
A Systematic Review of the Effects of Capsaicin on Alzheimer’s Disease
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Search Results and Study Characteristics
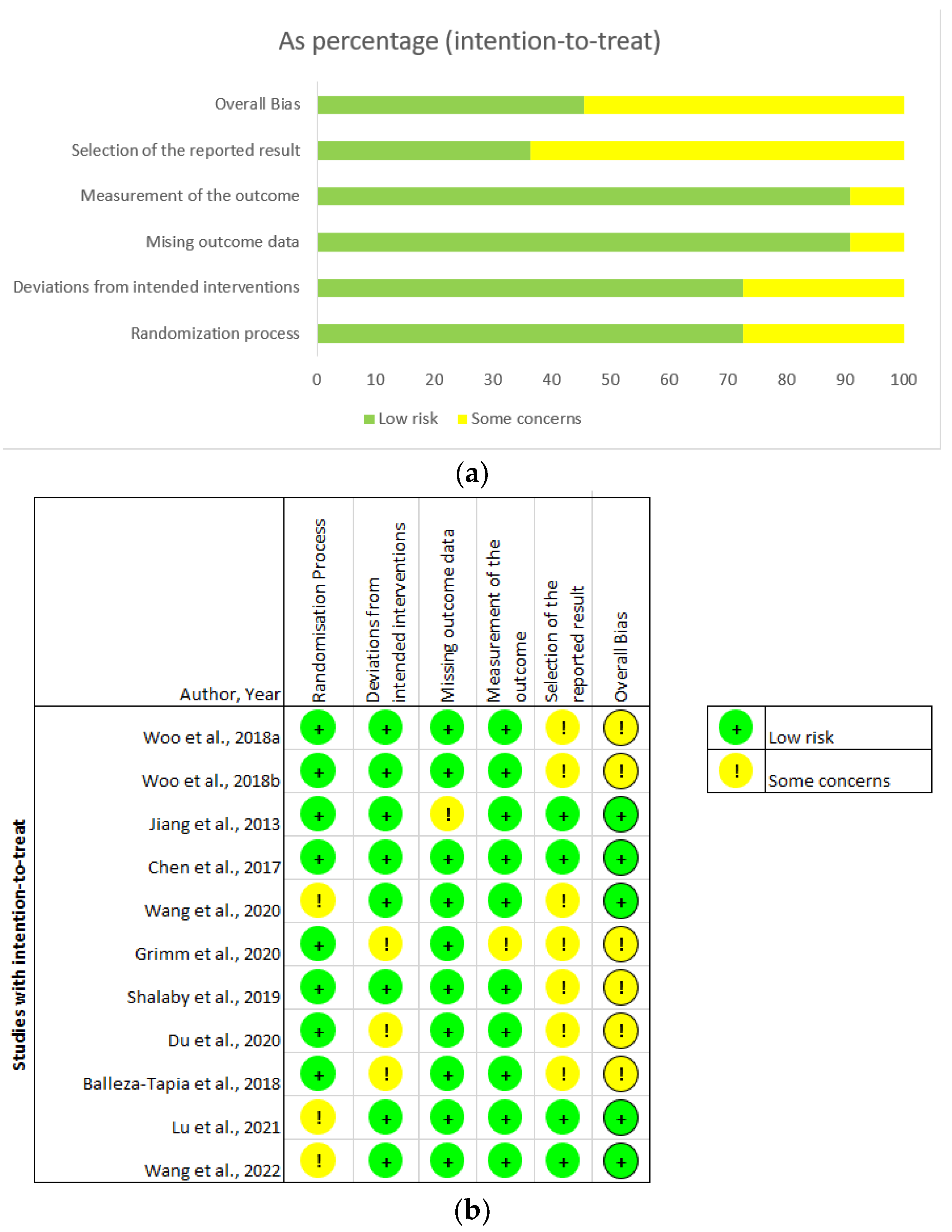
3.2. Critical Appraisal
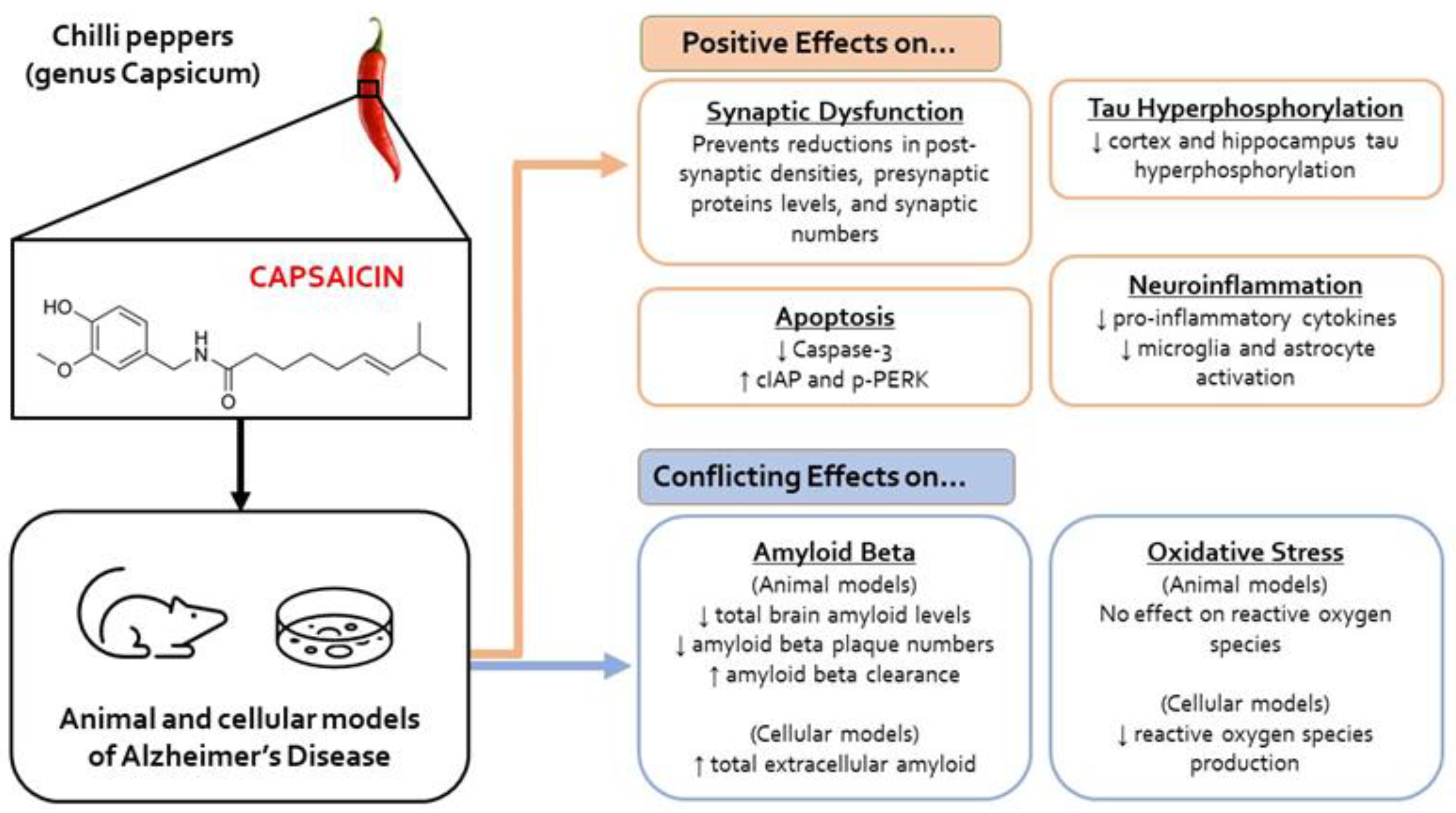
3.3. Molecular Outcomes
3.3.1. Oxidative Stress
3.3.2. Tau Hyperphosphorylation
3.3.3. Amyloid β
3.3.4. Neuroinflammation
3.3.5. Electrophysiological and Synapse Changes
3.3.6. Apoptosis
3.4. Behavioural and Cognitive Outcomes
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Fact Sheets of Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 25 May 2023).
- Gauthier, S.; Webster, C.; Servaes, S.; Morais, J.A.; Rosa-Neto, P. World Alzheimer Report 2022: Life after Diagnosis: Navigating Treatment, Care and Support; Alzheimer’s Disease International: London, UK, 2022. [Google Scholar]
- Gotz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Commins, S.; Kirby, B.P. The complexities of behavioural assessment in neurodegenerative disorders: A focus on Alzheimer’s disease. Pharmacol. Res. 2019, 147, 104363. [Google Scholar] [CrossRef]
- Bali, J.; Gheinani, A.H.; Zurbriggen, S.; Rajendran, L. Role of genes linked to sporadic Alzheimer’s disease risk in the production of beta-amyloid peptides. Proc. Natl. Acad. Sci. USA 2012, 109, 15307–15311. [Google Scholar] [CrossRef]
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol. Genet. 2020, 6, e512. [Google Scholar] [CrossRef] [PubMed]
- Shea, Y.-F.; Pan, Y.; Mak, H.K.-F.; Bao, Y.; Lee, S.-C.; Chiu, P.K.-C.; Chan, H.-W.F. A systematic review of atypical Alzheimer’s disease including behavioural and psychological symptoms. Psychogeriatrics 2021, 21, 396–406. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Koedam, E.L.; Lauffer, V.; van der Vlies, A.E.; van der Flier, W.M.; Scheltens, P.; Pijnenburg, Y.A. Early-versus late-onset Alzheimer’s disease: More than age alone. J. Alzheimers Dis. 2010, 19, 1401–1408. [Google Scholar] [CrossRef]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Lam, B.; Khan, A.; Keith, J.; Rogaeva, E.; Bilbao, J.; St George-Hyslop, P.; Ghani, M.; Freedman, M.; Stuss, D.T.; Chow, T.; et al. Characterizing familial corticobasal syndrome due to Alzheimer’s disease pathology and PSEN1 mutations. Alzheimers Dement. 2017, 13, 520–530. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Barber, I.; Lincoln, S.J.; Murray, M.E.; Camsari, G.B.; Khan, Q.U.A.; Nguyen, T.; Ma, L.; Bisceglio, G.D.; Crook, J.E.; et al. Evaluating pathogenic dementia variants in posterior cortical atrophy. Neurobiol. Aging 2016, 37, 38–44. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Tabuas-Pereira, M.; Baldeiras, I.; Duro, D.; Santiago, B.; Ribeiro, M.H.; Leitao, M.J.; Oliveira, C.; Santana, I. Prognosis of Early-Onset vs. Late-Onset Mild Cognitive Impairment: Comparison of Conversion Rates and Its Predictors. Geriatrics 2016, 1, 11. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. [Google Scholar] [CrossRef]
- Scheffer, S.; Hermkens, D.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M.J.A.P. Vascular Hypothesis of Alzheimer Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Giraldo, E.; Badìa, M.C.; Monllor, P.; Lloret, A.; Schininà, M.E.; Giorgi, A.; Cini, C.; Tramutola, A.; et al. Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic. Biol. Med. 2016, 91, 1–9. [Google Scholar] [CrossRef]
