scholar] [CrossRef]
- Ko, C.Y.; Tsai, M.Y.; Tseng, W.F.; Cheng, C.H.; Huang, C.R.; Wu, J.S.; Chung, H.Y.; Hsieh, C.S.; Sun, C.K.; Hwang, S.P.; et al. Integration of CNS survival and differentiation by HIF2alpha. Cell. Death Differ. 2011, 18, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Eschenhagen, T.; Geisslinger, G.; Scholich, K. Capturing adenylyl cyclases as potential drug targets. Nat. Rev. Drug. Discov. 2009, 8, 321–335. [Google Scholar] [CrossRef]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 14. [Google Scholar] [CrossRef]
- Bollen, E.; Prickaerts, J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life 2012, 64, 965–970. [Google Scholar] [CrossRef]
- Seaayfan, E.; Nasrah, S.; Quell, L.; Radi, A.; Kleim, M.; Schermuly, R.T.; Weber, S.; Laghmani, K.; Komhoff, M. Reciprocal Regulation of MAGED2 and HIF-1alpha Augments Their Expression under Hypoxia: Role of cAMP and PKA Type II. Cells 2022, 11, 3424. [Google Scholar] [CrossRef] [PubMed]
- Simko, V.; Iuliano, F.; Sevcikova, A.; Labudova, M.; Barathova, M.; Radvak, P.; Pastorekova, S.; Pastorek, J.; Csaderova, L. Hypoxia induces cancer-associated cAMP/PKA signalling through HIF-mediated transcriptional control of adenylyl cyclases VI and VII. Sci. Rep. 2017, 7, 10121. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J. The bioanalytical molecular pharmacology of the N-methyl-D-aspartate (NMDA) receptor nexus and the oxygen-responsive transcription factor HIF-1alpha: Putative mechanisms and regulatory pathways unravel the intimate hypoxia connection. Curr. Mol. Pharmacol. 2013, 6, 104–135. [Google Scholar] [CrossRef] [PubMed]
- Shahoha, M.; Cohen, R.; Ben-Simon, Y.; Ashery, U. cAMP-Dependent Synaptic Plasticity at the Hippocampal Mossy Fiber Terminal. Front. Synaptic Neurosci. 2022, 14, 861215. [Google Scholar] [CrossRef]
- Benito, E.; Barco, A. CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci. 2010, 33, 230–240. [Google Scholar] [CrossRef]
- Signorile, A.; De Rasmo, D. Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging. Antioxidants 2023, 12, 221. [Google Scholar] [CrossRef]
- Yang, J.; Jia, Z.; Song, X.; Shi, J.; Wang, X.; Zhao, X.; He, K. Proteomic and clinical biomarkers for acute mountain sickness in a longitudinal cohort. Commun. Biol. 2022, 5, 548. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Lenormand, P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur. J. Biochem. 2003, 270, 3291–3299. [Google Scholar] [CrossRef]
- Varela-Nallar, L.; Rojas-Abalos, M.; Abbott, A.C.; Moya, E.A.; Iturriaga, R.; Inestrosa, N.C. Chronic hypoxia induces the activation of the Wnt/beta-catenin signaling pathway and stimulates hippocampal neurogenesis in wild-type and APPswe-PS1DeltaE9 transgenic mice in vivo. Front. Cell. Neurosci. 2014, 8, 17. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Schmid, T.; Frank, R.; Brune, B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation. J. Biol. Chem. 2004, 279, 13506–13513. [Google Scholar] [CrossRef] [PubMed]
- Arsham, A.M.; Plas, D.R.; Thompson, C.B.; Simon, M.C. Phosphatidylinositol 3-kinase/Akt signaling is neither required for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J. Biol. Chem. 2002, 277, 15162–15170. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Sun, L.; Tu, L. GABAB Receptor-Mediated PI3K/Akt Signaling Pathway Alleviates Oxidative Stress and Neuronal Cell Injury in a Rat Model of Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, C.; Yin, X.H.; Zhang, G.Y. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology 2008, 54, 1029–1040. [Google Scholar] [CrossRef]
- Lv, M.R.; Li, B.; Wang, M.G.; Meng, F.G.; Yu, J.J.; Guo, F.; Li, Y. Activation of the PI3K-Akt pathway promotes neuroprotection of the delta-opioid receptor agonist against cerebral ischemia-reperfusion injury in rat models. Biomed. Pharmacother. 2017, 93, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, X.; Wan, B.; Wang, D.; Li, M.; Cheng, K.; Luo, Q.; Wang, D.; Lu, Y.; Zhu, L. Caveolin-1 accelerates hypoxia-induced endothelial dysfunction in high-altitude cerebral edema. Cell. Commun. Signal. 2022, 20, 160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lin, J.; Sieck, G.; Haddad, G.G. Neuroprotective Role of Akt in Hypoxia Adaptation in Andeans. Front. Neurosci. 2020, 14, 607711. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M.; Laskowski, D.; Erzurum, S.C. Nitric oxide in adaptation to altitude. Free. Radic. Biol. Med. 2012, 52, 1123–1134. [Google Scholar] [CrossRef]
- Rodriguez-Miguelez, P.; Lima-Cabello, E.; Martinez-Florez, S.; Almar, M.; Cuevas, M.J.; Gonzalez-Gallego, J. Hypoxia-inducible factor-1 modulates the expression of vascular endothelial growth factor and endothelial nitric oxide synthase induced by eccentric exercise. J. Appl. Physiol. 2015, 118, 1075–1083. [Google Scholar] [CrossRef]
- Heiss, C.; Rodriguez-Mateos, A.; Kelm, M. Central role of eNOS in the maintenance of endothelial homeostasis. Antioxid. Redox Signal. 2015, 22, 1230–1242. [Google Scholar] [CrossRef]
- Kuriyama, K.; Ohkuma, S. Role of nitric oxide in central synaptic transmission: Effects on neurotransmitter release. Jpn. J. Pharmacol. 1995, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7, 190. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, U.; Vollenweider, L.; Delabays, A.; Savcic, M.; Eichenberger, U.; Kleger, G.R.; Fikrle, A.; Ballmer, P.E.; Nicod, P.; Bartsch, P. Inhaled nitric oxide for high-altitude pulmonary edema. N. Engl. J. Med. 1996, 334, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Beleslin-Cokic, B.B.; Cokic, V.P.; Yu, X.; Weksler, B.B.; Schechter, A.N.; Noguchi, C.T. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004, 104, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Carraway, M.S.; Suliman, H.B.; Jones, W.S.; Chen, C.W.; Babiker, A.; Piantadosi, C.A. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 2010, 106, 1722–1730. [Google Scholar] [CrossRef]
