

ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries



Abstract
1. Introduction
2. The Structure and Physiological Function of ACSL4
2.1. The Structure of ACSL4
2.2. The Physiological Function of ACSL4
2.3. The Regulation Mechanism of ACSL4
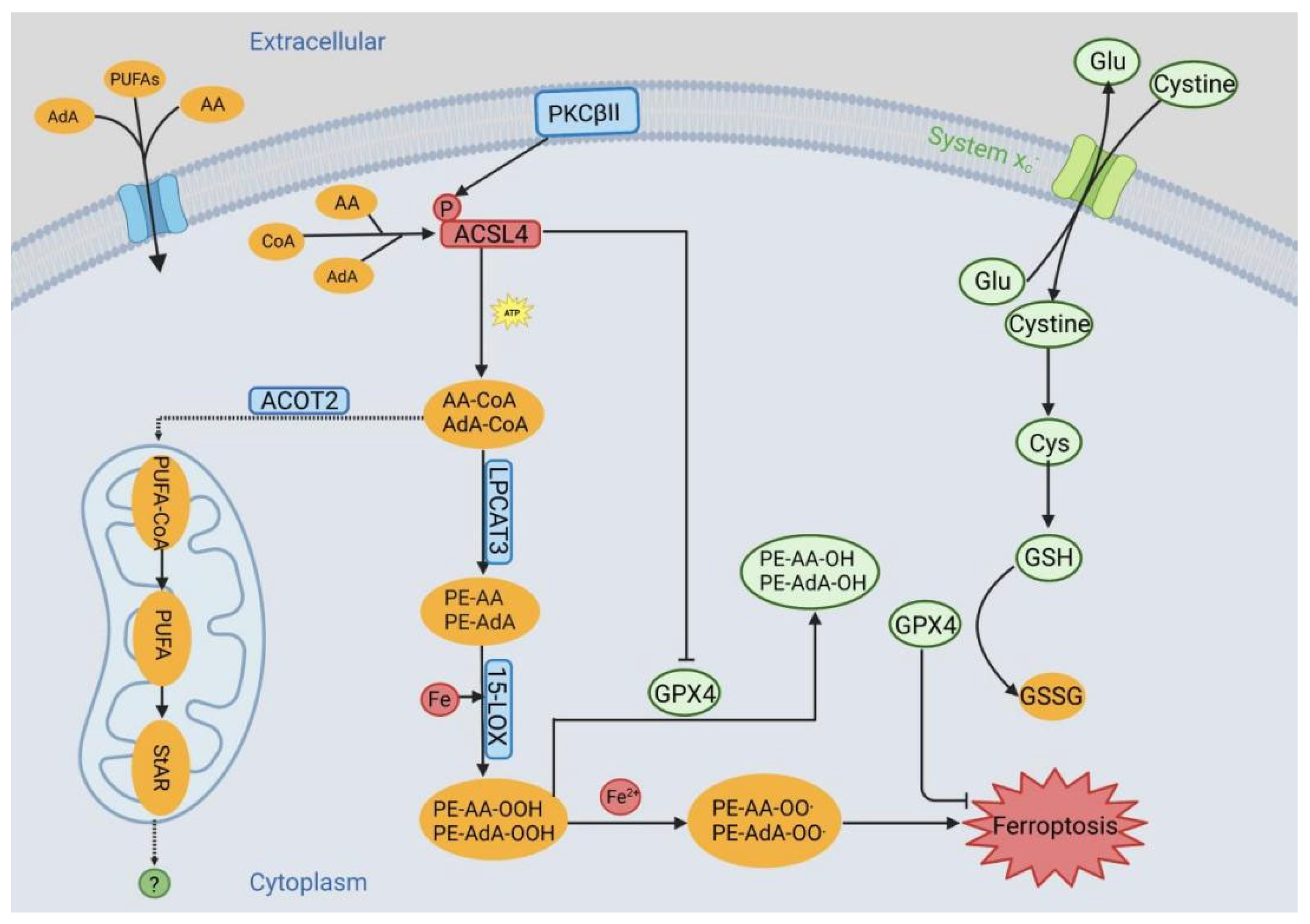
3. ACSL4 in Ferroptosis
4. ACSL4 in Neurological Diseases and Injuries
4.1. ACSL4 in Brain Injury
4.2. ACSL4 in Stroke
4.2.1. ACSL4 in Ischemic Stroke
4.2.2. ACSL4 in Hemorrhagic Stroke
4.3. ACSL4 in Alzheimer’s Disease
4.4. ACSL4 in Parkinson’s Disease
4.5. ACSL4 in Spinal Cord Diseases
4.6. ACSL4 in Multiple Sclerosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.; Lu, G. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363. [Google Scholar] [CrossRef] [PubMed]
- Radif, Y.; Ndiaye, H.; Kalantzi, V.; Jacobs, R.; Hall, A.; Minogue, S.; Waugh, M.G. The endogenous subcellular localisations of the long chain fatty acid-activating enzymes ACSL3 and ACSL4 in sarcoma and breast cancer cells. Mol. Cell. Biochem. 2018, 448, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Jiang, C.; Wen, X.; Li, C.; Xiong, S.; Yue, T.; Long, P.; Shi, J.; Zhang, Z. ACSL4 as a Potential Target and Biomarker for Anticancer: From Molecular Mechanisms to Clinical Therapeutics. Front. Pharmacol. 2022, 13, 949863. [Google Scholar] [CrossRef]
- Fujino, T.; Kang, M.; Suzuki, H.; Iijima, H.; Yamamoto, T. Molecular Characterization and Expression of Rat Acyl-CoA Synthetase 3. J. Biol. Chem. 1996, 271, 16748–16752. [Google Scholar] [CrossRef]
- Cao, Y.; Traer, E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Cloning, Expression, and Chromosomal Localization of Human Long-Chain Fatty Acid-CoA Ligase 4 (FACL4). Genomics 1998, 49, 327–330. [Google Scholar] [CrossRef]
- Trautenberg, L.C.; Brankatschk, M.; Shevchenko, A.; Wigby, S.; Reinhardt, K. Ecological lipidology. Elife 2022, 11, e79288. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Itabe, H.; Kinoshita, T.; Homma, K.J.; Onoduka, J.; Mori, M.; Yamaguchi, S.; Makita, M.; Higashi, Y.; Yamashita, A.; et al. Involvement of ACSL in local synthesis of neutral lipids in cytoplasmic lipid droplets in human hepatocyte HuH7. J. Lipid Res. 2007, 48, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, H.; Fan, X.; Yu, X.; Huai, J. Lipid Dyshomeostasis and Inherited Cerebellar Ataxia. Mol. Neurobiol. 2022, 59, 3800–3828. [Google Scholar] [CrossRef]
- Ri, K.; Lee-Okada, H.; Yokomizo, T. Omega-6 highly unsaturated fatty acids in Leydig cells facilitate male sex hormone production. Commun. Biol. 2022, 5, 1001. [Google Scholar] [CrossRef]
- Castillo, A.F.; Orlando, U.D.; Maloberti, P.M.; Prada, J.G.; Dattilo, M.A.; Solano, A.R.; Bigi, M.M.; Ríos Medrano, M.A.; Torres, M.T.; Indo, S.; et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell. Mol. Life Sci. 2021, 78, 2893–2910. [Google Scholar] [CrossRef]
- Killion, E.A.; Reeves, A.R.; El Azzouny, M.A.; Yan, Q.; Surujon, D.; Griffin, J.D.; Bowman, T.A.; Wang, C.; Matthan, N.R.; Klett, E.L.; et al. A role for long-chain acyl-CoA synthetase-4 (ACSL4) in diet-induced phospholipid remodeling and obesity-associated adipocyte dysfunction. Mol. Metab. 2018, 9, 43–56. [Google Scholar] [CrossRef]
- Golej, D.L.; Askari, B.; Kramer, F.; Barnhart, S.; Vivekanandan-Giri, A.; Pennathur, S.; Bornfeldt, K.E. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E2 release from human arterial smooth muscle cells. J. Lipid Res. 2011, 52, 782–793. [Google Scholar] [CrossRef]