- Chen, J.J.; Thiyagarajah, M.; Song, J.; Chen, C.; Herrmann, N.; Gallagher, D.; Rapoport, M.J.; Black, S.E.; Ramirez, J.; Andreazza, A.C.; et al. Altered central and blood glutathione in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Alzheimer’s Res. Ther. 2022, 14, 23. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Baquero, M.; Vento, M.; Cháfer-Pericás, C. Free radicals in Alzheimer’s disease: Lipid peroxidation biomarkers. Clin. Chim. Acta 2019, 491, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Walters, A.; Phillips, E.; Zheng, R.; Biju, M.; Kuruvilla, T. Evidence for neuroinflammation in Alzheimer’s disease. Prog. Neurol. Psychiatry 2016, 20, 25–31. [Google Scholar] [CrossRef]
- López-Sanz, D.; Bruña, R.; Delgado-Losada, M.L.; López-Higes, R.; Marcos-Dolado, A.; Maestú, F.; Walter, S. Electrophysiological brain signatures for the classification of subjective cognitive decline: Towards an individual detection in the preclinical stages of dementia. Alzheimer’s Res. Ther. 2019, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Singh, T.G.; Singh, S.; Garg, N.; Dhiman, S. Apoptotic Pathways and Alzheimer’s Disease: Probing Therapeutic Potential. Neurochem. Res. 2021, 46, 3103–3122. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef]
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Leelakanok, N.; D’Cunha, R.R. Association between polypharmacy and dementia—A systematic review and metaanalysis. Aging Ment. Health 2019, 23, 932–941. [Google Scholar] [CrossRef]
- Maher, R.L.; Hanlon, J.; Hajjar, E.R. Clinical consequences of polypharmacy in elderly. Expert Opin. Drug Saf. 2014, 13, 57–65. [Google Scholar] [CrossRef]
- Chang, A.; Rosani, A.; Quick, J. Capsaicin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Reyes-Escogido Mde, L.; Gonzalez-Mondragon, E.G.; Vazquez-Tzompantzi, E. Chemical and pharmacological aspects of capsaicin. Molecules 2011, 16, 1253–1270. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.; Brock, C.; Olesen, A.E.; Andresen, T.; Nilsson, M.; Dickenson, A.H. Unravelling the mystery of capsaicin: A tool to understand and treat pain. Pharmacol. Rev. 2012, 64, 939–971. [Google Scholar] [CrossRef] [PubMed]
- Rollyson, W.D.; Stover, C.A.; Brown, K.C.; Perry, H.E.; Stevenson, C.D.; McNees, C.A.; Ball, J.G.; Valentovic, M.A.; Dasgupta, P. Bioavailability of capsaicin and its implications for drug delivery. J. Control. Release 2014, 196, 96–105. [Google Scholar] [CrossRef]
- Govindarajan, V.S.; Sathyanarayana, M.N. Capsicum--production, technology, chemistry, and quality. Part V. Impact on physiology, pharmacology, nutrition, and metabolism; structure, pungency, pain, and desensitization sequences. Crit. Rev. Food Sci. Nutr. 1991, 29, 435–474. [Google Scholar] [CrossRef]
- Fattori, V.; Hohmann, M.S.; Rossaneis, A.C.; Pinho-Ribeiro, F.A.; Verri, W.A. Capsaicin: Current Understanding of Its Mechanisms and Therapy of Pain and Other Pre-Clinical and Clinical Uses. Molecules 2016, 21, 844. [Google Scholar] [CrossRef]
- Meghvansi, M.K.; Siddiqui, S.; Khan, M.H.; Gupta, V.K.; Vairale, M.G.; Gogoi, H.K.; Singh, L. Naga chilli: A potential source of capsaicinoids with broad-spectrum ethnopharmacological applications. J. Ethnopharmacol. 2010, 132, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shang, K.; Amna, T.; Amina, M.; Al-Musayeib, N.M.; Al-Deyab, S.S.; Hwang, I. Influence of Capsaicin on Inflammatory Cytokines Induced by Lipopolysaccharide in Myoblast Cells Under In vitro Environment. Pharmacogn. Mag. 2017, 13, S26–S32. [Google Scholar] [CrossRef]