- Aboouf, M.A.; Guscetti, F.; von Buren, N.; Armbruster, J.; Ademi, H.; Ruetten, M.; Melendez-Rodriguez, F.; Rulicke, T.; Seymer, A.; Jacobs, R.A.; et al. Erythropoietin receptor regulates tumor mitochondrial biogenesis through iNOS and pAKT. Front. Oncol. 2022, 12, 976961. [Google Scholar] [CrossRef]
- Lipton, A.J.; Johnson, M.A.; Macdonald, T.; Lieberman, M.W.; Gozal, D.; Gaston, B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature 2001, 413, 171–174. [Google Scholar] [CrossRef]
- Gozal, D.; Gozal, E.; Torres, J.E.; Gozal, Y.M.; Nuckton, T.J.; Hornby, P.J. Nitric oxide modulates ventilatory responses to hypoxia in the developing rat. Am. J. Respir. Crit. Care Med. 1997, 155, 1755–1762. [Google Scholar] [CrossRef]
- Comellas, A.P.; Dada, L.A.; Lecuona, E.; Pesce, L.M.; Chandel, N.S.; Quesada, N.; Budinger, G.R.; Strous, G.J.; Ciechanover, A.; Sznajder, J.I. Hypoxia-mediated degradation of Na,K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ. Res. 2006, 98, 1314–1322. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Petrushanko, I.Y.; Poluektov, Y.M.; Burnysheva, K.M.; Lakunina, V.A.; Anashkina, A.A.; Makarov, A.A. Basal Glutathionylation of Na,K-ATPase alpha-Subunit Depends on Redox Status of Cells during the Enzyme Biosynthesis. Oxid. Med. Cell. Longev. 2016, 2016, 9092328. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Mei, Y.; Weisbrod, R.M.; Silver, M.; Shukla, P.C.; Bolotina, V.M.; Cohen, R.A.; Tong, X. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J. Biol. Chem. 2014, 289, 19907–19916. [Google Scholar] [CrossRef]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed]
- Bisgard, G.E. Increase in carotid body sensitivity during sustained hypoxia. Biol. Signals 1995, 4, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Arias-Reyes, C.; Soliz, J.; Joseph, V. Mice and Rats Display Different Ventilatory, Hematological, and Metabolic Features of Acclimatization to Hypoxia. Front. Physiol. 2021, 12, 647822. [Google Scholar] [CrossRef]
- Busch, M.A.; Bisgard, G.E.; Forster, H.V. Ventilatory acclimatization to hypoxia is not dependent on arterial hypoxemia. J. Appl. Physiol. 1985, 58, 1874–1880. [Google Scholar] [CrossRef]
- Smith, C.A.; Bisgard, G.E.; Nielsen, A.M.; Daristotle, L.; Kressin, N.A.; Forster, H.V.; Dempsey, J.A. Carotid bodies are required for ventilatory acclimatization to chronic hypoxia. J. Appl. Physiol. 1986, 60, 1003–1010. [Google Scholar] [CrossRef]
- Hodson, E.J.; Nicholls, L.G.; Turner, P.J.; Llyr, R.; Fielding, J.W.; Douglas, G.; Ratnayaka, I.; Robbins, P.A.; Pugh, C.W.; Buckler, K.J.; et al. Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J. Physiol. 2016, 594, 1179–1195. [Google Scholar] [CrossRef]
- Kline, D.D.; Peng, Y.J.; Manalo, D.J.; Semenza, G.L.; Prabhakar, N.R. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 821–826. [Google Scholar] [CrossRef]
- Caravagna, C.; Soliz, J. PI3K and MEK1/2 molecular pathways are involved in the erythropoietin-mediated regulation of the central respiratory command. Respir. Physiol. Neurobiol. 2015, 206, 36–40. [Google Scholar] [CrossRef]
- Soliz, J.; Thomsen, J.J.; Soulage, C.; Lundby, C.; Gassmann, M. Sex-dependent regulation of hypoxic ventilation in mice and humans is mediated by erythropoietin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1837–R1846. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Barneo, J.; Ortega-Saenz, P.; Gonzalez-Rodriguez, P.; Fernandez-Aguera, M.C.; Macias, D.; Pardal, R.; Gao, L. Oxygen-sensing by arterial chemoreceptors: Mechanisms and medical translation. Mol. Aspects Med. 2016, 47, 90–108. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.M.; Salman, S.; Nurse, C.A. Sensory Processing and Integration at the Carotid Body Tripartite Synapse: Neurotransmitter Functions and Effects of Chronic Hypoxia. Front. Physiol. 2018, 9, 225. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R. Neurotransmitters in the carotid body. Adv. Exp. Med. Biol. 1994, 360, 57–69. [Google Scholar]
- Ortega-Saenz, P.; Lopez-Barneo, J. Physiology of the Carotid Body: From Molecules to Disease. Annu. Rev. Physiol. 2020, 82, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Arias-Reyes, C.; Laouafa, S.; Zubieta-DeUrioste, N.; Joseph, V.; Bairam, A.; Schneider Gasser, E.M.; Soliz, J. Erythropoietin Produces a Dual Effect on Carotid Body Chemoreception in Male Rats. Front. Pharmacol. 2021, 12, 727326. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, J.; Guan, Y.; Ji, X. The role of hypoxia in stem cell regulation of the central nervous system: From embryonic development to adult proliferation. CNS Neurosci. Ther. 2021, 27, 1446–1457. [Google Scholar] [CrossRef]
- Chatzi, C.; Schnell, E.; Westbrook, G.L. Localized hypoxia within the subgranular zone determines the early survival of newborn hippocampal granule cells. eLife 2015, 4, e08722. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Shrivastava, V.; Joshi, D.; Singal, C.M.S.; Tyagi, S.; Bhat, M.A.; Jaiswal, P.; Sharma, J.B.; Palanichamy, J.K.; Sinha, S.; et al. Hypoxia Induces Early Neurogenesis in Human Fetal Neural Stem Cells by Activating the WNT Pathway. Mol. Neurobiol. 2023, 60, 2910–2921. [Google Scholar] [CrossRef]