- Askari, B.; Kanter, J.E.; Sherrid, A.M.; Golej, D.L.; Bender, A.T.; Liu, J.; Hsueh, W.A.; Beavo, J.A.; Coleman, R.A.; Bornfeldt, K.E. Rosiglitazone Inhibits Acyl-CoA Synthetase Activity and Fatty Acid Partitioning to Diacylglycerol and Triacylglycerol via a Peroxisome Proliferator–Activated Receptor-γ–Independent Mechanism in Human Arterial Smooth Muscle Cells and Macrophages. Diabetes 2007, 56, 1143–1152. [Google Scholar] [CrossRef]
- Küch, E.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brügger, B.; Stremmel, W.; Ehehalt, R.; Poppelreuther, M.; Füllekrug, J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hao, X.; Han, L.; Yan, Z.; Shen, W.; Dong, D.; Hasbargen, K.; Bittner, S.; Cortez, Y.; Greenberg, A.S.; et al. Tissue-Specific Ablation of ACSL4 Results in Disturbed Steroidogenesis. Endocrinology 2019, 160, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Cornejo Maciel, F.; Maloberti, P.; Neuman, I.; Cano, F.; Castilla, R.; Castillo, F.; Paz, C.; Podestá, E.J. An arachidonic acid-preferring acyl-CoA synthetase is a hormone-dependent and obligatory protein in the signal transduction pathway of steroidogenic hormones. J. Mol. Endocrinol. 2005, 34, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Szczuko, M.; Kikut, J.; Komorniak, N.; Bilicki, J.; Celewicz, Z.; Zietek, M. The Role of Arachidonic and Linoleic Acid Derivatives in Pathological Pregnancies and the Human Reproduction Process. Int. J. Mol. Sci. 2020, 21, 9628. [Google Scholar] [CrossRef]
- Kuwata, H.; Hara, S. Inhibition of long-chain acyl-CoA synthetase 4 facilitates production of 5, 11-dihydroxyeicosatetraenoic acid via the cyclooxygenase-2 pathway. Biochem. Biophys. Res. Commun. 2015, 465, 528–533. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Qu, X.F.; Liang, T.Y.; Wu, D.G.; Lai, N.S.; Deng, R.M.; Ma, C.; Li, X.; Li, H.Y.; Liu, Y.Z.; Shen, H.T.; et al. Acyl-CoA synthetase long chain family member 4 plays detrimental role in early brain injury after subarachnoid hemorrhage in rats by inducing ferroptosis. CNS Neurosci. Ther. 2021, 27, 449–463. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Cho, Y.; Kang, M.; Sone, H.; Suzuki, T.; Abe, M.; Igarashi, M.; Tokunaga, T.; Ogawa, S.; Takei, Y.A.; Miyazawa, T.; et al. Abnormal Uterus with Polycysts, Accumulation of Uterine Prostaglandins, and Reduced Fertility in Mice Heterozygous for Acyl-CoA Synthetase 4 Deficiency. Biochem. Biophys. Res. Commun. 2001, 284, 993–997. [Google Scholar] [CrossRef]
- Chen, W.; Wang, C.; Hung, Y.; Weng, T.; Yen, M.; Lai, M. Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer. PLoS ONE 2016, 11, e155660. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Goel, H.L.; Mercurio, A.M. The α6β4 integrin promotes resistance to ferroptosis. J. Cell Biol. 2017, 216, 4287–4297. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Mun, S.; Yang, J.; Lee, J.; Seok, O.; Kim, E.; Kim, D.; An, S.Y.; Seo, D.; Suh, J.; et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat. Cell Biol. 2022, 24, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Orlando, U.; Maloberti, P.; Podestá, E.J.; Maciel, F.C. Tyrosine phosphatase SHP2 regulates the expression of acyl-CoA synthetase ACSL4. J. Lipid Res. 2011, 52, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hu, B.; Li, Z.; Du, T.; Shan, J.; Ye, Z.; Peng, X.; Li, X.; Huang, Y.; Zhu, X.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Rui, T.; Wang, H.; Li, Q.; Cheng, Y.; Gao, Y.; Fang, X.; Ma, X.; Chen, G.; Gao, C.; Gu, Z.; et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal Res. 2021, 70, e12704. [Google Scholar] [CrossRef]