- Tang, J.; Luo, K.; Li, Y.; Chen, Q.; Tang, D.; Wang, D.; Xiao, J. Capsaicin attenuates LPS-induced inflammatory cytokine production by upregulation of LXRα. Int. Immunopharmacol. 2015, 28, 264–269. [Google Scholar] [CrossRef]
- Galano, A.; Martínez, A. Capsaicin, a tasty free radical scavenger: Mechanism of action and kinetics. J. Phys. Chem. B 2012, 116, 1200–1208. [Google Scholar] [CrossRef]
- Lu, M.; Chen, C.; Lan, Y.; Xiao, J.; Li, R.; Huang, J.; Huang, Q.; Cao, Y.; Ho, C.T. Capsaicin-the major bioactive ingredient of chili peppers: Bio-efficacy and delivery systems. Food Funct. 2020, 11, 2848–2860. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.E.; Sleem, A.A.; Sayed, M.; Youness, E.R.; Shaffie, N. Capsaicin Exerts Anti-convulsant and Neuroprotective Effects in Pentylenetetrazole-Induced Seizures. Neurochem. Res. 2020, 45, 1045–1061. [Google Scholar] [CrossRef] [PubMed]
- Khatibi, N.H.; Jadhav, V.; Charles, S.; Chiu, J.; Buchholz, J.; Tang, J.; Zhang, J.H. Capsaicin pre-treatment provides neurovascular protection against neonatal hypoxic-ischemic brain injury in rats. Acta Neurochir. Suppl. 2011, 111, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, J.; Ma, D.; Yuan, G.; Lu, Y.; Yang, Y. Capsaicin reduces Alzheimer-associated tau changes in the hippocampus of type 2 diabetes rats. PLoS ONE 2017, 12, e0172477. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Bu, X.L.; Wang, J.; Zhang, T.; Xiang, Y.; Shen, L.L.; Wang, Q.H.; Deng, B.; Wang, X.; Zhu, C.; et al. The Associations between a Capsaicin-Rich Diet and Blood Amyloid-β Levels and Cognitive Function. J. Alzheimer’s Dis. 2016, 52, 1081–1088. [Google Scholar] [CrossRef]
- Tian, D.Y.; Wang, J.; Sun, B.L.; Wang, Z.; Xu, W.; Chen, Y.; Shen, Y.Y.; Li, H.Y.; Chen, D.W.; Zhou, F.Y.; et al. Spicy food consumption is associated with cognition and cerebrospinal fluid biomarkers of Alzheimer disease. Chin. Med. J. 2020, 134, 173–177. [Google Scholar] [CrossRef]
- Shi, Z.; El-Obeid, T.; Riley, M.; Li, M.; Page, A.; Liu, J. High Chili Intake and Cognitive Function among 4582 Adults: An Open Cohort Study over 15 Years. Nutrients 2019, 11, 1183. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Arksey, H.; O’Malley, L. Scoping studies: Towards a methodological framework. Int. J. Soc. Res. Methodol. 2005, 8, 19–32. [Google Scholar] [CrossRef]
- Shalaby, M.A.; Nounou, H.A.; Deif, M.M. The potential value of capsaicin in modulating cognitive functions in a rat model of streptozotocin-induced Alzheimer’s disease. Egypt. J. Neurol. Psychiatry Neurosurg. 2019, 55, 48. [Google Scholar] [CrossRef]
- Jiang, X.; Jia, L.W.; Li, X.H.; Cheng, X.S.; Xie, J.Z.; Ma, Z.W.; Xu, W.J.; Liu, Y.; Yao, Y.; Du, L.L.; et al. Capsaicin ameliorates stress-induced Alzheimer’s disease-like pathological and cognitive impairments in rats. J. Alzheimer’s Dis. 2013, 35, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, Z.; Du, Y.; Fu, M.; Han, H.; Wang, Y.; Dong, Z. Capsaicin Attenuates Amyloid-β-Induced Synapse Loss and Cognitive Impairments in Mice. J. Alzheimer’s Dis. 2017, 59, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Balleza-Tapia, H.; Crux, S.; Andrade-Talavera, Y.; Dolz-Gaiton, P.; Papadia, D.; Chen, G.; Johansson, J.; Fisahn, A. TrpV1 receptor activation rescues neuronal function and network gamma oscillations from Aβ-induced impairment in mouse hippocampus in vitro. Elife 2018, 7, e37703. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.; Kim, M.J.; Song, Y.O. Bioactive Compounds in Kimchi Improve the Cognitive and Memory Functions Impaired by Amyloid Beta. Nutrients 2018, 10, 1554. [Google Scholar] [CrossRef]