- Carrica, L.; Li, L.; Newville, J.; Kenton, J.; Gustus, K.; Brigman, J.; Cunningham, L.A. Genetic inactivation of hypoxia inducible factor 1-alpha (HIF-1alpha) in adult hippocampal progenitors impairs neurogenesis and pattern discrimination learning. Neurobiol. Learn. Mem. 2019, 157, 79–85. [Google Scholar] [CrossRef]
- Li, G.; Zhao, M.; Cheng, X.; Zhao, T.; Feng, Z.; Zhao, Y.; Fan, M.; Zhu, L. FG-4592 Improves Depressive-Like Behaviors through HIF-1-Mediated Neurogenesis and Synapse Plasticity in Rats. Neurotherapeutics 2020, 17, 664–675. [Google Scholar] [CrossRef]
- Li, L.; Candelario, K.M.; Thomas, K.; Wang, R.; Wright, K.; Messier, A.; Cunningham, L.A. Hypoxia inducible factor-1alpha (HIF-1alpha) is required for neural stem cell maintenance and vascular stability in the adult mouse SVZ. J. Neurosci. 2014, 34, 16713–16719. [Google Scholar] [CrossRef]
- Leu, T.; Fandrey, J.; Schreiber, T. (H)IF applicable: Promotion of neurogenesis by induced HIF-2 signalling after ischaemia. Pflugers Arch. 2021, 473, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Wakhloo, D.; Scharkowski, F.; Curto, Y.; Butt, U.J.; Bansal, V.; Steixner-Kumar, A.A.; Wustefeld, L.; Rajput, A.; Arinrad, S.; Zillmann, M.R.; et al. Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat. Commun. 2020, 11, 1313. [Google Scholar] [CrossRef] [PubMed]
- Schneider Gasser, E.M.; Gassmann, M.; Thiersch, M. HIF-2: An important player in neuronal response to ischemia. Pflugers Arch. 2021, 473, 1175–1176. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fu, J.; Qu, Z.; Li, D.; Si, P.; Qiao, Q.; Zhang, W.; Xue, Y.; Zhen, J.; Wang, W. Chronic mild hypoxia promotes hippocampal neurogenesis involving Notch1 signaling in epileptic rats. Brain Res. 2019, 1714, 88–98. [Google Scholar] [CrossRef] [PubMed]
- d’Anglemont de Tassigny, X.; Sirerol-Piquer, M.S.; Gomez-Pinedo, U.; Pardal, R.; Bonilla, S.; Capilla-Gonzalez, V.; Lopez-Lopez, I.; De la Torre-Laviana, F.J.; Garcia-Verdugo, J.M.; Lopez-Barneo, J. Resistance of subventricular neural stem cells to chronic hypoxemia despite structural disorganization of the germinal center and impairment of neuronal and oligodendrocyte survival. Hypoxia 2015, 3, 15–33. [Google Scholar]
- Chauhan, G.; Kumar, G.; Roy, K.; Kumari, P.; Thondala, B.; Kishore, K.; Panjwani, U.; Ray, K. Hypobaric Hypoxia Induces Deficits in Adult Neurogenesis and Social Interaction via Cyclooxygenase-1/EP1 Receptor Pathway Activating NLRP3 Inflammasome. Mol. Neurobiol. 2022, 59, 2497–2519. [Google Scholar] [CrossRef]
- Ji, W.; Zhang, Y.; Ge, R.L.; Wan, Y.; Liu, J. NMDA Receptor-Mediated Excitotoxicity Is Involved in Neuronal Apoptosis and Cognitive Impairment Induced by Chronic Hypobaric Hypoxia Exposure at High Altitude. High Alt. Med. Biol. 2021, 22, 45–57. [Google Scholar] [CrossRef]
- Maiti, P.; Singh, S.B.; Mallick, B.; Muthuraju, S.; Ilavazhagan, G. High altitude memory impairment is due to neuronal apoptosis in hippocampus, cortex and striatum. J. Chem. Neuroanat. 2008, 36, 227–238. [Google Scholar] [CrossRef]
- Cramer, N.P.; Korotcov, A.; Bosomtwi, A.; Xu, X.; Holman, D.R.; Whiting, K.; Jones, S.; Hoy, A.; Dardzinski, B.J.; Galdzicki, Z. Neuronal and vascular deficits following chronic adaptation to high altitude. Exp. Neurol. 2019, 311, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qu, Y.; Li, J.; Xiong, Y.; Mao, M.; Mu, D. Relationship between HIF-1alpha expression and neuronal apoptosis in neonatal rats with hypoxia-ischemia brain injury. Brain Res. 2007, 1180, 133–139. [Google Scholar] [CrossRef]
- Deng, C.; Li, J.; Li, L.; Sun, F.; Xie, J. Effects of hypoxia ischemia on caspase-3 expression and neuronal apoptosis in the brain of neonatal mice. Exp. Ther. Med. 2019, 17, 4517–4521. [Google Scholar] [CrossRef] [PubMed]
- Tregub, P.; Malinovskaya, N.; Hilazheva, E.; Morgun, A.; Kulikov, V. Permissive hypercapnia and hypercapnic hypoxia inhibit signaling pathways of neuronal apoptosis in ischemic/hypoxic rats. Mol. Biol. Rep. 2023, 50, 2317–2333. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Bi, Y.; Xu, Y.; Wu, Y.; Du, X.; Mou, Y.; Chen, J. Suppression of CHOP Reduces Neuronal Apoptosis and Rescues Cognitive Impairment Induced by Intermittent Hypoxia by Inhibiting Bax and Bak Activation. Neural Plast. 2021, 2021, 4090441. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Zhang, L.; Zhou, Y.; Yang, L.; Yang, M. By targeting apoptosis facilitator BCL2L13, microRNA miR-484 alleviates cerebral ischemia/reperfusion injury-induced neuronal apoptosis in mice. Bioengineered 2021, 12, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Saito, A.; Hayashi, T.; Chan, P.H. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J. Neurosci. 2004, 24, 7879–7887. [Google Scholar] [CrossRef]
- Yu, S.Z.; Yan, L.; Wang, Q.; An, T.L.; Guan, X.Q. Effects of caspase-3 inhibitor on the neuronal apoptosis in rat cerebral cortex after ischemia-reperfusion injury. Zhonghua Bing Li Xue Za Zhi 2006, 35, 165–170. [Google Scholar]
- Wang, H.; Yu, Q.; Zhang, Z.L.; Ma, H.; Li, X.Q. Involvement of the miR-137-3p/CAPN-2 Interaction in Ischemia-Reperfusion-Induced Neuronal Apoptosis through Modulation of p35 Cleavage and Subsequent Caspase-8 Overactivation. Oxid. Med. Cell. Longev. 2020, 2020, 2616871. [Google Scholar] [CrossRef]
- Krajewski, S.; Krajewska, M.; Ellerby, L.M.; Welsh, K.; Xie, Z.; Deveraux, Q.L.; Salvesen, G.S.; Bredesen, D.E.; Rosenthal, R.E.; Fiskum, G.; et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 5752–5757. [Google Scholar] [CrossRef]