- He, X.; Li, M.; Ye, Z.; You, X.; Wang, J.; Xiao, X.; Zhu, G.; Wei, J.; Zha, Y. Identification of Piperlongumine as Potent Inhibitor of Necroptosis. Drug Des. Dev. Ther. 2023, 17, 1387–1394. [Google Scholar] [CrossRef]
- Qiu, Y.; Shi, Y.; Zhu, N.; Zhang, S.; Zhang, C.; Gu, J.; He, P.; Dai, A.; Qin, L. A Lipid Perspective on Regulated Pyroptosis. Int. J. Biol. Sci. 2023, 19, 2333–2348. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kang, R.; Tang, D. Alkaliptosis: A new weapon for cancer therapy. Cancer Gene Ther. 2020, 27, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Song, Y.; Chen, H.; Li, Q.; Gao, Y.; Lu, G.; Luo, C. Ferroptosis Mediated by Lipid Reactive Oxygen Species: A Possible Causal Link of Neuroinflammation to Neurological Disorders. Oxidative Med. Cell. Longev. 2021, 2021, 5005136. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Yi-di, Z.; Ya-xuan, R.; Jun, Z.; Chu-zhao, L.; Rui-hua, D. The Role and Research Progress of ACSL4 Gene. China Cattle Sci. 2020, 46, 52–58. [Google Scholar] [CrossRef]
- Zhou, L.; Li, F.; Xu, H.; Luo, C.; Wu, H.; Zhu, M.; Lu, W.; Ji, X.; Zhou, Q.; Zhu, D. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat. Med. 2010, 16, 1439–1443. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Kuwata, H.; Yoshimura, M.; Sasaki, Y.; Yoda, E.; Nakatani, Y.; Kudo, I.; Hara, S. Role of long-chain acyl-coenzyme A synthetases in the regulation of arachidonic acid metabolism in interleukin 1beta-stimulated rat fibroblasts. Biochim. Biophys. Acta 2014, 1841, 44–53. [Google Scholar] [CrossRef]
- Cheng, J.; Fan, Y.Q.; Liu, B.H.; Zhou, H.; Wang, J.M.; Chen, Q.X. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol. Rep. 2019, 43, 147–158. [Google Scholar] [CrossRef]
- Tuo, Q.Z.; Liu, Y.; Xiang, Z.; Yan, H.F.; Zou, T.; Shu, Y.; Ding, X.L.; Zou, J.J.; Xu, S.; Tang, F.; et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal. Transduct. Target. Ther. 2022, 7, 59. [Google Scholar] [CrossRef]
- Jin, Z.; Gao, W.; Liao, S.; Yu, T.; Shi, Q.; Yu, S.; Cai, Y. Paeonol inhibits the progression of intracerebral haemorrhage by mediating the HOTAIR/UPF1/ACSL4 axis. ASN Neuro 2021, 13, 166567834. [Google Scholar] [CrossRef]
- Magtanong, L.; Mueller, G.D.; Williams, K.J.; Billmann, M.; Chan, K.; Armenta, D.A.; Pope, L.E.; Moffat, J.; Boone, C.; Myers, C.L.; et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem. Biol. 2022, 29, 1409–1418. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Shui, S.; Zhao, Z.; Wang, H.; Conrad, M.; Liu, G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 2021, 45, 102056. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Liu, X.; Wang, X.; Shi, X.; Ma, L.; Zhang, Y.; Zhou, T.; Zhao, C.; Zhang, X.; Fan, B.; et al. Edaravone Modulates Neuronal GPX4/ACSL4/5-LOX to Promote Recovery after Spinal Cord Injury. Front. Cell Dev. Biol. 2022, 10, 849854. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Meng, Z.; Yu, S.; Li, J.; Wang, Y.; Yang, W.; Wu, H. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem. Biol. Interact. 2022, 366, 110137. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, D.; Gao, C.; Teng, H.; Zhao, Y.; He, Z.; Chen, W.; Zong, Y.; Du, R. Seco-Lupane Triterpene Derivatives Induce Ferroptosis through GPX4/ACSL4 Axis and Target Cyclin D1 to Block the Cell Cycle. J. Med. Chem. 2022, 65, 10014–10044. [Google Scholar] [CrossRef]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 2018, 1–18. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Iaccarino, C.; Carretta, A.; Nicolosi, F.; Morselli, C. Epidemiology of severe traumatic brain injury. J. Neurosurg. Sci. 2018, 62, 535–541. [Google Scholar] [CrossRef]