- Woo, M.; Noh, J.S.; Cho, E.J.; Song, Y.O. Bioactive Compounds of Kimchi Inhibit Apoptosis by Attenuating Endoplasmic Reticulum Stress in the Brain of Amyloid β-Injected Mice. J. Agric. Food Chem. 2018, 66, 4883–4890. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Fu, M.; Huang, Z.; Tian, X.; Li, J.; Pang, Y.; Song, W.; Tian Wang, Y.; Dong, Z. TRPV1 activation alleviates cognitive and synaptic plasticity impairments through inhibiting AMPAR endocytosis in APP23/PS45 mouse model of Alzheimer’s disease. Aging Cell 2020, 19, e13113. [Google Scholar] [CrossRef]
- Wang, J.; Sun, B.L.; Xiang, Y.; Tian, D.Y.; Zhu, C.; Li, W.W.; Liu, Y.H.; Bu, X.L.; Shen, L.L.; Jin, W.S.; et al. Capsaicin consumption reduces brain amyloid-beta generation and attenuates Alzheimer’s disease-type pathology and cognitive deficits in APP/PS1 mice. Transl. Psychiatry 2020, 10, 230. [Google Scholar] [CrossRef]
- Lu, J.; Zhou, W.; Dou, F.; Wang, C.; Yu, Z. TRPV1 sustains microglial metabolic reprogramming in Alzheimer’s disease. EMBO Rep. 2021, 22, e52013. [Google Scholar] [CrossRef]
- Wang, C.; Huang, W.; Lu, J.; Chen, H.; Yu, Z. TRPV1-Mediated Microglial Autophagy Attenuates Alzheimer’s Disease-Associated Pathology and Cognitive Decline. Front. Pharmacol. 2022, 12, 763866. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Blümel, T.; Lauer, A.A.; Janitschke, D.; Stahlmann, C.; Mett, J.; Haupenthal, V.J.; Miederer, A.M.; Niemeyer, B.A.; Grimm, H.S.; et al. The impact of capsaicinoids on APP processing in Alzheimer’s disease in SH-SY5Y cells. Sci. Rep. 2020, 10, 9164. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015, 96, e52434. [Google Scholar] [CrossRef]
- Hoenig, M.C.; Bischof, G.N.; Seemiller, J.; Hammes, J.; Kukolja, J.; Onur, Ö.A.; Jessen, F.; Fliessbach, K.; Neumaier, B.; Fink, G.R.; et al. Networks of tau distribution in Alzheimer’s disease. Brain 2018, 141, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef]
- Smith, R.; Cullen, N.C.; Pichet Binette, A.; Leuzy, A.; Blennow, K.; Zetterberg, H.; Klein, G.; Borroni, E.; Ossenkoppele, R.; Janelidze, S.; et al. Tau-PET is superior to phospho-tau when predicting cognitive decline in symptomatic AD patients. Alzheimer’s Dement. 2022, 19, 2229–2756. [Google Scholar] [CrossRef]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020, 12, eaau5732. [Google Scholar] [CrossRef] [PubMed]
- Pákáski, M.; Hugyecz, M.; Sántha, P.; Jancsó, G.; Bjelik, A.; Domokos, A.; Janka, Z.; Kálmán, J. Capsaicin promotes the amyloidogenic route of brain amyloid precursor protein processing. Neurochem. Int. 2009, 54, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Lourenco, M.; Oliveira, M.; De Felice, F. Soluble amyloid-b oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Aires, I.D.; Ribeiro-Rodrigues, T.; Boia, R.; Ferreira-Rodrigues, M.; Girao, H.; Ambrosio, A.F.; Santiago, A.R. Microglial Extracellular Vesicles as Vehicles for Neurodegeneration Spreading. Biomolecules 2021, 11, 770. [Google Scholar] [CrossRef]
- Carrero, I.; Gonzalo, M.R.; Martin, B.; Sanz-Anquela, J.M.; Arévalo-Serrano, J.; Gonzalo-Ruiz, A. Oligomers of beta-amyloid protein (Aβ1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 2012, 236, 215–227. [Google Scholar] [CrossRef]