- Gao, S.; Mo, J.; Chen, L.; Wang, Y.; Mao, X.; Shi, Y.; Zhang, X.; Yu, R.; Zhou, X. Astrocyte GGTI-mediated Rac1 prenylation upregulates NF-kappaB expression and promotes neuronal apoptosis following hypoxia/ischemia. Neuropharmacology 2016, 103, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, X.; Ye, X.; Yu, R.; Deng, Y. Purpurogallin Reverses Neuronal Apoptosis and Enhances “M2” Polarization of Microglia Under Ischemia via Mediating the miR-124-3p/TRAF6/NF-kappaB Axis. Neurochem. Res. 2023, 48, 375–392. [Google Scholar] [CrossRef]
- Yan, C.; An, F.; Wang, J.; Shi, Y.; Yuan, L.; Lv, D.; Zhao, Y.; Liu, Y.; Wang, Y. Zhongfeng Capsules protects against cerebral ischemia-reperfusion injury via mediating the phosphoinositide 3-kinase/Akt and toll-like receptor 4/nuclear factor kappa B signaling pathways by regulating neuronal apoptosis and inflammation. Apoptosis 2022, 27, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-kappaB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar] [CrossRef]
- Liang, J.; Luan, Y.; Lu, B.; Zhang, H.; Luo, Y.N.; Ge, P. Protection of ischemic postconditioning against neuronal apoptosis induced by transient focal ischemia is associated with attenuation of NF-kappaB/p65 activation. PLoS ONE 2014, 9, e96734. [Google Scholar]
- Qiu, J.; Li, W.; Feng, S.; Wang, M.; He, Z. Transplantation of bone marrow-derived endothelial progenitor cells attenuates cerebral ischemia and reperfusion injury by inhibiting neuronal apoptosis, oxidative stress and nuclear factor-kappaB expression. Int. J. Mol. Med. 2013, 31, 91–98. [Google Scholar] [CrossRef][Green Version]
- He, F.; Zhang, N.; Lv, Y.; Sun, W.; Chen, H. Low-dose lipopolysaccharide inhibits neuronal apoptosis induced by cerebral ischemia/reperfusion injury via the PI3K/Akt/FoxO1 signaling pathway in rats. Mol. Med. Rep. 2019, 19, 1443–1452. [Google Scholar] [CrossRef]
- Hu, X.; Xie, C.; He, S.; Zhang, Y.; Li, Y.; Jiang, L. Remifentanil postconditioning improves global cerebral ischemia-induced spatial learning and memory deficit in rats via inhibition of neuronal apoptosis through the PI3K signaling pathway. Neurol. Sci. 2013, 34, 1955–1962. [Google Scholar] [CrossRef]
- Wu, Q.; Mao, Z.; Liu, J.; Huang, J.; Wang, N. Ligustilide Attenuates Ischemia Reperfusion-Induced Hippocampal Neuronal Apoptosis via Activating the PI3K/Akt Pathway. Front. Pharmacol. 2020, 11, 979. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, J.; Liu, J.; Liu, H.; Li, J. Effects of stem cell-derived exosomes on neuronal apoptosis and inflammatory cytokines in rats with cerebral ischemia-reperfusion injury via PI3K/AKT pathway-mediated mitochondrial apoptosis. Immunopharmacol. Immunotoxicol. 2021, 43, 731–740. [Google Scholar] [CrossRef]
- Chen, Y.F.; Tsai, H.Y.; Wu, K.J.; Siao, L.R.; Wood, W.G. Pipoxolan ameliorates cerebral ischemia via inhibition of neuronal apoptosis and intimal hyperplasia through attenuation of VSMC migration and modulation of matrix metalloproteinase-2/9 and Ras/MEK/ERK signaling pathways. PLoS ONE 2013, 8, e75654. [Google Scholar] [CrossRef]
- Zhou, L.; Ao, L.Y.; Yan, Y.Y.; Li, W.T.; Ye, A.Q.; Li, C.Y.; Shen, W.Y.; Liang, B.W.; Xiong, Z.; Li, Y.M. JLX001 Ameliorates Ischemia/Reperfusion Injury by Reducing Neuronal Apoptosis via Down-Regulating JNK Signaling Pathway. Neuroscience 2019, 418, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Song, J.; Guo, Y.; Wang, T.; Zhou, Z. Astragalus injection protects cerebral ischemic injury by inhibiting neuronal apoptosis and the expression of JNK3 after cerebral ischemia reperfusion in rats. Behav. Brain Funct. 2013, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, L.; Xu, M.; Wu, J.; Chen, L.; Li, J.; Liu, Z.; Lu, G.; Wang, Y.; Qiao, L. AMPK activates FOXO3a and promotes neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Brain Res. Bull. 2017, 132, 1–9. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, H.; Liu, W.; Liu, X.; Jiang, X.; Li, X.; Wu, Q.; Xu, Z.; Zhao, Q. Arctium lappa L. roots ameliorates cerebral ischemia through inhibiting neuronal apoptosis and suppressing AMPK/mTOR-mediated autophagy. Phytomedicine 2021, 85, 153526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, P.; Deng, C. miR-378a-5p regulates CAMKK2/AMPK pathway to contribute to cerebral ischemia/reperfusion injury-induced neuronal apoptosis. Folia Histochem. Cytobiol. 2021, 59, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhang, Y.; Doycheva, D.M.; Ding, Y.; Zhang, Y.; Tang, J.; Guo, H.; Zhang, J.H. Adiponectin attenuates neuronal apoptosis induced by hypoxia-ischemia via the activation of AdipoR1/APPL1/LKB1/AMPK pathway in neonatal rats. Neuropharmacology 2018, 133, 415–428. [Google Scholar] [CrossRef]
- Yu, C.H.; Moon, C.T.; Sur, J.H.; Chun, Y.I.; Choi, W.H.; Yhee, J.Y. Serial expression of hypoxia inducible factor-1alpha and neuronal apoptosis in hippocampus of rats with chronic ischemic brain. J. Korean Neurosurg. Soc. 2011, 50, 481–485. [Google Scholar] [CrossRef]
- Abdel-Latif, R.G.; Rifaai, R.A.; Amin, E.F. Empagliflozin alleviates neuronal apoptosis induced by cerebral ischemia/reperfusion injury through HIF-1alpha/VEGF signaling pathway. Arch. Pharm. Res. 2020, 43, 514–525. [Google Scholar] [CrossRef]
- Li, J.; Tao, T.; Xu, J.; Liu, Z.; Zou, Z.; Jin, M. HIF-1alpha attenuates neuronal apoptosis by upregulating EPO expression following cerebral ischemia-reperfusion injury in a rat MCAO model. Int. J. Mol. Med. 2020, 45, 1027–1036. [Google Scholar]
- Vangeison, G.; Carr, D.; Federoff, H.J.; Rempe, D.A. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J. Neurosci. 2008, 28, 1988–1993. [Google Scholar] [CrossRef]