- Majdan, M.; Plancikova, D.; Maas, A.; Polinder, S.; Feigin, V.; Theadom, A.; Rusnak, M.; Brazinova, A.; Haagsma, J. Years of life lost due to traumatic brain injury in Europe: A cross-sectional analysis of 16 countries. PLoS Med. 2017, 14, e1002331. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Yin, P.; Ning, P.; Wang, L.; Cheng, X.; Liu, Y.; Schwebel, D.C.; Liu, J.; Qi, J.; Hu, G.; et al. Trends in traumatic brain injury mortality in China, 2006–2013: A population-based longitudinal study. PLoS Med. 2017, 14, e1002332. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.; Guo, Z.; Guo, R.; Ye, R.; Zhu, W.; Yan, B. Ferroptosis and traumatic brain injury. Brain Res. Bull. 2021, 172, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.R.; Phan, J.H.; Alvarado-Velez, M.; Wang, M.D.; Bellamkonda, R.V.; Fernández, F.M.; LaPlaca, M.C. Discovery of Lipidome Alterations Following Traumatic Brain Injury via High-Resolution Metabolomics. J. Proteome Res. 2018, 17, 2131–2143. [Google Scholar] [CrossRef]
- Xiao, X.; Jiang, Y.; Liang, W.; Wang, Y.; Cao, S.; Yan, H.; Gao, L.; Zhang, L. miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol. Brain 2019, 12, 78. [Google Scholar] [CrossRef]
- Kenny, E.M.; Fidan, E.; Yang, Q.; Anthonymuthu, T.S.; New, L.A.; Meyer, E.A.; Wang, H.; Kochanek, P.M.; Dixon, C.E.; Kagan, V.E.; et al. Ferroptosis Contributes to Neuronal Death and Functional Outcome After Traumatic Brain Injury. Crit. Care Med. 2019, 47, 410–418. [Google Scholar] [CrossRef]
- Rui, T.; Li, Q.; Song, S.; Gao, Y.; Luo, C. Ferroptosis-relevant mechanisms and biomarkers for therapeutic interventions in traumatic brain injury. Histol. Histopathol. 2020, 35, 1105–1113. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Zheng, B.; Zhou, X.; Pang, L.; Che, Y.; Qi, X. Baicalin suppresses autophagy-dependent ferroptosis in early brain injury after subarachnoid hemorrhage. Bioengineered 2021, 12, 7794–7804. [Google Scholar] [CrossRef]
- Chen, S.; Hsu, C.; Huang, W.; Wang, J. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 2008, 155, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, B.; Cheng, Y.; Song, Y.; Li, Q.; Luo, C. Targeting Molecular Mediators of Ferroptosis and Oxidative Stress for Neurological Disorders. Oxidative Med. Cell. Longev. 2022, 2022, 3999083. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, L.; Geng, L.; He, J.; Chen, L.; Sun, Q.; Zhao, J.; Wang, X. Inhibition of Acyl-CoA Synthetase Long-Chain Family Member 4 Facilitates Neurological Recovery after Stroke by Regulation Ferroptosis. Front. Cell. Neurosci. 2021, 15, 632354. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Increased Lipid Peroxidation Precedes Amyloid Plaque Formation in an Animal Model of Alzheimer Amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, J.; Wu, Q.; Wang, S.; Yang, S.; Li, X.; Chen, N.; Li, L.; Zhang, L. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int. Immunopharmacol. 2021, 99, 108002. [Google Scholar] [CrossRef] [PubMed]