- Kong, W.L.; Peng, Y.Y.; Peng, B.W. Modulation of neuroinflammation: Role and therapeutic potential of TRPV1 in the neuro-immune axis. Brain Behav. Immun. 2017, 64, 354–366. [Google Scholar] [CrossRef]
- Annunziato, L.; Boscia, F.; Pignataro, G. Ionic transporter activity in astrocytes, microglia, and oligodendrocytes during brain ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 969–982. [Google Scholar] [CrossRef]
- Huang, W.X.; Yu, F.; Sanchez, R.M.; Liu, Y.Q.; Min, J.W.; Hu, J.J.; Bsoul, N.B.; Han, S.; Yin, J.; Liu, W.H.; et al. TRPV1 promotes repetitive febrile seizures by pro-inflammatory cytokines in immature brain. Brain Behav. Immun. 2015, 48, 68–77. [Google Scholar] [CrossRef]
- Yang, X.-L.; Wang, X.; Shao, L.; Jiang, G.-T.; Min, J.-W.; Mei, X.-Y.; He, X.-H.; Liu, W.-H.; Huang, W.-X.; Peng, B.-W. TRPV1 mediates astrocyte activation and interleukin-1β release induced by hypoxic ischemia (HI). J. Neuroinflamm. 2019, 16, 114. [Google Scholar] [CrossRef]
- Bok, E.; Chung, Y.C.; Kim, K.S.; Baik, H.H.; Shin, W.H.; Jin, B.K. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Tsai, S.-T.; Wei, T.-H.; Yang, Y.-W.; Lu, M.-K.; San, S.; Tsai, C.-H.; Lin, Y.-W. Transient receptor potential V1 modulates neuroinflammation in Parkinson’s disease dementia: Molecular implications for electroacupuncture and rivastigmine. Iran. J. Basic Med. Sci. 2021, 24, 1336–1345. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce an Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef]
- Mahmmoud, R.R.; Sase, S.; Aher, Y.D.; Sase, A.; Gröger, M.; Mokhtar, M.; Höger, H.; Lubec, G. Spatial and Working Memory Is Linked to Spine Density and Mushroom Spines. PLoS ONE 2015, 10, e0139739. [Google Scholar] [CrossRef]
- Zhou, C.-N.; Chao, F.-L.; Zhang, Y.; Jiang, L.; Zhang, L.; Fan, J.-H.; Wu, Y.-X.; Dou, X.-Y.; Tang, Y. Fluoxetine delays the cognitive function decline and synaptic changes in a transgenic mouse model of early Alzheimer’s disease. J. Comp. Neurol. 2019, 527, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Kurudenkandy, F.R.; Zilberter, M.; Biverstål, H.; Presto, J.; Honcharenko, D.; Strömberg, R.; Johansson, J.; Winblad, B.; Fisahn, A. Amyloid-β-Induced Action Potential Desynchronization and Degradation of Hippocampal Gamma Oscillations Is Prevented by Interference with Peptide Conformation Change and Aggregation. J. Neurosci. 2014, 34, 11416. [Google Scholar] [CrossRef] [PubMed]
- Kanta, V.; Pare, D.; Headley, D.B. Closed-loop control of gamma oscillations in the amygdala demonstrates their role in spatial memory consolidation. Nat. Commun. 2019, 10, 3970. [Google Scholar] [CrossRef] [PubMed]
- Etter, G.; van der Veldt, S.; Manseau, F.; Zarrinkoub, I.; Trillaud-Doppia, E.; Williams, S. Optogenetic gamma stimulation rescues memory impairments in an Alzheimer’s disease mouse model. Nat. Commun. 2019, 10, 5322. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef]
- Martorell, A.J.; Paulson, A.L.; Suk, H.-J.; Abdurrob, F.; Drummond, G.T.; Guan, W.; Young, J.Z.; Kim, D.N.-W.; Kritskiy, O.; Barker, S.J.; et al. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019, 177, 256–271.e222. [Google Scholar] [CrossRef]
- Prieto, G.A.; Tong, L.; Smith, E.D.; Cotman, C.W. TNFα and IL-1β but not IL-18 Suppresses Hippocampal Long-Term Potentiation Directly at the Synapse. Neurochem. Res. 2019, 44, 49–60. [Google Scholar] [CrossRef]