- Thiersch, M.; Lange, C.; Joly, S.; Heynen, S.; Le, Y.Z.; Samardzija, M.; Grimm, C. Retinal neuroprotection by hypoxic preconditioning is independent of hypoxia-inducible factor-1 alpha expression in photoreceptors. Eur. J. Neurosci. 2009, 29, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Heynen, S.R.; Tanimoto, N.; Thiersch, M.; Le, Y.Z.; Meneau, I.; Seeliger, M.W.; Samardzija, M.; Caprara, C.; Grimm, C. Normoxic activation of hypoxia-inducible factors in photoreceptors provides transient protection against light-induced retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5872–5880. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Tong, L.J.; Cai, D.S. Sevoflurane inhibits neuronal apoptosis and expressions of HIF-1 and HSP70 in brain tissues of rats with cerebral ischemia/reperfusion injury. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5082–5090. [Google Scholar] [PubMed]
- Chen, C.; Hu, Q.; Yan, J.; Lei, J.; Qin, L.; Shi, X.; Luan, L.; Yang, L.; Wang, K.; Han, J.; et al. Multiple effects of 2ME2 and D609 on the cortical expression of HIF-1alpha and apoptotic genes in a middle cerebral artery occlusion-induced focal ischemia rat model. J. Neurochem. 2007, 102, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Halterman, M.W.; Federoff, H.J. HIF-1alpha and p53 promote hypoxia-induced delayed neuronal death in models of CNS ischemia. Exp. Neurol. 1999, 159, 65–72. [Google Scholar] [CrossRef]
- Vetrovoy, O.; Sarieva, K.; Lomert, E.; Nimiritsky, P.; Eschenko, N.; Galkina, O.; Lyanguzov, A.; Tyulkova, E.; Rybnikova, E. Pharmacological HIF1 Inhibition Eliminates Downregulation of the Pentose Phosphate Pathway and Prevents Neuronal Apoptosis in Rat Hippocampus Caused by Severe Hypoxia. J. Mol. Neurosci. 2020, 70, 635–646. [Google Scholar] [CrossRef]
- Helton, R.; Cui, J.; Scheel, J.R.; Ellison, J.A.; Ames, C.; Gibson, C.; Blouw, B.; Ouyang, L.; Dragatsis, I.; Zeitlin, S.; et al. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J. Neurosci. 2005, 25, 4099–4107. [Google Scholar] [CrossRef]
- Barteczek, P.; Li, L.; Ernst, A.S.; Bohler, L.I.; Marti, H.H.; Kunze, R. Neuronal HIF-1alpha and HIF-2alpha deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow. Metab. 2017, 37, 291–306. [Google Scholar] [CrossRef]
- Baranova, O.; Miranda, L.F.; Pichiule, P.; Dragatsis, I.; Johnson, R.S.; Chavez, J.C. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J. Neurosci. 2007, 27, 6320–6332. [Google Scholar] [CrossRef]
- Ebner, L.J.A.; Samardzija, M.; Storti, F.; Todorova, V.; Karademir, D.; Behr, J.; Simpson, F.; Thiersch, M.; Grimm, C. Transcriptomic analysis of the mouse retina after acute and chronic normobaric and hypobaric hypoxia. Sci. Rep. 2021, 11, 16666. [Google Scholar] [CrossRef]
- Grimm, C.; Hermann, D.M.; Bogdanova, A.; Hotop, S.; Kilic, U.; Wenzel, A.; Kilic, E.; Gassmann, M. Neuroprotection by hypoxic preconditioning: HIF-1 and erythropoietin protect from retinal degeneration. Semin. Cell. Dev. Biol. 2005, 16, 531–538. [Google Scholar] [CrossRef]
- Grimm, C.; Wenzel, A.; Groszer, M.; Mayser, H.; Seeliger, M.; Samardzija, M.; Bauer, C.; Gassmann, M.; Reme, C.E. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat. Med. 2002, 8, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Jochmans-Lemoine, A.; Shahare, M.; Soliz, J.; Joseph, V. HIF1alpha and physiological responses to hypoxia are correlated in mice but not in rats. J. Exp. Biol. 2016, 219, 3952–3961. [Google Scholar] [PubMed]
- Kasischke, K.A.; Vishwasrao, H.D.; Fisher, P.J.; Zipfel, W.R.; Webb, W.W. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 2004, 305, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.F.; Patel, T.B.; Clark, J.B. The development of enzymes of energy metabolism in the brain of a precocial (guinea pig) and non-precocial (rat) species. J. Neurochem. 1980, 34, 17–25. [Google Scholar] [CrossRef]
- Kreisman, N.R.; Sick, T.J.; LaManna, J.C.; Rosenthal, M. Local tissue oxygen tension-cytochrome a,a3 redox relationships in rat cerebral cortex in vivo. Brain Res. 1981, 218, 161–174. [Google Scholar] [CrossRef]
- Milusheva, E.A.; Doda, M.; Baranyi, M.; Vizi, E.S. Effect of hypoxia and glucose deprivation on ATP level, adenylate energy charge and [Ca2+]o-dependent and independent release of [3H]dopamine in rat striatal slices. Neurochem. Int. 1996, 28, 501–507. [Google Scholar] [CrossRef]
- Paschen, W.; Djuricic, B. Comparison of in vitro ischemia-induced disturbances in energy metabolism and protein synthesis in the hippocampus of rats and gerbils. J. Neurochem. 1995, 65, 1692–1697. [Google Scholar] [CrossRef]
- Hong, S.S.; Gibney, G.T.; Esquilin, M.; Yu, J.; Xia, Y. Effect of protein kinases on lactate dehydrogenase activity in cortical neurons during hypoxia. Brain Res. 2004, 1009, 195–202. [Google Scholar] [CrossRef]
- Pena, F.; Ramirez, J.M. Hypoxia-induced changes in neuronal network properties. Mol. Neurobiol. 2005, 32, 251–283. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A.; West, C.A.; Reid, K.H.; Tseng, M.T.; Reiss, S.J.; Rigor, B.M. Increased glucose improves recovery of neuronal function after cerebral hypoxia in vitro. Brain Res. 1987, 421, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.F.; Baker, A.J. Glycolysis prevents anoxia-induced synaptic transmission damage in rat hippocampal slices. J. Neurophysiol. 2000, 83, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.Y.; Brucklacher, R.M.; Vannucci, R.C. Cerebral oxidative metabolism and redox state during hypoxia-ischemia and early recovery in immature rats. Am. J. Physiol. 1991, 261, H1102–H1108. [Google Scholar] [CrossRef] [PubMed]