- Song, L.M.; Xiao, Z.X.; Zhang, N.; Yu, X.Q.; Cui, W.; Xie, J.X.; Xu, H.M. Apoferritin improves motor deficits in MPTP-treated mice by regulating brain iron metabolism and ferroptosis. IScience 2021, 24, 102431. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Y.; Zhang, B.; Han, G.; Yu, J.; Yu, Q.; Zhang, L. Ketone Body β-Hydroxybutyric Acid Ameliorates Dopaminergic Neuron Injury through Modulating Zinc Finger Protein 36/Acyl-CoA Synthetase Long-Chain Family Member Four Signaling Axis-Mediated Ferroptosis. Neuroscience 2023, 509, 157–172. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, X.; Xie, Y.; Zan, J.; Tan, W. Isosteviol Sodium Protects Against Permanent Cerebral Ischemia Injury in Mice via Inhibition of NF-κB–Mediated Inflammatory and Apoptotic Responses. J. Stroke Cerebrovasc. Dis. 2017, 26, 2603–2614. [Google Scholar] [CrossRef]
- Blakeley, J.O.; Llinas, R.H. Thrombolytic therapy for acute ischemic stroke. J. Neurol. Sci. 2007, 261, 55–62. [Google Scholar] [CrossRef]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Elsherbiny, N.M.; Haque, R.; Khan, M.B.; Ishrat, T.; Shah, Z.A.; Khan, M.M.; Ali, M.; Jamal, A.; Katare, D.P.; et al. Sesamin attenuates neurotoxicity in mouse model of ischemic brain stroke. Neurotoxicology 2014, 45, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hailin, Z.; Yong, C.; Yongfeng, B.A.; Chao, L.U.; Wei, H.U.; Renpeng, Z. Research progress on ferroptosis regulators and their role in stroke. Chin. J. Clin. Pharmacol. Ther. 2021, 26, 1320–1327. [Google Scholar] [CrossRef]
- Xichen, Y.; Simei, Z.; Yongdan, C.; Li, Z.; Chengcai, Z.; Pengyue, Z.; Yaju, J. Research Progress on the Mechanism of Ferroptosis in Ischemic Stroke. J. Yunnan Univ. Chin. Med. 2022, 45, 97–102. [Google Scholar] [CrossRef]
- Morotti, A.; Boulouis, G.; Dowlatshahi, D.; Li, Q.; Shamy, M.; Al-Shahi, S.R.; Rosand, J.; Cordonnier, C.; Goldstein, J.N.; Charidimou, A. Intracerebral haemorrhage expansion: Definitions, predictors, and prevention. Lancet Neurol. 2023, 22, 159–171. [Google Scholar] [CrossRef]
- You, M.; Long, C.; Wan, Y.; Guo, H.; Shen, J.; Li, M.; He, Q.; Hu, B. Neuron derived fractalkine promotes microglia to absorb hematoma via CD163/HO-1 after intracerebral hemorrhage. Cell. Mol. Life Sci. 2022, 79, 224. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Z.; Yu, F.; Liu, H.; Ma, C.; Xie, D.; Hu, X.; Leak, R.K.; Chou, S.; Stetler, R.A.; et al. IL-4/STAT6 signaling facilitates innate hematoma resolution and neurological recovery after hemorrhagic stroke in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32679–32690. [Google Scholar] [CrossRef]
- Xie, L.; Wang, Y.; Chen, Z. LncRNA Blnc1 mediates the permeability and inflammatory response of cerebral hemorrhage by regulating the PPAR-γ/SIRT6/FoxO3 pathway. Life Sci. 2021, 267, 118942. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D.; Liu, L. Paeonol: Pharmacological effects and mechanisms of action. Int. Immunopharmacol. 2019, 72, 413–421. [Google Scholar] [CrossRef]
- Liu, H.; Schwarting, J.; Terpolilli, N.A.; Nehrkorn, K.; Plesnila, N. Scavenging Free Iron Reduces Arteriolar Microvasospasms after Experimental Subarachnoid Hemorrhage. Stroke 2021, 52, 4033–4042. [Google Scholar] [CrossRef]