- Frank, R.A.; Komiyama, N.H.; Ryan, T.J.; Zhu, F.; O’Dell, T.J.; Grant, S.G. NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat. Commun. 2016, 7, 11264. [Google Scholar] [CrossRef]
- Canu, N.; Calissano, P. In vitro cultured neurons for molecular studies correlating apoptosis with events related to Alzheimer disease. Cerebellum 2003, 2, 270–278. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; De Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011, 14, 69–76. [Google Scholar] [CrossRef]
- Ostapchenko, V.G.; Snir, J.; Suchy, M.; Fan, J.; Cobb, M.R.; Chronik, B.A.; Kovacs, M.; Prado, V.F.; Hudson, R.H.E.; Pasternak, S.H.; et al. Detection of Active Caspase-3 in Mouse Models of Stroke and Alzheimer’s Disease with a Novel Dual Positron Emission Tomography/Fluorescent Tracer 68Ga-TC3-OGDOTA. Contrast Media Mol. Imaging 2019, 2019, 6403274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Webster, J.D.; Dugger, D.L.; Goncharov, T.; Roose-Girma, M.; Hung, J.; Kwon, Y.C.; Vucic, D.; Newton, K.; Dixit, V.M. Ubiquitin Ligases cIAP1 and cIAP2 Limit Cell Death to Prevent Inflammation. Cell Rep. 2019, 27, 2679–2689.e3. [Google Scholar] [CrossRef]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- d’Isa, R.; Comi, G.; Leocani, L. Apparatus design and behavioural testing protocol for the evaluation of spatial working memory in mice through the spontaneous alternation T-maze. Sci. Rep. 2021, 11, 21177. [Google Scholar] [CrossRef]
- Possin, K.L.; Sanchez, P.E.; Anderson-Bergman, C.; Fernandez, R.; Kerchner, G.A.; Johnson, E.T.; Davis, A.; Lo, I.; Bott, N.T.; Kiely, T.; et al. Cross-species translation of the Morris maze for Alzheimer’s disease. J. Clin. Investig. 2016, 126, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yan, H.; Tang, N.; Li, X.; Pang, P.; Li, H.; Chen, W.; Guo, Y.; Shu, S.; Cai, Y.; et al. Impairments of spatial memory in an Alzheimer’s disease model via degeneration of hippocampal cholinergic synapses. Nat. Commun. 2017, 8, 1676. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.E.; Holtzer, R.; Lipton, R.B.; Hall, C.; Verghese, J. Egocentric and exocentric navigation skills in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 1356–1363. [Google Scholar] [CrossRef]
- Tangen, G.G.; Nilsson, M.H.; Stomrud, E.; Palmqvist, S.; Hansson, O. Spatial Navigation and Its Association with Biomarkers and Future Dementia in Memory Clinic Patients Without Dementia. Neurology 2022, 99, e2081. [Google Scholar] [CrossRef] [PubMed]
- Castel, A.D.; Balota, D.A.; McCabe, D.P. Memory efficiency and the strategic control of attention at encoding: Impairments of value-directed remembering in Alzheimer’s disease. Neuropsychology 2009, 23, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Cleal, M.; Fontana, B.D.; Ranson, D.C.; McBride, S.D.; Swinny, J.D.; Redhead, E.S.; Parker, M.O. The Free-movement pattern Y-maze: A cross-species measure of working memory and executive function. Behav. Res. Methods 2021, 53, 536–557. [Google Scholar] [CrossRef]
- Lara, A.H.; Wallis, J.D. The Role of Prefrontal Cortex in Working Memory: A Mini Review. Front. Syst. Neurosci. 2015, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, 126, e55718. [Google Scholar] [CrossRef]
- Peters, C.; Bascuñán, D.; Opazo, C.; Aguayo, L.G. Differential Membrane Toxicity of Amyloid-β Fragments by Pore Forming Mechanisms. J. Alzheimer’s Dis. 2016, 51, 689–699. [Google Scholar] [CrossRef]
