
Parkinson’s Disease: From Genetics and Epigenetics to Treatment, a miRNA-Based Strategy

Abstract
1. Introduction
2. Parkinson’s Disease: Genetics and Epigenetics
2.1. Parkinson’s Genetics
2.2. Parkinson’s Epigenetics
Epigenetic Alterations in Parkinson’s: The Critical Role of miRNAs
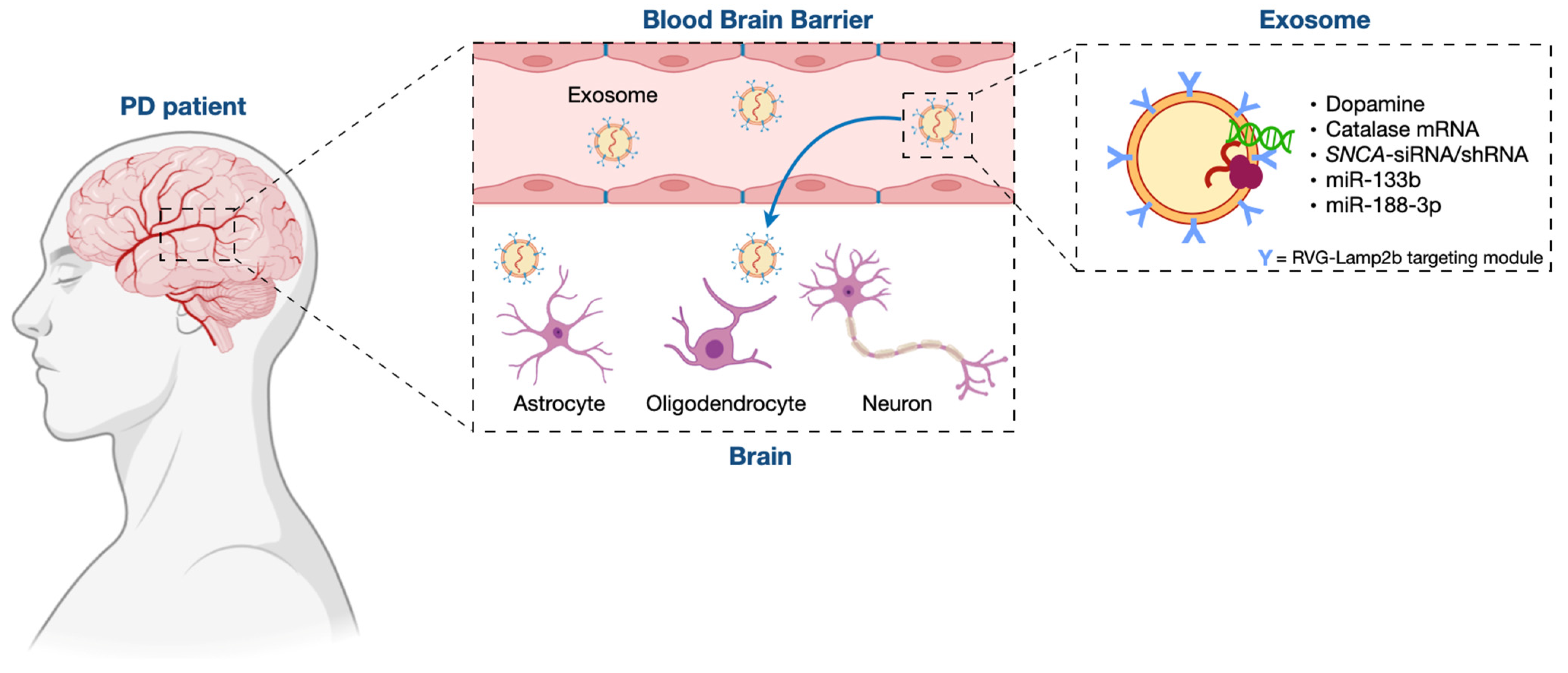
3. Exosome/miRNA Network: Not Only a Role in the Pathogenesis of PD but Also a Clinical Potential for Its Treatment
3.1. Exosomes in PD Treatment
3.2. Mesenchymal Stem Cell (MSC)-Derived Exosomes as Therapeutic Vehicles for miRNA Delivery in PD Treatment
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parkinson, J. An Essay on the Shaking Palsy; Whittingham & Rowland: London, UK, 1817. [Google Scholar]
- Goetz, C.G. The history of parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a0088622011. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Kurz, A.; Arzberger, T.; Giese, A.; Höglinger, G.U. The Differential Diagnosis and Treatment of Atypical Parkinsonism. Dtsch. Arztebl. Int. 2016, 113, 61–99. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W. Is cell death primary or secondary in the pathophysiology of idiopathic Parkinson’s disease? Biomolecules 2015, 5, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. Gene-environment interactions in Parkinson’s disease: Specific evidence in humans and mammalian models. Neurobiol. Dis. 2013, 57, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-Synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef]
- Hansen, C.; Li, J.Y. Beyond α-Synuclein transfer: Pathology propagation in Parkinson’s disease. Trends Mol. Med. 2012, 18, 248–255. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-synuclein: From early synaptic dysfunction to neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef]
- Gasser, T. Genetics of Parkinson’s disease. Clin. Genet. 1998, 54, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Calne, D.B.; Mizuno, Y. The neuromythology of Parkinson’s Disease. Park. Relat. Disord. 2004, 10, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Gwinn, K.; Singleton, A.; Hardy, J. Parkinson’s disease and alpha-synuclein. Mov. Disord. 2011, 26, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.L.; Chen, Y.; Zhang, C.H.; Wang, Y.X.; Fernandez-Funez, P. Genetics of Parkinson’s disease and related disorders. J. Med. Genet. 2018, 55, 73–80. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha- synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.G.; Feany, M.B. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51d alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. α-Synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Jiang, X.; Forno, L.S.; Di Monte, D.A.; Tanner, C.M.; Langston, J.W. Absence of mutations in the coding region of the alpha-synuclein gene in pathologically proven Parkinson’s disease. Neurology 1998, 50, 1136–1137. [Google Scholar] [CrossRef]
- Farrer, M.; Wavrant-De Vrieze, F.; Crook, R.; Boles, L.; Perez-Tur, J.; Hardy, J.; Johnson, W.G.; Steele, J.; Maraganore, D.; Gwinn, K.; et al. Low frequency of alpha-synuclein mutations in familial Parkinson’s disease. Ann. Neurol. 1998, 43, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.R.; Farrer, M.J.; Wszolek, Z.K.; Gasser, T.; Durr, A.; Agid, Y.; Bonifati, V.; DeMichele, G.; Volpe, G.; Lincoln, S.; et al. Sequencing of the α- synuclein gene in a large series of cases of familial Parkinson’s disease fails to reveal any further mutations. The European Consortium on Genetic Susceptibility in Parkinson’s Disease (GSPD). Hum. Mol. Genet. 1998, 7, 751–753. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Curran, M.D.; Wallace, A.; Middleton, D.; Murgatroyd, C.; Curtis, A.; Perry, R.; Jaros, E. Mutation screening in exons 3 and 4 of alpha-synuclein in sporadic Parkinson’s and sporadic and familial dementia with Lewy bodies cases. Neuroreport 1998, 9, 3925–3927. [Google Scholar] [CrossRef]