- Kuang, H.; Wang, T.; Liu, L.; Tang, C.; Li, T.; Liu, M.; Wang, T.; Zhong, W.; Wang, Y. Treatment of early brain injury after subarachnoid hemorrhage in the rat model by inhibiting p53-induced ferroptosis. Neurosci. Lett. 2021, 762, 136134. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Zhao, X.; Shen, J.; Chen, S.; Huang, H.; Zhou, X.; Han, Y.; Zhou, L.; Lu, X.; Wu, Q. Activation of SIRT1 Alleviates Ferroptosis in the Early Brain Injury after Subarachnoid Hemorrhage. Oxidative Med. Cell. Longev. 2022, 2022, 9069825. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, H.; Hu, Y.; Zhou, C.; Wu, J.; Wu, Y.; Wang, H.; Lenahan, C.; Huang, L.; Nie, S.; et al. Puerarin Attenuates Oxidative Stress and Ferroptosis via AMPK/PGC1α/Nrf2 Pathway after Subarachnoid Hemorrhage in Rats. Antioxidants 2022, 11, 1259. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; Maaser-Hecker, A.; Tanzi, R.E. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023, 15, 6. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics—1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathobiological Subtypes of Alzheimer Disease. Dement. Geriatr. Cogn. Disord. 2021, 49, 321–333. [Google Scholar] [CrossRef]
- Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, A.M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer’s Dement. 2017, 13, 1–7. [Google Scholar] [CrossRef]
- von Bartheld, C.S. Myths and truths about the cellular composition of the human brain: A review of influential concepts. J. Chem. Neuroanat. 2018, 93, 2–15. [Google Scholar] [CrossRef]
- Pratico, D.; Sung, S. Lipid peroxidation and oxidative imbalance: Early functional events in Alzheimer’s disease. J. Alzheimers Dis. 2004, 6, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, Y.; Gong, Y.; Jin, W.; Topchiy, E.; Turdi, S.; Gao, Y.; Culver, B.; Wang, S.; Ge, W.; et al. Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer’s disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol. Sin. 2022, 43, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Liang, Z.; Yongqun, L.; Qinchuan, X.; Shitong, Z.; Xiaoli, L.I. Ferroptosis regulatory signaling pathway and its research progress in re-lated diseases. Chin. J. Clin. Pharmacol. Ther. 2022, 27, 227–234. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, B.; Zhu, J.; Yu, J.; Hui, J.; Xia, S.; Ji, J. Emerging Mechanisms and Targeted Therapy of Ferroptosis in Neurological Diseases and Neuro-oncology. Int. J. Biol. Sci. 2022, 18, 4260–4274. [Google Scholar] [CrossRef]
- Tang, F.; Zhou, L.; Li, P.; Jiao, L.; Chen, K.; Guo, Y.; Ding, X.; He, S.; Dong, B.; Xu, R.; et al. Inhibition of ACSL4 Alleviates Parkinsonism Phenotypes by Reduction of Lipid Reactive Oxygen Species. Neurotherapeutics 2023. [Google Scholar] [CrossRef]
- Jia, R.; Liu, Y.; Shuai, K.; Zhou, C.; Chen, L.; Zhu, L.; Wu, X. The Relationship between Iron and LRRK2 in a 6-OHDA-Induced Parkinson’s Disease Model. Int. J. Mol. Sci. 2023, 24, 3709. [Google Scholar] [CrossRef]
- Lee, H.; Choi, C.; Lee, S. Membrane-bound α-Synuclein Has a High Aggregation Propensity and the Ability to Seed the Aggregation of the Cytosolic Form*. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, C.; Xu, H.H.; Liu, Y.; Zhou, F. Binding of α-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem. 2010, 104, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, D.J. Potential for Therapeutic-Loaded Exosomes to Ameliorate the Pathogenic Effects of α-Synuclein in Parkinson’s Disease. Biomedicines 2023, 11, 1187. [Google Scholar] [CrossRef] [PubMed]