- Scherder, E.; Eggermont, L.; Sergeant, J.; Boersma, F. Physical activity and cognition in Alzheimer’s disease: Relationship to vascular risk factors, executive functions and gait. Rev. Neurosci. 2007, 18, 149–158. [Google Scholar] [CrossRef]
- Serra-Añó, P.; Pedrero-Sánchez, J.F.; Hurtado-Abellán, J.; Inglés, M.; Espí-López, G.V.; López-Pascual, J. Mobility assessment in people with Alzheimer disease using smartphone sensors. J. Neuroeng. Rehabil. 2019, 16, 103. [Google Scholar] [CrossRef]
- Bailey, K.R.; Crawley, J.N. Anxiety-Related Behaviors in Mice. In Methods of Behavior Analysis in Neuroscience; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis, Taylor & Francis Group, LLC.: Boca Raton, FL, USA, 2009. [Google Scholar]
- Zhao, Q.F.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Xu, W.; Li, J.Q.; Wang, J.; Lai, T.J.; et al. The prevalence of neuropsychiatric symptoms in Alzheimer’ disease: Systematic review and meta-analysis. J. Affect. Disord. 2016, 190, 264–271. [Google Scholar] [CrossRef]
- Mendez, M.F. The Relationship between Anxiety and Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2021, 5, 171–177. [Google Scholar] [CrossRef]
- Barth, A.M.; Domonkos, A.; Fernandez-Ruiz, A.; Freund, T.F.; Varga, V. Hippocampal Network Dynamics during Rearing Episodes. Cell Rep. 2018, 23, 1706–1715. [Google Scholar] [CrossRef]
- Grabher, B.J. Effects of Alzheimer Disease on Patients and Their Family. J. Nucl. Med. Technol. 2018, 46, 335–340. [Google Scholar] [CrossRef]
- Yu, J.T.; Xu, W.; Tan, C.C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.W. Neurobiology of reproduction in the female rat. A fifty-year perspective. Monogr. Endocrinol. 1989, 32, 1–133. [Google Scholar] [PubMed]
- Coluccia, E.; Louse, G. Gender differences in spatial orientation: A review. J. Environ. Psychol. 2004, 24, 329–340. [Google Scholar] [CrossRef]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.M.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Type | Inclusion Criteria | Exclusion Criteria |
|---|---|---|
| Population | Patients with Alzheimer’s disease and experimental models (animals, placenta, cell culture, human) | Non-Alzheimer’s disease populations, Alzheimer’s Disease models with comorbidities |
| Intervention | chilli/hot pepper; capsaicin | No mention of capsaicin (capsicum included), use of other capsaicinoids |
| Control | Papers with and without controls | |
| Outcomes | Mention of measurable outcome of AD (progression, severity) and test to measure outcome | Non-English, articles ahead of print. |
| Study Type | Journal articles only | Other study types (conference abstracts, books, editorials) |
| Reference | Alzheimer’s Disease Model | Capsaicin Source | Capsaicin Dose |
|---|---|---|---|
| Jiang et al., 2013 [65] | Model of cold-water stress in Sprague Dawley rats | Sigma Chemical Co. (St. Louis, MO, USA) | 10 mg/kg/day for 7 days |
| Chen et al., 2017 [66] | ICV Aβ1-42 into C57BI/6 mice | Sigma Chemical Co. (St. Louis, MO, USA) | 1 mg/kg/day for 14 days |
| Balleza-Tapia et al., 2018 [67] | Hippocampal slices from C57BL/6 and TRPV1 knockout mice incubated in 50 nM Aβ | Tocris Bioscience (Bristol, UK) | 10 μM reduced to 5 μM for 15 min and 30 min |
| Woo et al., 2018 [68] | ICV Aβ25-35 injection into ICR mice | Sigma-Aldrich (St. Louis, MO, USA) | 10 mg/kg/day for 14 days |
| Woo et al., 2018 [69] | ICV Aβ25-35 injection into ICR mice | Sigma-Aldrich (St. Louis, MO, USA) | 10 mg/kg/day for 14 days |