- Pastor, P.; Muñoz, E.; Ezquerra, M.; Obach, V.; Martí, M.J.; Valldeoriola, F.; Tolosa, E.; Oliva, R. Analysis of the coding and the 5’ flanking regions of the alpha-synuclein gene in patients with Parkinson’s disease. Mov. Disord. 2001, 16, 1115–1119. [Google Scholar] [CrossRef]
- Hope, A.D.; Myhre, R.; Kachergus, J.; Lincoln, S.; Bisceglio, G.; Hulihan, M.; Farrer, M.J. Alpha-synuclein missense and multiplication mutations in autosomal dominant Parkinson’s disease. Neurosci. Lett. 2004, 367, 97–100. [Google Scholar] [CrossRef]
- Berg, D.; Niwar, M.; Maass, S.; Zimprich, A.; Möller, J.C.; Wuellner, U.; Schmitz-Hübsch, T.; Klein, C.; Tan, E.K.; Schöls, L.; et al. Alpha-synuclein and Parkinson’s disease: Implications from the screening of more than 1900 patients. Mov. Disord. 2005, 20, 1191–1194. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, P.; Bonnet, A.M.; Débarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Dürr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Hayashi, S.; Farrer, M.J.; Singleton, A.B.; Yoshino, H.; Imai, H.; Kitami, T.; Sato, K.; Kuroda, R.; Tomiyama, H.; et al. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann. Neurol. 2006, 59, 298–309. [Google Scholar] [CrossRef]
- Chiba-Falek, O.; Kowalak, J.A.; Smulson, M.E.; Nussbaum, R.L. Regulation of alpha-synuclein expression by poly (ADP ribose) polymerase-1 (PARP-1) binding to the NACP-Rep1 polymorphic site upstream of the SNCA gene. Am. J. Hum. Genet. 2005, 76, 478–492. [Google Scholar] [CrossRef][Green Version]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Mata, I.F.; Wedemeyer, W.J.; Farrer, M.J.; Taylor, J.P.; Gallo, K.A. LRRK2 in Parkinson’s disease: Protein domains and functional insights. Trends Neurosci. 2006, 29, 286–293. [Google Scholar] [CrossRef]
- Biskup, S.; West, A.B. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 625–633. [Google Scholar]
- Monfrini, E.; Di Fonzo, A. Kinase L-RR. (LRRK2) Genetics and Parkinson’s disease. Adv. Neurobiol. 2017, 14, 3–30. [Google Scholar]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef]
- Rajput, A.; Dickson, D.W.; Robinson, C.A.; Ross, O.A.; Dächsel, J.C.; Lincoln, S.J.; Cobb, S.A.; Rajput, M.L.; Farrer, M.J. Parkinsonism, LRRK2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef]
- Dächsel, J.C.; Ross, O.A.; Mata, I.F.; Kachergus, J.; Toft, M.; Cannon, A.; Baker, M.; Adamson, J.; Hutton, M.; Dickson, D.W.; et al. LRRK2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol. 2007, 113, 601–606. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto- Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Szu-Tu, C.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome- wide linkage and sequencing study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Yan, W.; Tang, B.; Zhou, X.; Lei, L.; Li, K.; Sun, Q.; Xu, Q.; Yan, X.; Guo, J.; Liu, Z. TMEM230 mutation analysis in Parkinson’s disease in a Chinese population. Neurobiol. Aging 2017, 49, 219.e1–219.e3. [Google Scholar] [CrossRef] [PubMed]