- Brekke, E.; Berger, H.R.; Wideroe, M.; Sonnewald, U.; Morken, T.S. Glucose and Intermediary Metabolism and Astrocyte-Neuron Interactions Following Neonatal Hypoxia-Ischemia in Rat. Neurochem. Res. 2017, 42, 115–132. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Vannucci, R.C. Glycogen metabolism in neonatal rat brain during anoxia and recovery. J. Neurochem. 1980, 34, 1100–1105. [Google Scholar] [CrossRef]
- Dombrowski, G.J., Jr.; Swiatek, K.R.; Chao, K.L. Lactate, 3-hydroxybutyrate, and glucose as substrates for the early postnatal rat brain. Neurochem. Res. 1989, 14, 667–675. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Clark, C.M.; Brown, W.D.; Stanley, C.; Stone, C.K.; Nickles, R.J.; Zhu, G.G.; Allen, P.S.; Holden, J.E. The brain at high altitude: Hypometabolism as a defense against chronic hypoxia? J. Cereb. Blood Flow. Metab. 1994, 14, 671–679. [Google Scholar] [CrossRef]
- Giffard, R.G.; Monyer, H.; Christine, C.W.; Choi, D.W. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 1990, 506, 339–342. [Google Scholar] [CrossRef]
- Tombaugh, G.C.; Sapolsky, R.M. Mild acidosis protects hippocampal neurons from injury induced by oxygen and glucose deprivation. Brain Res. 1990, 506, 343–345. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Clark, C.M.; Monge, C.; Stanley, C.; Brown, W.D.; Stone, C.K.; Nickles, R.J.; Holden, J.E. Sherpa brain glucose metabolism and defense adaptations against chronic hypoxia. J. Appl. Physiol. 1996, 81, 1355–1361. [Google Scholar] [CrossRef]
- Cheong, H.I.; Janocha, A.J.; Monocello, L.T.; Garchar, A.C.; Gebremedhin, A.; Erzurum, S.C.; Beall, C.M. Alternative hematological and vascular adaptive responses to high-altitude hypoxia in East African highlanders. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L172–L177. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Clark, C.M.; Matheson, G.O.; Brown, W.D.; Stone, C.K.; Nickles, R.J.; Holden, J.E. Effects on regional brain metabolism of high-altitude hypoxia: A study of six US marines. Am. J. Physiol. 1999, 277, R314–R319. [Google Scholar] [CrossRef] [PubMed]
- Pamenter, M.E.; Hogg, D.W.; Ormond, J.; Shin, D.S.; Woodin, M.A.; Buck, L.T. Endogenous GABA(A) and GABA(B) receptor-mediated electrical suppression is critical to neuronal anoxia tolerance. Proc. Natl. Acad. Sci. USA 2011, 108, 11274–11279. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.; Nilsson, G.E. Two decades of research on anoxia tolerance-mitochondria, -omics and physiological diversity. J. Exp. Biol. 2023, 226, 245584. [Google Scholar] [CrossRef]
- Larson, J.; Park, T.J. Extreme hypoxia tolerance of naked mole-rat brain. Neuroreport 2009, 20, 1634–1637. [Google Scholar] [CrossRef]
- Park, T.J.; Reznick, J.; Peterson, B.L.; Blass, G.; Omerbasic, D.; Bennett, N.C.; Kuich, P.; Zasada, C.; Browe, B.M.; Hamann, W.; et al. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 2017, 356, 307–311. [Google Scholar] [CrossRef]
- Cheng, H.; Pamenter, M.E. Naked mole-rat brain mitochondria tolerate in vitro ischaemia. J. Physiol. 2021, 599, 4671–4685. [Google Scholar] [CrossRef]
- Nathaniel, T.I.; Saras, A.; Umesiri, F.E.; Olajuyigbe, F. Tolerance to oxygen nutrient deprivation in the hippocampal slices of the naked mole rats. J. Integr. Neurosci. 2009, 8, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.L.; Park, T.J.; Larson, J. Adult naked mole-rat brain retains the NMDA receptor subunit GluN2D associated with hypoxia tolerance in neonatal mammals. Neurosci. Lett. 2012, 506, 342–345. [Google Scholar] [CrossRef]
- Puka-Sundvall, M.; Sandberg, M.; Hagberg, H. Brain injury after hypoxia-ischemia in newborn rats: Relationship to extracellular levels of excitatory amino acids and cysteine. Brain Res. 1997, 750, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.Y.; Brucklacher, R.M.; Vannucci, R.C. Cerebral energy metabolism during hypoxia-ischemia and early recovery in immature rats. Am. J. Physiol. 1992, 262, H672–H677. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Towfighi, J.; Vannucci, S.J. Secondary energy failure after cerebral hypoxia-ischemia in the immature rat. J. Cereb. Blood Flow. Metab. 2004, 24, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Varoqui, H.; Zhu, H.; Yao, D.; Ming, H.; Erickson, J.D. Cloning and functional identification of a neuronal glutamine transporter. J. Biol. Chem. 2000, 275, 4049–4054. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Qin, Y.A.; Dhillon, R.; Dowell, J.; Denu, J.M.; Pamenter, M.E. Metabolomic Analysis of Carbohydrate and Amino Acid Changes Induced by Hypoxia in Naked Mole-Rat Brain and Liver. Metabolites 2022, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Seong, H.H.; Jin, H.B.; Kim, Y.J. The Effect of Long-Term Environmental Enrichment in Chronic Cerebral Hypoperfusion-Induced Memory Impairment in Rats. Biol. Res. Nurs. 2017, 19, 278–286. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernandez, E.; Maestre, C.; Moncada, S.; Bolanos, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell. Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Xiao, B.; Wang, S.; Yang, G.; Sun, X.; Zhao, S.; Lin, L.; Cheng, J.; Yang, W.; Cong, W.; Sun, W.; et al. HIF-1alpha contributes to hypoxia adaptation of the naked mole rat. Oncotarget 2017, 8, 109941–109951. [Google Scholar] [CrossRef]
- Cheng, F.; Xie, S.; Guo, M.; Fang, H.; Li, X.; Yin, J.; Lu, G.; Li, Y.; Ji, X.; Yu, S. Altered glucose metabolism and preserved energy charge and neuronal structures in the brain of mouse intermittently exposed to hypoxia. J. Chem. Neuroanat. 2011, 42, 65–71. [Google Scholar] [CrossRef]