| Shalaby et al., 2019 [64] | ICV streptozotocin injection into adult albino rats (strain unidentified) | Dry ripe fruits of Capsicum frutescens cut from stalk | 10 mg/kg/day for 47 days |
| Du et al., 2020 [70] | APP23/PS45 transgenic mice | Sigma-Aldrich (St. Louis, MO, USA) | 1 mg/kg/day for 1 month |
| Grimm et al., 2020 [74] | In vitro SH-SY5Y/COS7 APP695 and SH-SY5Y C99 cell lines | Sigma-Aldrich (Darmstadt, Germany) | 10 μM for 24 h |
| Wang et al., 2020 [71] | In vitro human SH-SY5Y-APP695 cell line and APP/PS1 transgenic mice | Sigma-Aldrich (St. Louis, MO, USA) | Animal: 30 mg/kg/day for 6 months Cell cultures: 0.1, 1, 5, 10, 50 μM for 24 h |
| Du et al., 2020 [60] | APP23/PS45 transgenic mice | Sigma-Aldrich (St. Louis, MO, USA) | 1 mg/kg/day |
| Balleza-Tapia et al., 2018 [67] | Hippocampal slices from C57BL/6 and TRPV1 knockout mice incubated in 50 nM Aβ | Tocris Bioscience (Bristol, UK) | 10 μM reduced to 5 μM for 15 min and 30 min |
| Lu et al., 2021 [72] | APP/PS1 transgenic mice and primary cortical microglia from newborn C57BL/6 mice incubated with 5 μM Aβ oligomer | Target Molecule Corp. (Shanghai, China) | Animal: 0.01% chow for 4 weeks Cell cultures: 10 μM for 24 h |
| Wang et al., 2022 [73] | 3xTg triple transgenic mice, in vitro murine microglial BV2 cell line and primary mixed glial cells from newborn C57BL/6J wild type incubated with 2 μg/mL Aβ1-42 | Target Molecule Corp. (Shanghai, China) | Animal: 1 mg/kg; single intraperitoneal injection/day for 1 month Cell cultures: 10 μM for 24 h |
| Test | Study Outcome |
|---|---|
| Morris Water Maze (MWM) | Capsaicin group had improved spatial memory compared to controls in all studies [65,66,68,70,71,72,73] except Woo et al., 2018 [69]. |
| Barnes Maze | Capsaicin group was found to have improved spatial memory and learning compared to controls [70]. |
| T-maze | Capsaicin had no significant effect on spatial perception abilities and spatial cognition compared with controls [68]. |
| Y-maze | Capsaicin group had improved working memory, compared to controls [71,73]. |
| Novel Object Recognition Test | Capsaicin group demonstrated improved cognitive abilities, particularly episodic memory, when compared to AD controls [65,66], while Woo et al., (2018) [68], found no difference. |
| Passive Avoidance Test | Capsaicin group had improved episodic memory [64]. |
| Open Field Test | Capsaicin group had greater anxiety-like behaviours, seen in an increased amount of rearing and more time spent in peripheral areas compared with central areas [71]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inyang, D.; Saumtally, T.; Nnadi, C.N.; Devi, S.; So, P.-W. A Systematic Review of the Effects of Capsaicin on Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 10176. https://doi.org/10.3390/ijms241210176
Inyang D, Saumtally T, Nnadi CN, Devi S, So P-W. A Systematic Review of the Effects of Capsaicin on Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(12):10176. https://doi.org/10.3390/ijms241210176
Chicago/Turabian StyleInyang, Deborah, Tasneem Saumtally, Chinelo Nonyerem Nnadi, Sharmila Devi, and Po-Wah So. 2023. "A Systematic Review of the Effects of Capsaicin on Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 12: 10176. https://doi.org/10.3390/ijms241210176
APA StyleInyang, D., Saumtally, T., Nnadi, C. N., Devi, S., & So, P.-W. (2023). A Systematic Review of the Effects of Capsaicin on Alzheimer’s Disease. International Journal of Molecular Sciences, 24(12), 10176. https://doi.org/10.3390/ijms241210176