
Dietary (Poly)phenols in Traumatic Brain Injury

 ,
,  ,
,  ,
,  , and
, and 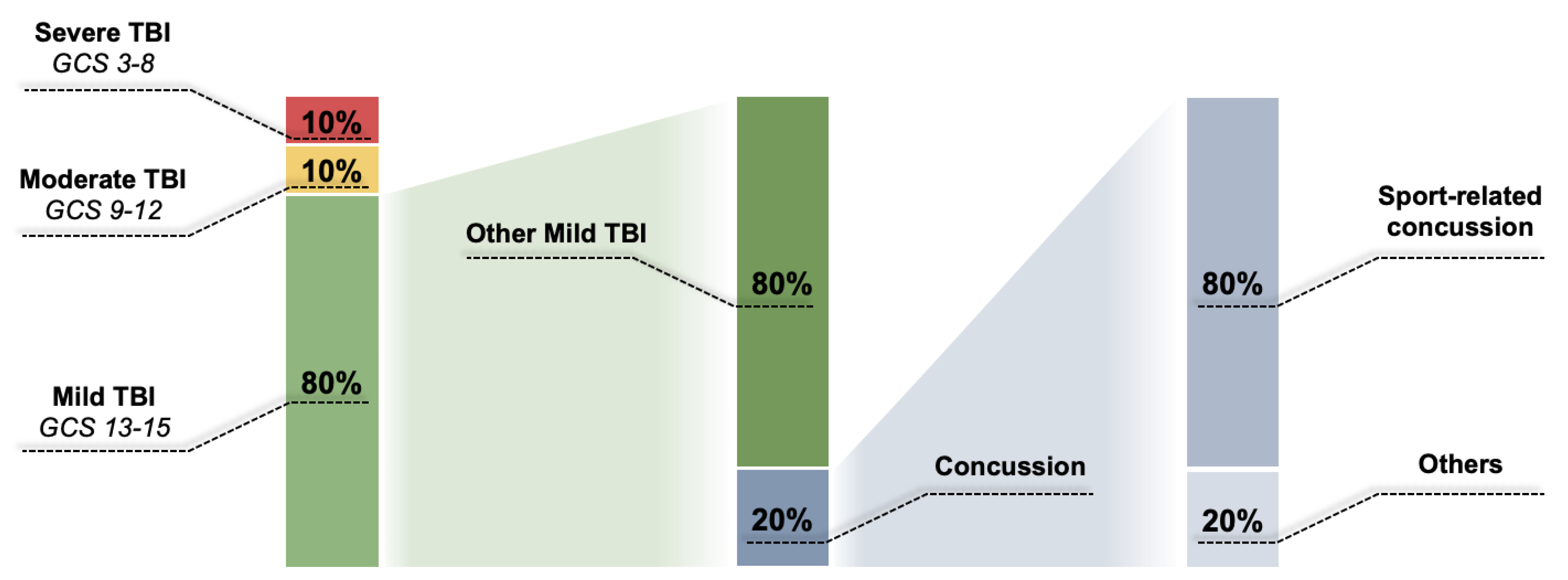{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Different Types of TBI
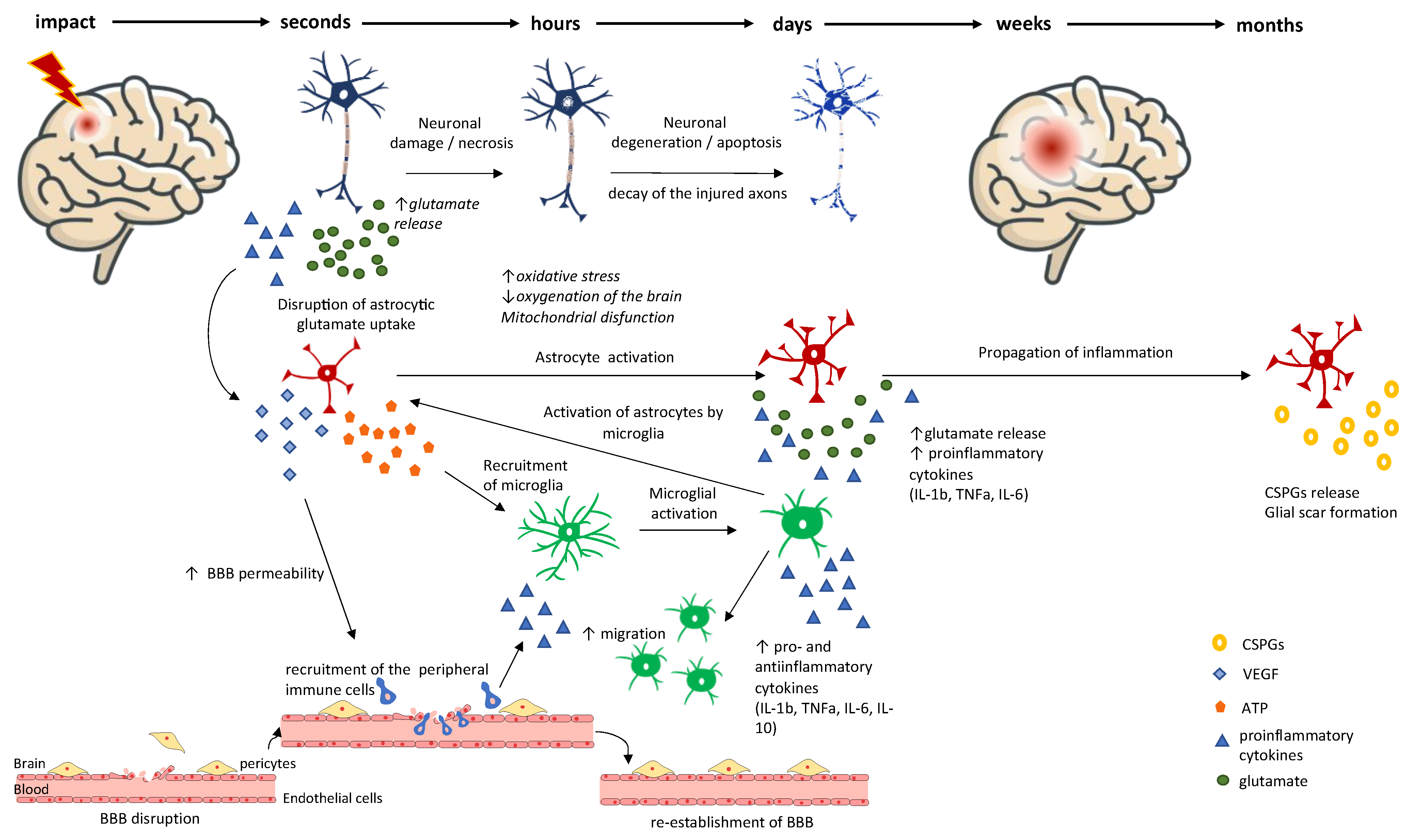
2. Primary vs. Secondary Brain Injury
Current Pharmacological Approaches for Treating TBI Patients
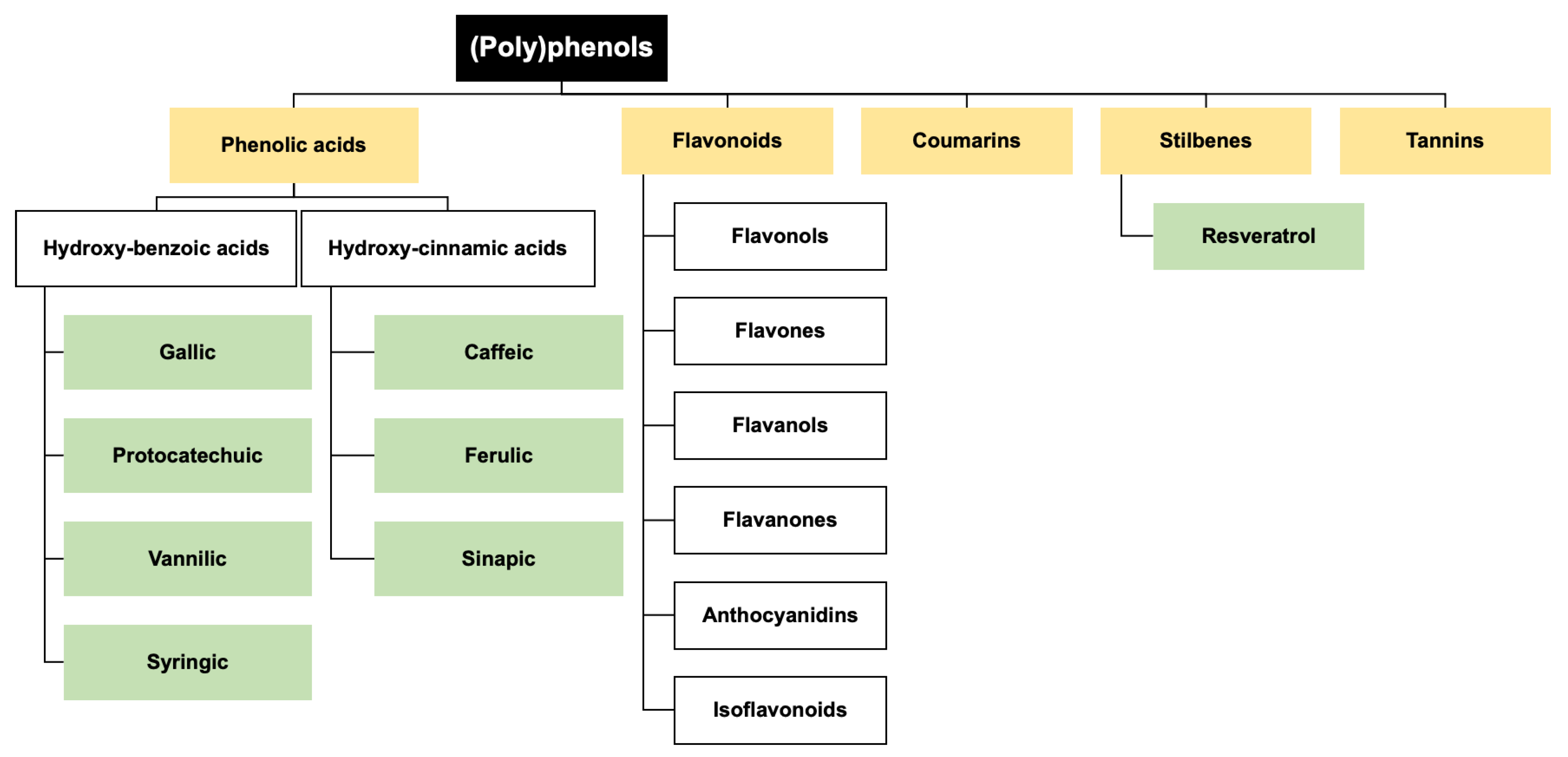
3. Dietary (Poly)phenols in TBI Prevention and Management
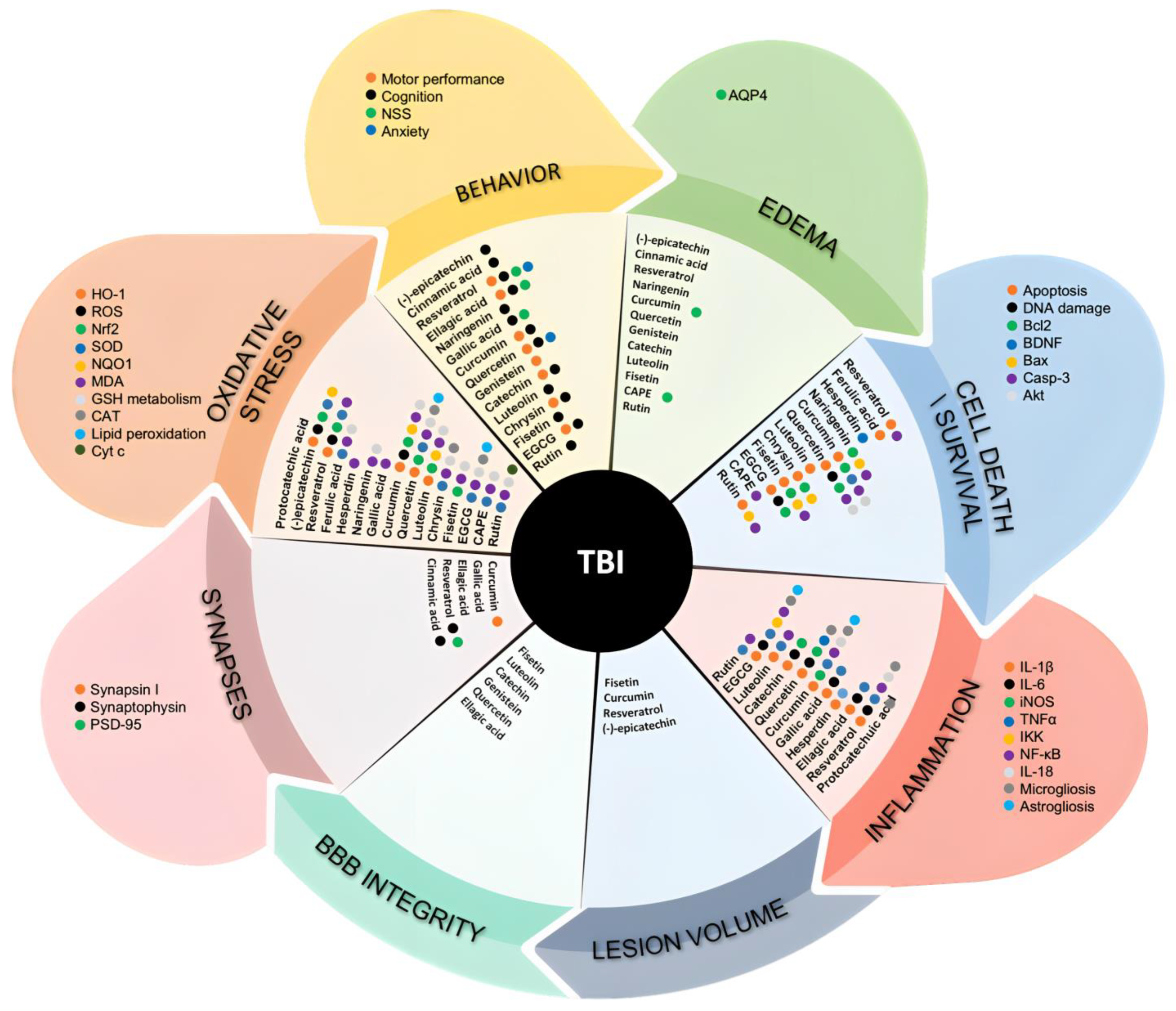
3.1. (Poly)phenols in TBI State-of-the-Art from the In Vivo Models
3.1.1. Flavanols
3.1.2. Flavanones
3.1.3. Flavonols
3.1.4. Flavones
3.1.5. Isoflavones
3.1.6. Curcuminoids
3.1.7. Stilbenes
3.1.8. Phenolic Acids
3.1.9. Ellagitannins
3.2. (Poly)phenols Potential against TBI in Humans
4. Gaps and Further Research Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Williamson, C.; Rajajee, V. Traumatic Brain Injury: Epidemiology, Classification, and Pathophysiology. Available online: www.uptodate.com/contents/traumatic-brain-injury-epidemiology-classification-and-pathophysiology/print (accessed on 22 March 2023).