- LaManna, J.C.; Vendel, L.M.; Farrell, R.M. Brain adaptation to chronic hypobaric hypoxia in rats. J. Appl. Physiol. 1992, 72, 2238–2243. [Google Scholar] [CrossRef]
- Harik, S.I.; Behmand, R.A.; LaManna, J.C. Hypoxia increases glucose transport at blood-brain barrier in rats. J. Appl. Physiol. 1994, 77, 896–901. [Google Scholar] [CrossRef]
- Harik, S.I.; Lust, W.D.; Jones, S.C.; Lauro, K.L.; Pundik, S.; LaManna, J.C. Brain glucose metabolism in hypobaric hypoxia. J. Appl. Physiol. 1995, 79, 136–140. [Google Scholar] [CrossRef]
- Chavez, J.C.; Pichiule, P.; Boero, J.; Arregui, A. Reduced mitochondrial respiration in mouse cerebral cortex during chronic hypoxia. Neurosci. Lett. 1995, 193, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Morken, T.S.; Brekke, E.; Haberg, A.; Wideroe, M.; Brubakk, A.M.; Sonnewald, U. Altered astrocyte-neuronal interactions after hypoxia-ischemia in the neonatal brain in female and male rats. Stroke 2014, 45, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Yu, W.; Lou, Z.; Mu, D.; Wang, Y.; Shen, B.; Qi, S. Reperfusion promotes mitochondrial dysfunction following focal cerebral ischemia in rats. PLoS ONE 2012, 7, e46498. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front. Mol. Neurosci. 2020, 13, 28. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Bundgaard, A.; James, A.M.; Gruszczyk, A.V.; Martin, J.; Murphy, M.P.; Fago, A. Metabolic adaptations during extreme anoxia in the turtle heart and their implications for ischemia-reperfusion injury. Sci. Rep. 2019, 9, 2850. [Google Scholar] [CrossRef]
- Dahl, H.A.; Johansen, A.; Nilsson, G.E.; Lefevre, S. The Metabolomic Response of Crucian Carp (Carassius carassius) to Anoxia and Reoxygenation Differs between Tissues and Hints at Uncharacterized Survival Strategies. Metabolites 2021, 11, 435. [Google Scholar] [CrossRef]
- Pamenter, M.E.; Lau, G.Y.; Richards, J.G.; Milsom, W.K. Naked mole rat brain mitochondria electron transport system flux and H(+) leak are reduced during acute hypoxia. J. Exp. Biol. 2018, 221, jeb171397. [Google Scholar] [CrossRef]
- Pena, E.; El Alam, S.; Siques, P.; Brito, J. Oxidative Stress and Diseases Associated with High-Altitude Exposure. Antioxidants 2022, 11, 267. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Takano, N.; Xiang, L.; Gilkes, D.M.; Luo, W.; Semenza, G.L. Hypoxia-inducible factors enhance glutamate signaling in cancer cells. Oncotarget 2014, 5, 8853–8868. [Google Scholar] [CrossRef] [PubMed]
- Park, E.C.; Ghose, P.; Shao, Z.Y.; Ye, Q.; Kang, L.J.; Xu, X.Z.S.; Powell-Coffman, J.A.; Rongo, C. Hypoxia regulates glutamate receptor trafficking through an HIF-independent mechanism. Embo J. 2012, 31, 1618–1619. [Google Scholar] [CrossRef][Green Version]
- Hsieh, C.H.; Lin, Y.J.; Chen, W.L.; Huang, Y.C.; Chang, C.W.; Cheng, F.C.; Liu, R.S.; Shyu, W.C. HIF-1alpha triggers long-lasting glutamate excitotoxicity via system x(c)(-) in cerebral ischaemia-reperfusion. J. Pathol. 2017, 241, 337–349. [Google Scholar] [CrossRef]
- Dallas, M.; Boycott, H.E.; Atkinson, L.; Miller, A.; Boyle, J.P.; Pearson, H.A.; Peers, C. Hypoxia suppresses glutamate transport in astrocytes. J. Neurosci. 2007, 27, 3946–3955. [Google Scholar] [CrossRef]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Bowery, N.G. GABAB receptor pharmacology. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 109–147. [Google Scholar] [CrossRef]
- Anju, T.R.; Abraham, P.M.; Antony, S.; Paulose, C.S. Alterations in cortical GABAB receptors in neonatal rats exposed to hypoxic stress: Role of glucose, oxygen, and epinephrine resuscitation. Mol. Cell. Biochem. 2010, 343, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Ham, S.; Lee, S.E.; Lee, Y.; Lee, G.H. Hypoxia regulates the level of glutamic acid decarboxylase enzymes and interrupts inhibitory synapse stability in primary cultured neurons. Neurotoxicology 2018, 65, 221–230. [Google Scholar] [CrossRef]
- Ramamoorthy, P.; Shi, H. Ischemia induces different levels of hypoxia inducible factor-1alpha protein expression in interneurons and pyramidal neurons. Acta Neuropathol. Commun. 2014, 2, 51. [Google Scholar] [CrossRef] [PubMed]
- Moya, E.A.; Go, A.; Kim, C.B.; Fu, Z.; Simonson, T.S.; Powell, F.L. Neuronal HIF-1alpha in the nucleus tractus solitarius contributes to ventilatory acclimatization to hypoxia. J. Physiol. 2020, 598, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Juarez Olguin, H.; Calderon Guzman, D.; Hernandez Garcia, E.; Barragan Mejia, G. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxid. Med. Cell. Longev. 2016, 2016, 9730467. [Google Scholar] [CrossRef]
- Witten, L.; Sager, T.; Thirstrup, K.; Johansen, J.L.; Larsen, D.B.; Montezinho, L.P.; Mork, A. HIF prolyl hydroxylase inhibition augments dopamine release in the rat brain in vivo. J. Neurosci. Res. 2009, 87, 1686–1694. [Google Scholar] [CrossRef]
- Johansen, J.L.; Sager, T.N.; Lotharius, J.; Witten, L.; Mork, A.; Egebjerg, J.; Thirstrup, K. HIF prolyl hydroxylase inhibition increases cell viability and potentiates dopamine release in dopaminergic cells. J. Neurochem. 2010, 115, 209–219. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.J.; Yang, Z.H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.L.; Zhong, M.L.; Wang, T.; Li, J.Y.; et al. Deferoxamine-mediated up-regulation of HIF-1alpha prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef]
- Lim, J.; Kim, H.I.; Bang, Y.; Seol, W.; Choi, H.S.; Choi, H.J. Hypoxia-inducible factor-1alpha upregulates tyrosine hydroxylase and dopamine transporter by nuclear receptor ERRgamma in SH-SY5Y cells. Neuroreport 2015, 26, 380–386. [Google Scholar] [CrossRef]