- Sudhaman, S.; Muthane, U.B.; Behari, M.; Govindappa, S.T.; Juyal, R.C.; Thelma, B.K. Evidence of mutations in RIC3 acetylcholine receptor chaperone as a novel cause of autosomal-dominant Parkinson’s disease with non-motor phenotypes. J. Med. Genet. 2016, 53, 559–566. [Google Scholar] [CrossRef]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Foroud, T.; Uniacke, S.K.; Liu, L.; Pankratz, N.; Rudolph, A.; Halter, C.; Shults, C.; Marder, K.; Conneally, P.M.; Nichols, W.C.; et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003, 60, 796–801. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef]
- Farrer, M.; Chan, P.; Chen, R.; Tan, L.; Lincoln, S.; Hernandez, D.; Forno, L.; Gwinn-Hardy, K.; Petrucelli, L.; Hussey, J.; et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann. Neurol. 2001, 50, 293–300. [Google Scholar]
- Sun, M.; Latourelle, J.C.; Wooten, G.F.; Lew, M.F.; Klein, C.; Shill, H.A.; Golbe, L.I.; Mark, M.H.; Racette, B.A.; Perlmutter, J.S.; et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: The GenePD study. Arch. Neurol. 2006, 63, 826–832. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Kann, M.; Jacobs, H.; Mohrmann, K.; Schumacher, K.; Hedrich, K.; Garrels, J.; Wiegers, K.; Schwinger, E.; Pramstaller, P.P.; Breakefield, X.O.; et al. Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann. Neurol. 2002, 51, 621–625. [Google Scholar] [CrossRef]
- Nichols, W.C.; Pankratz, N.; Uniacke, S.K.; Pauciulo, M.W.; Halter, C.; Rudolph, A.; Conneally, P.M.; Foroud, T. Linkage stratification and mutation analysis at the parkin locus identifies mutation positive Parkinson’s disease families. J. Med. Genet. 2002, 39, 489–492. [Google Scholar] [CrossRef]
- Hedrich, K.; Eskelson, C.; Wilmot, B.; Marder, K.; Harris, J.; Garrels, J.; Meija-Santana, H.; Vieregge, P.; Jacobs, H.; Bressman, S.B.; et al. Distribution, type, and origin of parkin mutations: Review and case studies. Mov. Disord. 2004, 19, 1146–1157. [Google Scholar] [CrossRef]
- Van de Warrenburg, B.P.; Lammens, M.; Lücking, C.B.; Denèfle, P.; Wesseling, P.; Booij, J.; Praamstra, P.; Quinn, N.; Brice, A.; Horstink, M.W. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 2001, 56, 555–557. [Google Scholar] [CrossRef]
- Lohmann, E.; Periquet, M.; Bonifati, V.; Wood, N.W.; De Michele, G.; Bonnet, A.M.; Fraix, V.; Broussolle, E.; Horstink, M.W.; Vidailhet, M.; et al. How much phenotypic variation can be attributed to parkin genotype? Ann. Neurol. 2003, 54, 176–185. [Google Scholar] [CrossRef]
- Kirola, L.; Behari, M.; Shishir, C.; Thelma, B.K. Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile parkinsonism. Park. Relat. Disord. 2016, 31, 124–128. [Google Scholar] [CrossRef]
- Rauschendorf, M.A.; Jost, M.; Stock, F.; Zimmer, A.; Rösler, B.; Rijntjes, M.; Piroth, T.; Coenen, V.A.; Reinacher, P.C.; Meyer, P.T.; et al. Novel compound heterozygous synaptojanin-1 mutation causes l-dopa-responsive dystonia-parkinsonism syndrome. Mov. Disord. 2017, 32, 478–480. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Tanaka, K. The PINK1-Parkin axis: An Overview. Neurosci. Res. 2020, 159, 9–15. [Google Scholar] [CrossRef]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e3622004. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, M.; Wilson, M.A.; Petsko, G.A.; Fink, A.L. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J. Mol. Biol. 2006, 356, 1036–1048. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Hague, S.; Rogaeva, E.; Hernandez, D.; Gulick, C.; Singleton, A.; Hanson, M.; Johnson, J.; Weiser, R.; Gallardo, M.; Ravina, B.; et al. Early-onset Parkinson’s disease caused by a compound heterozygous DJ-1 mutation. Ann. Neurol. 2003, 54, 271–274. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Healy, D.G.; Quinn, N.; Lees, A.J.; Wood, N.W. The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann. Neurol. 2003, 54, 283–286. [Google Scholar] [CrossRef]