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the Global Incidence of Traumatic Brain Injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position Statement: Definition of Traumatic Brain Injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Lončarević-Vasiljković, N.; Pešić, V.; Tanić, N.; Milanović, D.; Popić, J.; Kanazir, S.; Ruždijić, S. Changes in Markers of Neuronal and Glial Plasticity after Cortical Injury Induced by Food Restriction. Exp. Neurol. 2009, 220, 198–206. [Google Scholar] [CrossRef] [PubMed]
- di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant Therapies in Traumatic Brain Injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef]
- Menon, D.K.; Maas, A.I.R. Progress, Failures and New Approaches for TBI Research. Nat. Rev. Neurol. 2015, 11, 71–72. [Google Scholar] [CrossRef]
- Teasdale, G.; Maas, A.; Lecky, F.; Manley, G.; Stocchetti, N.; Murray, G. The Glasgow Coma Scale at 40 Years: Standing the Test of Time. Lancet Neurol. 2014, 13, 844–854. [Google Scholar] [CrossRef]
- Teasdale, G.; Jennett, B. Assessment of Coma and Impaired Consciousness. Lancet 1974, 304, 81–84. [Google Scholar] [CrossRef]
- Bodanapally, U.K.; Sours, C.; Zhuo, J.; Shanmuganathan, K. Imaging of Traumatic Brain Injury. Radiol. Clin. N. Am. 2015, 53, 695–715. [Google Scholar] [CrossRef]