- Koeglsperger, T.; Rumpf, S.L.; Schliesser, P.; Struebing, F.L.; Brendel, M.; Levin, J.; Trenkwalder, C.; Hoglinger, G.U.; Herms, J. Neuropathology of incidental Lewy body & prodromal Parkinson’s disease. Mol. Neurodegener. 2023, 18, 32. [Google Scholar] [CrossRef]
- Fecchio, C.; Palazzi, L.; de Laureto, P.P. α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules 2018, 23, 1531. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Ma, J.J.; Li, X.H.; Fan, Y.Y.; Yang, D.W.; Gu, Q.; Li, D.S.; Chen, S.Y.; Wu, S.P.; Zheng, J.H.; Yang, H.Q.; et al. miR-494-3p Promotes Erastin-Induced Ferroptosis by Targeting REST to Activate the Interplay between SP1 and ACSL4 in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2022, 2022, 7671324. [Google Scholar] [CrossRef]
- Kuruppu, A.I.; Turyanska, L.; Bradshaw, T.D.; Manickam, S.; Galhena, B.P.; Paranagama, P.; De Silva, R. Apoferritin and Dps as drug delivery vehicles: Some selected examples in oncology. Biochim. Biophys. Acta-Gen. Subj. 2022, 1866, 130067. [Google Scholar] [CrossRef]
- Anjum, A.; Yazid, M.D.I.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef]
- Cozzolino, M.; Ferri, A.; Teresa Carrì, M. Amyotrophic Lateral Sclerosis: From Current Developments in the Laboratory to Clinical Implications. Antioxid. Redox Signal. 2008, 10, 405–444. [Google Scholar] [CrossRef]
- Matsuzono, K.; Suzuki, M.; Miura, K.; Ozawa, T.; Mashiko, T.; Koide, R.; Tanaka, R.; Fujimoto, S. Higher incidence of cervical spinal cord compression in amyotrophic lateral sclerosis: A single-institute cohort study. Neurol. Sci. 2022, 43, 1079–1086. [Google Scholar] [CrossRef]
- Sykova, E.; Cizkova, D.; Kubinova, S. Mesenchymal Stem Cells in Treatment of Spinal Cord Injury and Amyotrophic Lateral Sclerosis. Front. Cell Dev. Biol. 2021, 9, 695900. [Google Scholar] [CrossRef]
- Spasić, S.; Nikolić-Kokić, A.; Miletić, S.; Oreščanin-Dušić, Z.; Spasić, M.B.; Blagojević, D.; Stević, Z. Edaravone May Prevent Ferroptosis in ALS. Curr. Drug Targets 2020, 21, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, C.; Zhao, C.; Hao, J.; Zhang, Y.; Fan, B.; Li, B.; Duan, H.; Liu, C.; Kong, X.; et al. Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res. 2019, 1706, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef]
- Cha, S.J.; Kim, K. Effects of the Edaravone, a Drug Approved for the Treatment of Amyotrophic Lateral Sclerosis, on Mitochondrial Function and Neuroprotection. Antioxidants 2022, 11, 195. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, R.; Lv, M.; Xu, X.; Liu, W.; Li, X.; Gao, Y.; Zhao, Z.; Zhang, Z.; Li, Y.; et al. ACSL4 promotes microglia-mediated neuroinflammation by regulating lipid metabolism and VGLL4 expression. Brain Behav. Immun. 2023, 109, 331–343. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Doshi, A.; Chataway, J. Multiple sclerosis, a treatable disease. Clin. Med. 2016, 16, s53–s59. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R. What Is Multiple Sclerosis? JAMA 2022, 328, 2078. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Luoqian, J.; Yang, W.; Ding, X.; Tuo, Q.Z.; Xiang, Z.; Zheng, Z.; Guo, Y.J.; Li, L.; Guan, P.; Ayton, S.; et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell. Mol. Immunol. 2022, 19, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef]
- Yue, T.; Guo-dong, L.; Yue-Ming, J. Studies on fatty acid activation mediated by ACSL4 and tumor. J. Toxicol. 2018, 32, 160–164. [Google Scholar]