- Hering, R.; Strauss, K.M.; Tao, X.; Bauer, A.; Woitalla, D.; Mietz, E.M.; Petrovic, S.; Bauer, P.; Schaible, W.; Müller, T.; et al. Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7). Hum. Mutat. 2004, 24, 321–329. [Google Scholar] [CrossRef]
- Waragai, M.; Wei, J.; Fujita, M.; Nakai, M.; Ho, G.J.; Masliah, E.; Akatsu, H.; Yamada, T.; Hashimoto, M. Increased level of DJ-1 in the cerebro- spinal fluids of sporadic Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 345, 967–972. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Outeiro, T.F. Epigenetics in Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 978, 363–390. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Wu, L.; Zhao, Z.B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.F.; Chen, S.D. Methylation of alpha-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Park. Relat. Disord. 2014, 20, 308–313. [Google Scholar] [CrossRef]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef]
- de Boni, L.; Riedel, L.; Schmitt, I.; Kraus, T.F.J.; Kaut, O.; Piston, D.; Akbarian, S.; Wüllner, U. DNA methylation levels of α-synuclein intron 1 in the aging brain. Neurobiol. Aging 2015, 36, 3334.e7–3334.e11. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Pieper, H.C.; Evert, B.O.; Kaut, O.; Riederer, P.F.; Waha, A.; Wüllner, U. Different methylation of the TNF-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability. Neurobiol Dis. 2008, 32, 521–527. [Google Scholar] [CrossRef]
- Mouradian, M.M. MicroRNAs in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 279–284. [Google Scholar] [CrossRef]
- Kanagaraj, N.; Beiping, H.; Dheen, S.T.; Tay, S.S. Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 2014, 272, 167–179. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Fragkouli, A.; Doxakis, E. miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front. Cell Neurosci. 2014, 8, 182. [Google Scholar] [CrossRef]
- Titze-de-Almeida, S.S.; Soto-Sánchez, C.; Fernandez, E.; Koprich, J.B.; Brotchie, J.M.; Titze-de-Almeida, R. The Promise and Challenges of Developing miRNA-Based Therapeutics for Parkinson’s Disease. Cells 2020, 9, 841. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Piperi, C. miR-124 and Parkinson’s disease: A biomarker with therapeutic potential. Pharm. Res. 2019, 150, 104515. [Google Scholar] [CrossRef] [PubMed]
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’Neill, M.J.; Wong, L.F.; et al. Loss of MicroRNA-7 Regulation Leads to alpha-Synuclein Accumulation and Dopaminergic Neuronal Loss In Vivo. Mol. Ther. 2017, 25, 2404–2414. [Google Scholar] [CrossRef]
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed]
- Minones-Moyano, E.; Porta, S.; Escaramis, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Marti, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef]
- Soreq, H.; Wolf, Y. NeurimmiRs: MicroRNAs in the neuroimmune interface. Trends Mol. Med. 2011, 17, 548–555. [Google Scholar] [CrossRef]
- Sonntag, K.C. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 2010, 1338, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Conaco, C.; Xuan, Z.; Schmidt, B.H.; Parker, J.S.; Mandel, G.; Hannon, G.J. A biochemical approach to identifying microRNA targets. Proc. Natl. Acad. Sci. USA 2007, 104, 19291–19296. [Google Scholar] [CrossRef]