- Young, L.; Rule, G.T.; Bocchieri, R.T.; Walilko, T.J.; Burns, J.M.; Ling, G. When Physics Meets Biology: Low and High-Velocity Penetration, Blunt Impact, and Blast Injuries to the Brain. Front. Neurol. 2015, 6, 89. [Google Scholar] [CrossRef]
- Risdall, J.E.; Menon, D.K. Traumatic Brain Injury. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 241–250. [Google Scholar] [CrossRef]
- Najem, D.; Rennie, K.; Ribecco-Lutkiewicz, M.; Ly, D.; Haukenfrers, J.; Liu, Q.; Nzau, M.; Fraser, D.D.; Bani-Yaghoub, M. Traumatic Brain Injury: Classification, Models, and Markers. Biochem. Cell Biol. 2018, 96, 391–406. [Google Scholar] [CrossRef]
- Gardner, A.J.; Zafonte, R. Neuroepidemiology of Traumatic Brain Injury. Handb. Clin. Neurol. 2016, 138, 207–223. [Google Scholar]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early Management of Severe Traumatic Brain Injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic Brain Injury: Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Web-Based Injury Statistics Query and Reporting System (WISQARS) (2003). National Center for Injury Prevention and Control, Centers for Disease Control and Prevention. Available online: www.cdc.gov/injury/wisqars (accessed on 28 February 2023).