- Bence, M.; Kereszturi, E.; Mozes, V.; Sasvari-Szekely, M.; Keszler, G. Hypoxia-induced transcription of dopamine D3 and D4 receptors in human neuroblastoma and astrocytoma cells. BMC Neurosci. 2009, 10, 92. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Eddahibi, S.; Fabre, V.; Boni, C.; Martres, M.P.; Raffestin, B.; Hamon, M.; Adnot, S. Induction of serotonin transporter by hypoxia in pulmonary vascular smooth muscle cells. Relationship with the mitogenic action of serotonin. Circ. Res. 1999, 84, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Sarkar, P.; Shrivastava, S.; Chattopadhyay, A. Effect of Hypoxia on the Function of the Human Serotonin(1A) Receptor. ACS Chem. Neurosci. 2022, 13, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.; Maruyama, H.; Li, C.; Pastuhov, S.I.; Nix, P.; Bastiani, M.; Hisamoto, N.; Matsumoto, K. Axotomy-induced HIF-serotonin signalling axis promotes axon regeneration in C. elegans. Nat. Commun. 2016, 7, 10388. [Google Scholar] [CrossRef]
- Booth, R.F.; Harvey, S.A.; Clark, J.B. Effects of in vivo hypoxia on acetylcholine synthesis by rat brain synaptosomes. J. Neurochem. 1983, 40, 106–110. [Google Scholar] [CrossRef]
- Kim, D.K.; Prabhakar, N.R.; Kumar, G.K. Acetylcholine release from the carotid body by hypoxia: Evidence for the involvement of autoinhibitory receptors. J. Appl. Physiol. 2004, 96, 376–383. [Google Scholar] [CrossRef]
- Gibson, G.E.; Duffy, T.E. Impaired synthesis of acetylcholine by mild hypoxic hypoxia or nitrous oxide. J. Neurochem. 1981, 36, 28–33. [Google Scholar] [CrossRef]
- He, Y.; Su, H.; Li, N.; Zhang, Y.; Zhang, P.; Zhang, Y.; Ye, Y.; Zhang, Y.; Tang, J.; Xu, Z. In utero hypoxia attenuated acetylcholine-mediated vasodilatation via CHRM3/p-NOS3 in fetal sheep MCA: Role of ROS/ERK1/2. Hypertens. Res. 2022, 45, 1168–1182. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Polak, J. Hypoxia. 4. Hypoxia and ion channel function. Am. J. Physiol. Cell. Physiol. 2011, 300, C951–C967. [Google Scholar] [CrossRef]
- Hyllienmark, L.; Brismar, T. Effect of hypoxia on membrane potential and resting conductance in rat hippocampal neurons. Neuroscience 1999, 91, 511–517. [Google Scholar] [CrossRef]
- Mironov, S.L.; Langohr, K.; Haller, M.; Richter, D.W. Hypoxia activates ATP-dependent potassium channels in inspiratory neurones of neonatal mice. J. Physiol. 1998, 509, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, M.; Butt, A.M.; Lewis, A. A critical role for the inward rectifying potassium channel Kir7.1 in oligodendrocytes of the mouse optic nerve. Brain Struct. Funct. 2020, 225, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Doll, C.J.; Hochachka, P.W.; Reiner, P.B. Channel arrest: Implications from membrane resistance in turtle neurons. Am. J. Physiol. 1991, 261, R1321–R1324. [Google Scholar] [CrossRef]
- Zivkovic, G.; Buck, L.T. Regulation of AMPA receptor currents by mitochondrial ATP-sensitive K+ channels in anoxic turtle neurons. J. Neurophysiol. 2010, 104, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.H.; Chapman, P.F.; Kairiss, E.W.; Keenan, C.L. Long-Term Synaptic Potentiation. Science 1988, 242, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Sathyanesan, M.; Watt, M.J.; Haiar, J.M.; Scholl, J.L.; Davies, S.R.; Paulsen, R.T.; Wiederin, J.; Ciborowski, P.; Newton, S.S. Carbamoylated erythropoietin modulates cognitive outcomes of social defeat and differentially regulates gene expression in the dorsal and ventral hippocampus. Transl. Psychiat 2018, 8, 113. [Google Scholar] [CrossRef]
- Tazangi, P.E.; Moosavi, S.M.S.; Shabani, M.; Haghani, M. Erythropoietin improves synaptic plasticity and memory deficits by decrease of the neurotransmitter release probability in the rat model of Alzheimer’s disease. Pharmacol. Biochem. Behav. 2015, 130, 15–21. [Google Scholar] [CrossRef]
- Corcoran, A.; Kunze, R.; Harney, S.C.; Breier, G.; Marti, H.H.; O’Connor, J.J. A Role for Prolyl Hydroxylase Domain Proteins in Hippocampal Synaptic Plasticity. Hippocampus 2013, 23, 861–872. [Google Scholar] [CrossRef]
- Winning, S.; Fandrey, J. Dendritic Cells under Hypoxia: How Oxygen Shortage Affects the Linkage between Innate and Adaptive Immunity. J. Immunol. Res. 2016, 2016, 5134329. [Google Scholar] [CrossRef]
- Nakayama, K. cAMP-response element-binding protein (CREB) and NF-kappaB transcription factors are activated during prolonged hypoxia and cooperatively regulate the induction of matrix metalloproteinase MMP1. J. Biol. Chem. 2013, 288, 22584–22595. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboouf, M.A.; Thiersch, M.; Soliz, J.; Gassmann, M.; Schneider Gasser, E.M. The Brain at High Altitude: From Molecular Signaling to Cognitive Performance. Int. J. Mol. Sci. 2023, 24, 10179. https://doi.org/10.3390/ijms241210179
Aboouf MA, Thiersch M, Soliz J, Gassmann M, Schneider Gasser EM. The Brain at High Altitude: From Molecular Signaling to Cognitive Performance. International Journal of Molecular Sciences. 2023; 24(12):10179. https://doi.org/10.3390/ijms241210179
Chicago/Turabian StyleAboouf, Mostafa A., Markus Thiersch, Jorge Soliz, Max Gassmann, and Edith M. Schneider Gasser. 2023. "The Brain at High Altitude: From Molecular Signaling to Cognitive Performance" International Journal of Molecular Sciences 24, no. 12: 10179. https://doi.org/10.3390/ijms241210179
APA StyleAboouf, M. A., Thiersch, M., Soliz, J., Gassmann, M., & Schneider Gasser, E. M. (2023). The Brain at High Altitude: From Molecular Signaling to Cognitive Performance. International Journal of Molecular Sciences, 24(12), 10179. https://doi.org/10.3390/ijms241210179