- Harischandra, D.S.; Ghaisas, S.; Rokad, D.; Zamanian, M.; Jin, H.; Anantharam, V.; Kimber, M.; Kanthasamy, A.; Kanthasamy, A.G. Environmental neurotoxicant manganese regulates exosome- mediated extracellular miRNAs in cell culture model of Parkinson’s disease: Relevance to α-synuclein misfolding in metal neurotoxicity. Neurotoxicology 2018, 64, 267–277. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, C.; Sun, Q.; Pan, H.; Huang, P.; Ding, J.; Chen, S. MicroRNA- 4639 is a regulator of DJ-1 expression and a potential early diagnostic marker for Parkinson’s disease. Front. Aging Neurosci. 2017, 9, 232. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, J.; Chen, L.; Jin, Y.; Zhang, G.; Lin, Z.; Du, S.; Fu, Z.; Chen, T.; Qin, Y.; et al. Serum secreted miR-137-containing exosomes affects oxidative stress of neurons by regulating OXR1 in Parkinson’s disease. Brain Res. 2019, 1722, 146331. [Google Scholar] [CrossRef]
- Müller, T. Drug therapy in patients with Parkinson’s disease. Transl. Neurodegener. 2012, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Breakefield, X. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front. Physiol. 2012, 3, 228. [Google Scholar] [CrossRef]
- Mirzaei, H.; Sahebkar, A.; Jaafari, M.R.; Goodarzi, M.; Mirzaei, H.R. Diagnostic and therapeutic potential of exosomes in cancer: The beginning of a new tale? J. Cell. Physiol. 2017, 232, 3251–3260. [Google Scholar] [CrossRef]
- Saeedi Borujeni, M.J.; Esfandiary, E.; Taheripak, G.; Codoñer-Franch, P.; Alonso- Iglesias, E.; Mirzaei, H. Molecular aspects of diabetes mellitus: Resistin, microRNA, and exosome. J. Cell. Biochem. 2018, 119, 1257–1272. [Google Scholar] [CrossRef]
- Pourhanifeh, M.H.; Mahjoubin-Tehran, M.; Shafiee, A.; Hajighadimi, S.; Moradizarmehri, S.; Mirzaei, H.; Asemi, Z. MicroRNAs and exosomes: Small molecules with big actions in multiple myeloma pathogenesis. IUBMB Life 2019, 72, 314–333. [Google Scholar] [CrossRef]
- van der Meel, R.; Fens, M.H.; Vader, P.; van Solinge, W.W.; Eniola-Adefeso, O.; Schiffelers, R.M. Extracellular vesicles as drug delivery systems: Lessons from the liposome field. J. Control. Release 2014, 195, 72–85. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Tomlinson, P.R.; Zheng, Y.; Fischer, R.; Heidasch, R.; Gardiner, C.; Evetts, S.; Hu, M.; Wade-Martins, R.; Turner, M.R.; Morris, J.; et al. Identification of distinct circulating exosomes in Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 353–361. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Sánchez-Madrid, F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell. Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef]
- Katsuda, T.; Kosaka, N.; Takeshita, F.; Ochiya, T. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics 2013, 13, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Savitz, S.I.; Chopp, M.; Deans, R.; Carmichael, T.; Phinney, D.; Wechsler, L.; STEPS Participants. Stem Cell Therapy as an Emerging Paradigm for Stroke (STEPS) II. Stroke 2011, 42, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Janowski, M.; Wagner, D.C.; Boltze, J. Stem Cell-Based Tissue Replacement After Stroke: Factual Necessity or Notorious Fiction? Stroke 2015, 46, 2354–2363. [Google Scholar] [CrossRef]