- Mustafa, A.G.; Alshboul, O.A. Pathophysiology of Traumatic Brain Injury. Neurosciences 2013, 18, 222–234. [Google Scholar]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Martinez, B.I.; Stabenfeldt, S.E. Current Trends in Biomarker Discovery and Analysis Tools for Traumatic Brain Injury. J. Biol. Eng. 2019, 13, 16. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Frati, A.; Cerretani, D.; Fiaschi, A.; Frati, P.; Gatto, V.; La Russa, R.; Pesce, A.; Pinchi, E.; Santurro, A.; Fraschetti, F.; et al. Diffuse Axonal Injury and Oxidative Stress: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2600. [Google Scholar] [CrossRef]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. [Google Scholar] [CrossRef]
- Kermer, P.; Klöcker, N.; Labes, M.; Thomsen, S.; Srinivasan, A.; Bähr, M. Activation of Caspase-3 in Axotomized Rat Retinal Ganglion Cells in Vivo. FEBS Lett. 1999, 453, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Thompson, B.; Gao, X.; Hall, E. Temporal Relationship of Peroxynitrite-Induced Oxidative Damage, Calpain-Mediated Cytoskeletal Degradation and Neurodegeneration after Traumatic Brain Injury. Exp. Neurol. 2007, 205, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Deng-Bryant, Y.; Singh, I.N.; Carrico, K.M.; Hall, E.D. Neuroprotective Effects of Tempol, a Catalytic Scavenger of Peroxynitrite-Derived Free Radicals, in a Mouse Traumatic Brain Injury Model. J. Cereb. Blood Flow Metab. 2008, 28, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Oda, Y.; Wei, E.P.; Povlishock, J.T. The Combination of Either Tempol or FK506 with Delayed Hypothermia: Implications for Traumatically Induced Microvascular and Axonal Protection. J. Neurotrauma 2011, 28, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.G.; Singh, I.N.; Wang, J.; Carrico, K.M.; Hall, E.D. Mitochondrial Protection after Traumatic Brain Injury by Scavenging Lipid Peroxyl Radicals. J. Neurochem. 2010, 114, 271–280. [Google Scholar] [CrossRef]
- Mustafa, A.G.; Wang, J.A.; Carrico, K.M.; Hall, E.D. Pharmacological Inhibition of Lipid Peroxidation Attenuates Calpain-Mediated Cytoskeletal Degradation after Traumatic Brain Injury. J. Neurochem. 2011, 117, 579–588. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal Pathology in Traumatic Brain Injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef]
- Loane, D.J.; Byrnes, K.R. Role of Microglia in Neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef]
- Chen, X.-H.; Johnson, V.E.; Uryu, K.; Trojanowski, J.Q.; Smith, D.H. A Lack of Amyloid Beta Plaques despite Persistent Accumulation of Amyloid Beta in Axons of Long-Term Survivors of Traumatic Brain Injury. Brain Pathol. 2009, 19, 214–223. [Google Scholar] [CrossRef]
- Gentleman, S.M.; Leclercq, P.D.; Moyes, L.; Graham, D.I.; Smith, C.; Griffin, W.S.T.; Nicoll, J.A.R. Long-Term Intracerebral Inflammatory Response after Traumatic Brain Injury. Forensic Sci. Int. 2004, 146, 97–104. [Google Scholar] [CrossRef]
- Stoica, B.A.; Faden, A.I. Cell Death Mechanisms and Modulation in Traumatic Brain Injury. Neurotherapeutics 2010, 7, 3–12. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Hanafy, K.A. Cell Death and Recovery in Traumatic Brain Injury. Neurotherapeutics 2020, 17, 446–456. [Google Scholar] [CrossRef]
- Vella, M.A.; Crandall, M.L.; Patel, M.B. Acute Management of Traumatic Brain Injury. Surg. Clin. N. Am. 2017, 97, 1015–1030. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.-R. Traumatic Brain Injury. Cell Transplant 2017, 26, 1118–1130. [Google Scholar] [CrossRef]
- Czekajlo, M.S.; Milbrandt, E.B. Corticosteroids Increased Short and Long-Term Mortality in Adults with Traumatic Head Injury. Crit. Care 2005, 9, E21. [Google Scholar] [CrossRef]
- CRASH Trial Collaborators. Effect of Intravenous Corticosteroids on Death within 14 Days in 10,008 Adults with Clinically Significant Head Injury (MRC CRASH Trial): Randomised Placebo-Controlled Trial. Lancet 2004, 364, 1321–1328. [Google Scholar] [CrossRef]
- Alderson, P.; Roberts, I. Corticosteroids for Acute Traumatic Brain Injury. Cochrane Database Syst. Rev. 2005, 200, A40–A41. [Google Scholar] [CrossRef]
- Khellaf, A.; Khan, D.Z.; Helmy, A. Recent Advances in Traumatic Brain Injury. J. Neurol. 2019, 266, 2878–2889. [Google Scholar] [CrossRef]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and Severe Traumatic Brain Injury in Adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Nwafor, D.; Goeckeritz, J.; Hasanpour, Z.; Davidson, C.; Lucke-Wold, B. Nutritional Support Following Traumatic Brain Injury: A Comprehensive Review. Explor. Res. Hypothesis Med. 2022. [Google Scholar] [CrossRef]