- Ye, X.; Zhang, Y.; Wang, X.; Li, Y.; Gao, Y. Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in gastric cancer. IUBMB Life 2016, 68, 320–327. [Google Scholar] [CrossRef]
- Jiang, X.; Guo, S.; Zhang, Y.; Zhao, Y.; Li, X.; Jia, Y.; Xu, Y.; Ma, B. LncRNA NEAT1 promotes docetaxel resistance in prostate cancer by regulating ACSL4 via sponging miR-34a-5p and miR-204-5p. Cell. Signal. 2020, 65, 109422. [Google Scholar] [CrossRef]
- Wu, X.; Zhi, F.; Lun, W.; Deng, Q.; Zhang, W. Baicalin inhibits PDGF-BB-induced hepatic stellate cell proliferation, apoptosis, invasion, migration and activation via the miR-3595/ACSL4 axis. Int. J. Mol. Med. 2018, 41, 1992–2002. [Google Scholar] [CrossRef]
- Park, S.; Oh, J.; Kim, Y.; Choe, S.; Chun, C.; Jin, E. Suppression of ABCD2 dysregulates lipid metabolism via dysregulation of miR-141:ACSL4 in human osteoarthritis. Cell Biochem. Funct. 2018, 36, 366–376. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Diseases | Biological Model | Intervention Measure | Consequence | Reference |
|---|---|---|---|---|
| Traumatic Brain Injury | Controlled cortical impact (CCI) | / | ACSL4 expression level increased | [66] |
| CCI | Baicalein | Decreased ferroptotic PE oxidation | [67] | |
| Ischemic stroke | Middle cerebral artery occlusion (MCAO) | / | ACSL4 increased after decreasing 1–3 h of ischemia | [46] |
| MCAO | liproxstatin-1/Rosiglitazo-ne/ | Lipid peroxidation index was significantly inhibited in comparison with untreated group | [73] | |
| Hemorrhagic stroke | Oxygen and glucose deprivation (OGD) | / | ACSL4 mRNA expression was significantly increased | [46] |
| OGD | Paeonol | Paeonol inhibited the expression of ACSL4 | [50] | |
| Alzheimer’s disease | APPswe transgenic mice | / | Aβ accumulates in brain tissue due to lipid peroxidation | [74] |
| APPswe/PSEN1dE9 (APP/PS1) double transgene mice | tetrahydroxy stilbene glycoside (TSG) | TSG inhibited the expression of ACSL4 | [75] | |
| Parkinson’s disease | PD mice model | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) | The expression of ACSL4 significantly increased | [76] |
| PD mice model | β-hydroxybutyrate (BHB) | BHB inhibits ferroptosis in PD model | [77] | |
| Spinal cord injury | Spinal cord | Edaravone | Reduces ACSL4 levels | [54] |
| contusion injury model | ||||
| Multiple sclerosis | Experimental autoimmune encephalitis (EAE) model | ACSL4-KO | Knocking down the ACSL4 gene considerably reduced the severity of EAE and the clinical score of EAE mice | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, B.; Li, J.; Song, Y.; Luo, C. ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. Int. J. Mol. Sci. 2023, 24, 10021. https://doi.org/10.3390/ijms241210021
Jia B, Li J, Song Y, Luo C. ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. International Journal of Molecular Sciences. 2023; 24(12):10021. https://doi.org/10.3390/ijms241210021
Chicago/Turabian StyleJia, Bowen, Jing Li, Yiting Song, and Chengliang Luo. 2023. "ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries" International Journal of Molecular Sciences 24, no. 12: 10021. https://doi.org/10.3390/ijms241210021
APA StyleJia, B., Li, J., Song, Y., & Luo, C. (2023). ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. International Journal of Molecular Sciences, 24(12), 10021. https://doi.org/10.3390/ijms241210021