- Fang, Y.; Gao, T.; Zhang, B.; Pu, J. Recent Advances: Decoding Alzheimer’s Disease with Stem Cells. Front. Aging Neurosci. 2018, 10, 77. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Liu, Z.; Wang, X.; Shang, X.; Cui, Y.; Zhang, Z.G.; Chopp, M. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells 2013, 31, 2737–2746. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Lv, Q.; Gao, J.; Hu, L.; He, Z. MicroRNA-21 Overexpression Promotes the Neuroprotective Efficacy of Mesenchymal Stem Cells for Treatment of Intracerebral Hemorrhage. Front. Neurol. 2018, 9, 931. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Kubota, K.; Kobayashi, E.; Chikenji, T.S.; Saito, Y.; Konari, N.; Fujimiya, M. Bone marrow-derived mesenchymal stem cells improve cognitive impairment in an Alzheimer’s disease model by increasing the expression of microRNA-146a in hippocampus. Sci. Rep. 2020, 10, 10772. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Verrilli, M.A.; Picou, F.; Court, F.A. Schwann cell-derived exosomes enhance axonal regeneration in the peripheral nervous system. Glia 2013, 61, 1795–1806. [Google Scholar] [CrossRef]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef]
- Cooper, J.M.; Wiklander, P.B.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef]
- Izco, M.; Blesa, J.; Schleef, M.; Schmeer, M.; Porcari, R.; Al-Shawi, R.; Ellmerich, S.; de Toro, M.; Gardiner, C.; Seow, Y.; et al. Systemic exosomal delivery of shRNA minicircles prevents Parkinsonian pathology. Mol. Ther. 2019, 27, 30371–30375. [Google Scholar] [CrossRef]
- Pushparaj, P.N.; Aarthi, J.J.; Kumar, S.D.; Manikandan, J. RNAi and RNAa--the yin and yang of RNAome. Bioinformation 2008, 2, 235–237. [Google Scholar] [CrossRef]
- Dalmizrak, A.; Dalmizrak, O. Mesenchymal stem cell-derived exosomes as new tools for delivery of miRNAs in the treatment of cancer. Front. Bioeng. Biotechnol. 2022, 10, 956563. [Google Scholar] [CrossRef]
- Oveili, E.; Vafaei, S.; Bazavar, H.; Eslami, Y.; Mamaghanizadeh, E.; Yasamineh, S.; Gholizadeh, O. The potential use of mesenchymal stem cells-derived exosomes as microRNAs delivery systems in different diseases. Cell. Commun. Signal. 2023, 21, 20. [Google Scholar] [CrossRef]
- Vilaca-Faria, H.; Salgado, A.J.; Teixeira, F.G. Mesenchymal stem cells- derived exosomes: A new possible therapeutic strategy for Parkinson’s disease? Cells 2019, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Z.; Xing, H.; Wang, Y.; Guo, Y. Exosomes derived from miR-188-3p-modified adipose-derived mesenchymal stem cells protect Parkinson’s disease. Mol. Nucleic. Acids 2021, 23, 1334–1344. [Google Scholar] [CrossRef]
- Bortolozzi, A.; Manashirov, S.; Chen, A.; Artigas, F. Oligonucleotides as therapeutic tools for brain disorders: Focus on major depressive disorder and Parkinson’s disease. Pharm. Ther. 2021, 227, 107873. [Google Scholar] [CrossRef] [PubMed]
- Marczak, S.; Richards, K.; Ramshani, Z.; Smith, E.; Senapati, S.; Hill, R.; Go, D.B.; Chang, H.C. Simultaneous Isolation and Preconcentration of Exosomes by Ion Concentration Polarization. Electrophoresis 2018, 39, 2029–2038. [Google Scholar] [CrossRef]
- Kang, Y.T.; Kim, Y.J.; Bu, J.; Cho, Y.H.; Han, S.W.; Moon, B.I. High-Purity Capture and Release of Circulating Exosomes Using an Exosome- Specific Dual-Patterned Immunofiltration (ExoDIF) Device. Nanoscale 2017, 9, 13495–13505. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, W.; Zhang, L.; Ban, L.; Chen, P.; Du, W.; Feng, X.; Liu, B.F. Chemically Edited Exosomes with Dual Ligand Purified by Microfluidic Device for Active Targeted Drug Delivery to Tumor Cells. ACS Appl. Mater. Interfaces 2017, 9, 27441–27452. [Google Scholar] [CrossRef]
- Woo, H.K.; Sunkara, V.; Park, J.; Kim, T.H.; Han, J.R.; Kim, C.J.; Choi, H.I.; Kim, Y.K.; Cho, Y.K. Exodisc for Rapid, Size-Selective, and Efficient Isolation and Analysis of Nanoscale Extracellular Vesicles from Biological Samples. ACS Nano. 2017, 11, 1360–1370. [Google Scholar] [CrossRef]