- Wang, X.; Dong, Y.; Han, X.; Qi, X.-Q.; Huang, C.-G.; Hou, L.-J. Nutritional Support for Patients Sustaining Traumatic Brain Injury: A Systematic Review and Meta-Analysis of Prospective Studies. PLoS ONE 2013, 8, e58838. [Google Scholar] [CrossRef] [PubMed]
- Bistrian, B.R.; Askew, W.; Erdman, J.W.; Oria, M.P. Nutrition and Traumatic Brain Injury. J. Parenter. Enter. Nutr. 2011, 35, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Perel, P.; Yanagawa, T.; Bunn, F.; Roberts, I.G.; Wentz, R. Nutritional Support for Head-Injured Patients. Cochrane Database Syst. Rev. 2006, 4. [Google Scholar] [CrossRef] [PubMed]
- Loncarevic-Vasiljkovic, N.; Pesic, V.; Todorovic, S.; Popic, J.; Smiljanic, K.; Milanovic, D.; Ruzdijic, S.; Kanazir, S. Caloric Restriction Suppresses Microglial Activation and Prevents Neuroapoptosis Following Cortical Injury in Rats. PLoS ONE 2012, 7, e37215. [Google Scholar] [CrossRef]
- Lončarević-Vasiljković, N.; Milanović, D.; Pešić, V.; Tešić, V.; Brkić, M.; Lazić, D.; Avramović, V.; Kanazir, S. Dietary Restriction Suppresses Apoptotic Cell Death, Promotes Bcl-2 and Bcl-Xl MRNA Expression and Increases the Bcl-2/Bax Protein Ratio in the Rat Cortex after Cortical Injury. Neurochem. Int. 2016, 96, 69–76. [Google Scholar] [CrossRef]
- Sharma, S.; Kaur, G. Dietary Restriction Enhances Kainate-Induced Increase in NCAM While Blocking the Glial Activation in Adult Rat Brain. Neurochem. Res. 2008, 33, 1178–1188. [Google Scholar] [CrossRef]
- Rubovitch, V.; Pharayra, A.; Har-Even, M.; Dvir, O.; Mattson, M.P.; Pick, C.G. Dietary Energy Restriction Ameliorates Cognitive Impairment in a Mouse Model of Traumatic Brain Injury. J. Mol. Neurosci. 2019, 67, 613–621. [Google Scholar] [CrossRef]
- Taguchi, C.; Kishimoto, Y.; Fukushima, Y.; Kondo, K.; Yamakawa, M.; Wada, K.; Nagata, C. Dietary Intake of Total Polyphenols and the Risk of All-Cause and Specific-Cause Mortality in Japanese Adults: The Takayama Study. Eur. J. Nutr. 2020, 59, 1263–1271. [Google Scholar] [CrossRef]
- Godos, J.; Rapisarda, G.; Marventano, S.; Galvano, F.; Mistretta, A.; Grosso, G. Association between Polyphenol Intake and Adherence to the Mediterranean Diet in Sicily, Southern Italy. NFS J. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (Poly)Phenolics in Human Health: Structures, Bioavailability, and Evidence of Protective Effects Against Chronic Diseases. Antioxid. Redox Signal 2013, 18, 1818–1892. [Google Scholar] [CrossRef]
- Carregosa, D.; Carecho, R.; Figueira, I.; Santos, N.C. Low-Molecular Weight Metabolites from Polyphenols as Effectors for Attenuating Neuroinflammation. J. Agric. Food Chem. 2020, 68, 1790–1807. [Google Scholar] [CrossRef]
- Carregosa, D.; Pinto, C.; Ávila-Gálvez, M.Á.; Bastos, P.; Berry, D.; Santos, C.N. A Look beyond Dietary (Poly)Phenols: The Low Molecular Weight Phenolic Metabolites and Their Concentrations in Human Circulation. Compr. Rev. Food Sci. Food Saf. 2022, 21, 3931–3962. [Google Scholar] [CrossRef]
- Carecho, R.; Carregosa, D.; dos Santos, C.N. Low Molecular Weight (Poly)Phenol Metabolites Across the Blood-Brain Barrier: The Underexplored Journey. Brain Plast. 2021, 6, 193–214. [Google Scholar] [CrossRef]
- Figueira, I.; Garcia, G.; Pimpão, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols Journey through Blood-Brain Barrier towards Neuronal Protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef]
- Pimpão, R.C.; Ventura, M.R.; Ferreira, R.B.; Williamson, G.; Santos, C.N. Phenolic Sulfates as New and Highly Abundant Metabolites in Human Plasma after Ingestion of a Mixed Berry Fruit Purée. Br. J. Nutr. 2015, 113, 454–463. [Google Scholar] [CrossRef]
- Scalbert, A.; Morand, C.; Manach, C.; Rémésy, C. Absorption and Metabolism of Polyphenols in the Gut and Impact on Health. Biomed. Pharmacother. 2002, 56, 276–282. [Google Scholar] [CrossRef]
- Angelino, D.; Carregosa, D.; Domenech-Coca, C.; Savi, M.; Figueira, I.; Brindani, N.; Jang, S.; Lakshman, S.; Molokin, A.; Urban, J.F.; et al. 5-(Hydroxyphenyl)-γ-Valerolactone-Sulfate, a Key Microbial Metabolite of Flavan-3-Ols, Is Able to Reach the Brain: Evidence from Different in Silico, In Vitro and In Vivo Experimental Models. Nutrients 2019, 11, 2678. [Google Scholar] [CrossRef]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of Microbial Metabolites of Dietary Polyphenols in Rats: Is the Brain Their Target Destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef]
- Shimazu, R.; Anada, M.; Miyaguchi, A.; Nomi, Y.; Matsumoto, H. Evaluation of Blood–Brain Barrier Permeability of Polyphenols, Anthocyanins, and Their Metabolites. J. Agric. Food Chem. 2021, 69, 11676–11686. [Google Scholar] [CrossRef]
- Velásquez-Jiménez, D.; Corella-Salazar, D.A.; Zuñiga-Martínez, B.S.; Domínguez-Avila, J.A.; Montiel-Herrera, M.; Salazar-López, N.J.; Rodrigo-Garcia, J.; Villegas-Ochoa, M.A.; González-Aguilar, G.A. Phenolic Compounds That Cross the Blood–Brain Barrier Exert Positive Health Effects as Central Nervous System Antioxidants. Food Funct. 2021, 12, 10356–10369. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Wang, J.; Wang, Q.; Ma, Q.; Chen, Y. Protocatechuic Acid Inhibits Inflammatory Responses in LPS-Stimulated BV2 Microglia via NF-ΚB and MAPKs Signaling Pathways. Neurochem. Res. 2015, 40, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Seong, A.-R.; Yoo, J.-Y.; Jin, C.-H.; Lee, Y.-H.; Kim, Y.J.; Lee, J.; Jun, W.J.; Yoon, H.-G. Gallic Acid, a Histone Acetyltransferase Inhibitor, Suppresses β-Amyloid Neurotoxicity by Inhibiting Microglial-Mediated Neuroinflammation. Mol. Nutr. Food Res. 2011, 55, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Nanni, S.; Figueira, I.; Ivanov, I.; McDougall, G.J.; Stewart, D.; Ferreira, R.B.; Pinto, P.; Silva, R.F.M.; Brites, D.; et al. Bioaccessible (Poly)Phenol Metabolites from Raspberry Protect Neural Cells from Oxidative Stress and Attenuate Microglia Activation. Food Chem. 2017, 215, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Javed, H.; Azimullah, S.; Abul Khair, S.B.; Haque, E. Neuroprotective Potential of Ferulic Acid in the Rotenone Model of Parkinson’s Disease. Drug Des. Devel. Ther. 2015, 9, 5499–5510. [Google Scholar] [CrossRef]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic Acid Attenuates Aβ1-42-Induced Oxidative Stress and Cognitive Impairment in Mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef]
- Carecho, R.; Figueira, I.; Terrasso, A.P.; Godinho-Pereira, J.; de Oliveira Sequeira, C.; Pereira, S.A.; Milenkovic, D.; Leist, M.; Brito, C.; Nunes dos Santos, C. Circulating (Poly)Phenol Metabolites: Neuroprotection in a 3D Cell Model of Parkinson’s Disease. Mol. Nutr. Food Res. 2022, 66, 2100959. [Google Scholar] [CrossRef]
- Kho, A.; Choi, B.; Lee, S.; Hong, D.; Lee, S.; Jeong, J.; Park, K.-H.; Song, H.; Choi, H.; Suh, S. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef]
- Wang, J.; Guo, Y.; Yue Zhang, S. Vanillic Acid Improve Neural Function after Focal Cerebral Ischemia-Reperfusion Rats. Int. J. Pharmacol. 2018, 14, 488–494. [Google Scholar] [CrossRef]
- Singh, J.C.H.; Kakalij, R.M.; Kshirsagar, R.P.; Kumar, B.H.; Komakula, S.S.B.; Diwan, P.V. Cognitive Effects of Vanillic Acid against Streptozotocin-Induced Neurodegeneration in Mice. Pharm. Biol. 2015, 53, 630–636. [Google Scholar] [CrossRef]
- Shi, G.-F.; An, L.-J.; Jiang, B.; Guan, S.; Bao, Y.-M. Alpinia Protocatechuic Acid Protects against Oxidative Damage in Vitro and Reduces Oxidative Stress in Vivo. Neurosci. Lett. 2006, 403, 206–210. [Google Scholar] [CrossRef]
- Rekha, K.R.; Selvakumar, G.P.; Sivakamasundari, R.I. Effects of Syringic Acid on Chronic MPTP/Probenecid Induced Motor Dysfunction, Dopaminergic Markers Expression and Neuroinflammation in C57BL/6 Mice. Biomed. Aging Pathol. 2014, 4, 95–104. [Google Scholar] [CrossRef]
- Gennarelli, T.A. Animate Models of Human Head Injury. J. Neurotrauma 1994, 11, 357–368. [Google Scholar] [CrossRef]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef]
- Nair, A.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose Translation from Animal to Human Studies Revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Itoh, T.; Imano, M.; Nishida, S.; Tsubaki, M.; Hashimoto, S.; Ito, A.; Satou, T. (−)-Epigallocatechin-3-Gallate Protects Against Neuronal Cell Death and Improves Cerebral Function After Traumatic Brain Injury in Rats. Neuromol. Med. 2011, 13, 300–309. [Google Scholar] [CrossRef]
- Itoh, T.; Imano, M.; Nishida, S.; Tsubaki, M.; Mizuguchi, N.; Hashimoto, S.; Ito, A.; Satou, T. (−)-Epigallocatechin-3-Gallate Increases the Number of Neural Stem Cells around the Damaged Area after Rat Traumatic Brain Injury. J. Neural. Transm. 2012, 119, 877–890. [Google Scholar] [CrossRef]
- Itoh, T.; Tabuchi, M.; Mizuguchi, N.; Imano, M.; Tsubaki, M.; Nishida, S.; Hashimoto, S.; Matsuo, K.; Nakayama, T.; Ito, A.; et al. Neuroprotective Effect of (–)-Epigallocatechin-3-Gallate in Rats When Administered Pre- or Post-Traumatic Brain Injury. J. Neural. Transm. 2013, 120, 767–783. [Google Scholar] [CrossRef]
- Sawmiller, D.; Li, S.; Shahaduzzaman, M.; Smith, A.; Obregon, D.; Giunta, B.; Borlongan, C.; Sanberg, P.; Tan, J. Luteolin Reduces Alzheimer’s Disease Pathologies Induced by Traumatic Brain Injury. Int. J. Mol. Sci. 2014, 15, 895–904. [Google Scholar] [CrossRef]