- Liu, F.; Vermesh, O.; Mani, V.; Ge, T.J.; Madsen, S.J.; Sabour, A.; Hsu, E.C.; Gowrishankar, G.; Kanada, M.; Jokerst, J.V.; et al. The Exosome Total Isolation Chip. ACS Nano. 2017, 11, 10712–10723. [Google Scholar] [CrossRef]
- Wunsch, B.H.; Smith, J.T.; Gifford, S.M.; Wang, C.; Brink, M.; Bruce, R.L.; Austin, R.H.; Stolovitzky, G.; Astier, Y. Nanoscale lateral displacement arrays for the separation of exosomes and colloids down to 20 nm. Nat. Nanotechnol. 2016, 11, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell. Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.M.; Vyas, A.D.; Qiu, Y.; Messer, K.S.; White, R.; Heller, M.J. Integrated Analysis of Exosomal Protein Biomarkers on Alternating Current Electrokinetic Chips Enables Rapid Detection of Pancreatic Cancer in Patient Blood. ACS Nano. 2018, 12, 3311–3320. [Google Scholar] [CrossRef]
- Zhang, P.; Yeo, J.C.; Lim, C.T. Advances in Technologies for Purification and Enrichment of Extracellular Vesicles. SLAS Technol. 2019, 24, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef]
- Bobrie, A.; Colombo, M.; Krumeich, S.; Raposo, G.; Théry, C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 2012, 1, 18397. [Google Scholar] [CrossRef]
- Sahu, S.S.; Gevari, M.T.; Nagy, Á.; Gestin, M.; Hååg, P.; Lewensohn, R.; Viktorsson, K.; Karlström, A.E.; Dev, A. Multi-marker profiling of extracellular vesicles using streaming current and sequential electrostatic labeling. Biosens. Bioelectron. 2023, 227, 115142. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Liu, C.; Su, C. Design strategies and application progress of therapeutic exosomes. Theranostics 2019, 9, 1015–1028. [Google Scholar] [CrossRef]
- Ingato, D.; Lee, J.U.; Sim, S.J.; Kwon, Y.J. Good things come in small packages: Overcoming challenges to harness extracellular vesicles for therapeutic delivery. J. Control. Release 2016, 241, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Furlán, M.; Vidal, M.; Colombo, M.I. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J. Biol. Chem. 2003, 278, 20083–20090. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular vesicles: Composition, biological relevance, and methods of study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, K.; Zhang, N.; Zhao, Y.; Ma, Q.; Shen, J.; Lin, Y.; Xiang, P.; Tang, Y.; Hu, X.; et al. Myocardial reparative functions of exosomes from mesenchymal stem cells are enhanced by hypoxia treatment of the cells via transferring microRNA-210 in an nSMase2-dependent way. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef]
- Wysoczynski, M.; Ratajczak, M.Z. Lung cancer secreted microvesicles: Underappreciated modulators of microenvironment in expanding tumors. Int. J. Cancer 2009, 125, 1595–1603. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Emam, S.E.; Ando, H.; Abu Lila, A.S.; Shimizu, T.; Ukawa, M.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.S.; Ishida, T. A novel strategy to increase the yield of exosomes (extracellular vesicles) for an expansion of basic research. Biol. Pharm. Bull. 2018, 41, 733–742. [Google Scholar] [CrossRef]
- Chen, T.S.; Arslan, F.; Yin, Y.; Tan, S.S.; Lai, R.C.; Choo, A.B.; Padmanabhan, J.; Lee, C.N.; de Kleijn, D.P.; Lim, S.K. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J. Transl. Med. 2011, 9, 47. [Google Scholar] [CrossRef]
- Phan, J.; Kumar, P.; Hao, D.; Gao, K.; Farmer, D.; Wang, A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 2018, 7, 1522236. [Google Scholar] [CrossRef] [PubMed]