- Sharma, S.; Zhuang, Y.; Ying, Z.; Wu, A.; Gomez-Pinilla, F. Dietary Curcumin Supplementation Counteracts Reduction in Levels of Molecules Involved in Energy Homeostasis after Brain Trauma. Neuroscience 2009, 161, 1037–1044. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary Curcumin Counteracts the Outcome of Traumatic Brain Injury on Oxidative Stress, Synaptic Plasticity, and Cognition. Exp. Neurol. 2006, 197, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Samini, F.; Samarghandian, S.; Borji, A.; Mohammadi, G.; Bakaian, M. Curcumin Pretreatment Attenuates Brain Lesion Size and Improves Neurological Function Following Traumatic Brain Injury in the Rat. Pharmacol. Biochem. Behav. 2013, 110, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.D.; Sukumari-Ramesh, S.; Swift, A.E.B.; Meiler, S.E.; Vender, J.R.; Dhandapani, K.M. Curcumin Attenuates Cerebral Edema Following Traumatic Brain Injury in Mice: A Possible Role for Aquaporin-4? J. Neurochem. 2010, 113, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Liu, X.; Li, G.; Wang, Y. Resveratrol Pretreatment Attenuates Traumatic Brain Injury in Rats by Suppressing NLRP3 Inflammasome Activation via SIRT1. Mol. Med. Rep. 2017, 17, 3212–3217. [Google Scholar] [CrossRef] [PubMed]
- Farbood, Y.; Sarkaki, A.; Dianat, M.; Khodadadi, A.; Haddad, M.K.; Mashhadizadeh, S. Ellagic Acid Prevents Cognitive and Hippocampal Long-Term Potentiation Deficits and Brain Inflammation in Rat with Traumatic Brain Injury. Life Sci. 2015, 124, 120–127. [Google Scholar] [CrossRef]
- Mirshekar, M.A.; Sarkaki, A.; Farbood, Y.; Gharib Naseri, M.K.; Badavi, M.; Mansouri, M.T.; Haghparast, A. Neuroprotective Effects of Gallic Acid in a Rat Model of Traumatic Brain Injury: Behavioral, Electrophysiological, and Molecular Studies. Iran J. Basic Med. Sci. 2018, 21, 1056–1063. [Google Scholar] [CrossRef]
- Sarkaki, A.; Farbood, Y.; Gharib-Naseri, M.K.; Badavi, M.; Mansouri, M.T.; Haghparast, A.; Mirshekar, M.A. Gallic Acid Improved Behavior, Brain Electrophysiology, and Inflammation in a Rat Model of Traumatic Brain Injury. Can. J. Physiol. Pharmacol. 2015, 93, 687–694. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, J.; Cai, Y.; Huang, J.; You, L. Catechin Attenuates Traumatic Brain Injury-Induced Blood-Brain Barrier Damage and Improves Longer-Term Neurological Outcomes in Rats. Exp. Physiol. 2017, 102, 1269–1277. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, W.; Li, Q.; Han, X.; Xing, J.; Qi, C.; Lan, X.; Wan, J.; Potts, A.; Guan, F.; et al. Cerebroprotection of Flavanol (-)-Epicatechin after Traumatic Brain Injury via Nrf2-Dependent and -Independent Pathways. Free Radic. Biol. Med. 2016, 92, 15–28. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, J. (−)-Epigallocatechin-3-Gallate Provides Neuroprotection via AMPK Activation against Traumatic Brain Injury in a Mouse Model. Naunyn Schmiedebergs Arch. Pharm. 2020, 393, 2209–2220. [Google Scholar] [CrossRef]
- Boonpawa, R.; Spenkelink, A.; Punt, A.; Rietjens, I.M.C.M. Physiologically Based Kinetic Modeling of Hesperidin Metabolism and Its Use to Predict in Vivo Effective Doses in Humans. Mol. Nutr. Food Res. 2017, 61, 1600894. [Google Scholar] [CrossRef]
- Deng, C.; Yi, R.; Fei, M.; Li, T.; Han, Y.; Wang, H. Naringenin Attenuates Endoplasmic Reticulum Stress, Reduces Apoptosis, and Improves Functional Recovery in Experimental Traumatic Brain Injury. Brain Res. 2021, 1769, 147591. [Google Scholar] [CrossRef]
- Almeida, A.F.; Borge, G.I.A.; Piskula, M.; Tudose, A.; Tudoreanu, L.; Valentová, K.; Williamson, G.; Santos, C.N. Bioavailability of Quercetin in Humans with a Focus on Interindividual Variation. Compr. Rev. Food Sci. Food Saf. 2018, 17, 714–731. [Google Scholar] [CrossRef]
- Song, J.; Du, G.; Wu, H.; Gao, X.; Yang, Z.; Liu, B.; Cui, S. Protective Effects of Quercetin on Traumatic Brain Injury Induced Inflammation and Oxidative Stress in Cortex through Activating Nrf2/HO-1 Pathway. Restor. Neurol. Neurosci. 2021, 39, 73–84. [Google Scholar] [CrossRef]
- Yang, T.; Kong, B.; Gu, J.-W.; Kuang, Y.-Q.; Cheng, L.; Yang, W.-T.; Xia, X.; Shu, H.-F. Anti-Apoptotic and Anti-Oxidative Roles of Quercetin After Traumatic Brain Injury. Cell Mol. Neurobiol. 2014, 34, 797–804. [Google Scholar] [CrossRef]
- Yuceli, S.; Yazici, G.N.; Mammadov, R.; Suleyman, H.; Kaya, M.; Ozdogan, S. The Effect of Rutin on Experimental Traumatic Brain Injury and Edema in Rats. In Vivo 2020, 34, 2453–2460. [Google Scholar] [CrossRef]
- Kumar, A.; Rinwa, P.; Dhar, H. Possible Nitric Oxide Modulation in the Protective Effects of Rutin against Experimental Head Trauma–Induced Cognitive Deficits: Behavioral, Biochemical, and Molecular Correlates. J. Surg. Res. 2014, 188, 268–279. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Gao, Y.; Li, L.; Tang, C.; Wen, G.; Zhou, Y.; Zhou, M.; Mao, L.; Fan, Y. Protective Effects of Quercetin on Mitochondrial Biogenesis in Experimental Traumatic Brain Injury via the Nrf2 Signaling Pathway. PLoS ONE 2016, 11, e0164237. [Google Scholar] [CrossRef]