- Jafari, D.; Shajari, S.; Jafari, R.; Mardi, N.; Gomari, H.; Ganji, F.; Forouzandeh Moghadam, M.; Samadikuchaksaraei, A. Designer exosomes: A new platform for biotechnology therapeutics. BioDrugs 2020, 34, 567–586. [Google Scholar] [CrossRef]
- Wang, J.; Bonacquisti, E.E.; Brown, A.D.; Nguyen, J. Boosting the biogenesis and secretion of mesenchymal stem cell-derived exosomes. Cells 2020, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bi, J.; Huang, J.; Tang, Y.; Du, S.; Li, P. Exosome: A Review of Its Classification, Isolation Techniques, Storage, Diagnostic and Targeted Therapy Applications. Int. J. Nanomed. 2020, 15, 6917–6934. [Google Scholar] [CrossRef]
- De Ronde, M.W.J.; Kok, M.G.M.; Moerland, P.D.; Van den Bossche, J.; Neele, A.E.; Halliani, A.; van der Made, I.; de Winther, M.P.J.; Meijers, J.C.M.; Creemers, E.E.; et al. High miR-124-3p expression identifies smoking individuals susceptible to atherosclerosis. Atherosclerosis 2017, 263, 377–384. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| miRNAs downregulated in PD | Target genes | Molecular mechanisms |
| miR-7 | SNCA [91] | miR-7 is responsible for downregulating SNCA gene expression, its depletion being associated with α-Syn accumulation and neuron loss |
| miR-153 | SNCA [92] | miR-153 is responsible for downregulating SNCA gene expression, its depletion inducing α-Syn accumulation and neuron loss |
| miR-34b/c | SNCA, Parkin and DU-1 [93] | miR-34b/c is responsibl for dowregulating SNCA gene expression, its depletion leading to both α-Syn deposition in PD brain tssues and downregulation of Parkin and DJ-1 gene expression |
| miR-133b | Pitx3 [94] | miR-133b is specifically expressed in midbrain, where it regulates both the maturation and function of midbrain DA nourons, its depletion being associated with a massive lost of DA neurons |
| miR-124 | Calpain 1, Bim, STAT3, Annexin A5, MEKK3 [95,96,97] | miR-124 regulates synapse morphology, neurotransmission, infammation, autophagy and mitochondrial function, its depletion being implicated in the core pathophysiologic mechanisms |
| miRNAs upregulated in PD | Target genes | Molecular mechanisms |
| miR-4639-5p | DJ-1 [99] | miR-4639-5p negatively regulates the post-transcription levels of DJ-1, its upregulation being responsible for a massive induction of oxidative stress and, consequently. neuronal death |
| miR-137 | OXR1 [100] | miR-137 is involved in the induction of oxidative stress in neurons, its upregulation being involved in a massive induction of oxidative stress and neuronal death |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paccosi, E.; Proietti-De-Santis, L. Parkinson’s Disease: From Genetics and Epigenetics to Treatment, a miRNA-Based Strategy. Int. J. Mol. Sci. 2023, 24, 9547. https://doi.org/10.3390/ijms24119547
Paccosi E, Proietti-De-Santis L. Parkinson’s Disease: From Genetics and Epigenetics to Treatment, a miRNA-Based Strategy. International Journal of Molecular Sciences. 2023; 24(11):9547. https://doi.org/10.3390/ijms24119547
Chicago/Turabian StylePaccosi, Elena, and Luca Proietti-De-Santis. 2023. "Parkinson’s Disease: From Genetics and Epigenetics to Treatment, a miRNA-Based Strategy" International Journal of Molecular Sciences 24, no. 11: 9547. https://doi.org/10.3390/ijms24119547
APA StylePaccosi, E., & Proietti-De-Santis, L. (2023). Parkinson’s Disease: From Genetics and Epigenetics to Treatment, a miRNA-Based Strategy. International Journal of Molecular Sciences, 24(11), 9547. https://doi.org/10.3390/ijms24119547