- Schültke, E.; Kamencic, H.; Zhao, M.; Tian, G.-F.; Baker, A.J.; Griebel, R.W.; Juurlink, B.H.J. Neuroprotection Following Fluid Percussion Brain Trauma: A Pilot Study Using Quercetin. J. Neurotrauma 2005, 22, 1475–1484. [Google Scholar] [CrossRef]
- Zhai, X.; Ding, Y.; Wang, Q.; Zhang, H.; Li, F. Rutin Acid Ameliorates Neural Apoptosis Induced by Traumatic Brain Injury via Mitochondrial Pathways in Mice. Neuroimmunomodulation 2016, 23, 179–187. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Zhou, Y.; Zhu, Y.; Fei, M. Fisetin Alleviates Oxidative Stress after Traumatic Brain Injury via the Nrf2-ARE Pathway. Neurochem. Int. 2018, 118, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Wen, G.; Li, L.; Gao, Y.; Zhuang, Z.; Zhou, M.; Mao, L.; Fan, Y. Neuroprotection by Quercetin via Mitochondrial Function Adaptation in Traumatic Brain Injury: PGC-1α Pathway as a Potential Mechanism. J. Cell Mol. Med. 2017, 22, 883–891. [Google Scholar] [CrossRef]
- Du, G.; Zhao, Z.; Chen, Y.; Li, Z.; Tian, Y.; Liu, Z.; Liu, B.; Song, J. Quercetin Protects Rat Cortical Neurons against Traumatic Brain Injury. Mol. Med. Rep. 2018, 17, 7859–7865. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, Z.; Chen, Y.; Li, Z.; Tian, Y.; Liu, Z.; Liu, B.; Song, J. Quercetin Attenuates Neuronal Autophagy and Apoptosis in Rat Traumatic Brain Injury Model via Activation of PI3K/Akt Signaling Pathway. Neurol. Res. 2016, 38, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, J.; Santhakumar, V.; Kannurpatti, S.S. Beneficial Effects of Kaempferol after Developmental Traumatic Brain Injury Is through Protection of Mitochondrial Function, Oxidative Metabolism, and Neural Viability. J. Neurotrauma 2019, 36, 1264–1278. [Google Scholar] [CrossRef]
- Kosari-Nasab, M.; Shokouhi, G.; Ghorbanihaghjo, A.; Mesgari-Abbasi, M.; Salari, A.-A. Quercetin Mitigates Anxiety-like Behavior and Normalizes Hypothalamus–Pituitary–Adrenal Axis Function in a Mouse Model of Mild Traumatic Brain Injury. Behav. Pharmacol. 2019, 30, 282–289. [Google Scholar] [CrossRef]
- Parent, M.; Chitturi, J.; Santhakumar, V.; Hyder, F.; Sanganahalli, B.G.; Kannurpatti, S.S. Kaempferol Treatment after Traumatic Brain Injury during Early Development Mitigates Brain Parenchymal Microstructure and Neural Functional Connectivity Deterioration at Adolescence. J. Neurotrauma 2020, 37, 966–974. [Google Scholar] [CrossRef]
- Dos Santos, C.N.; Menezes, R.; Carregosa, D.; Valentova, K.; Foito, A.; McDougall, G.; Stewart, D. Flavonols and Flavones. In Dietary Polyphenols; Wiley: Hoboken, NJ, USA, 2020; pp. 163–198. [Google Scholar]
- Xu, J.; Wang, H.; Lu, X.; Ding, K.; Zhang, L.; He, J.; Wei, W.; Wu, Y. Posttraumatic Administration of Luteolin Protects Mice from Traumatic Brain Injury: Implication of Autophagy and Inflammation. Brain Res. 2014, 1582, 237–246. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin Provides Neuroprotection in Models of Traumatic Brain Injury via the Nrf2–ARE Pathway. Free Radic. Biol. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef]
- Rashno, M.; Sarkaki, A.; Farbood, Y.; Rashno, M.; Khorsandi, L.; Naseri, M.K.G.; Dianat, M. Therapeutic Effects of Chrysin in a Rat Model of Traumatic Brain Injury: A Behavioral, Biochemical, and Histological Study. Life Sci. 2019, 228, 285–294. [Google Scholar] [CrossRef]
- Soltani, Z.; Khaksari, M.; Jafari, E.; Iranpour, M.; Shahrokhi, N. Is Genistein Neuroprotective in Traumatic Brain Injury? Physiol. Behav. 2015, 152, 26–31. [Google Scholar] [CrossRef]
- Sun, M.; McDonald, S.J.; Brady, R.D.; O’Brien, T.J.; Shultz, S.R. The Influence of Immunological Stressors on Traumatic Brain Injury. Brain Behav. Immun. 2018, 69, 618–628. [Google Scholar] [CrossRef]
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D.; et al. Curcumin Plays Neuroprotective Roles against Traumatic Brain Injury Partly via Nrf2 Signaling. Toxicol. Appl. Pharmacol. 2018, 346, 28–36. [Google Scholar] [CrossRef]
- Zhu, H.; Bian, C.; Yuan, J.; Chu, W.; Xiang, X.; Chen, F.; Wang, C.; Feng, H.; Lin, J. Curcumin Attenuates Acute Inflammatory Injury by Inhibiting the TLR4/MyD88/NF-ΚB Signaling Pathway in Experimental Traumatic Brain Injury. J. Neuroinflamm. 2014, 11, 59. [Google Scholar] [CrossRef]
- Sun, G.; Miao, Z.; Ye, Y.; Zhao, P.; Fan, L.; Bao, Z.; Tu, Y.; Li, C.; Chao, H.; Xu, X.; et al. Curcumin Alleviates Neuroinflammation, Enhances Hippocampal Neurogenesis, and Improves Spatial Memory after Traumatic Brain Injury. Brain Res. Bull. 2020, 162, 84–93. [Google Scholar] [CrossRef]
- Feng, Y.; Cui, Y.; Gao, J.-L.; Li, R.; Jiang, X.-H.; Tian, Y.-X.; Wang, K.-J.; Li, M.-H.; Zhang, H.-A.; Cui, J.-Z. Neuroprotective Effects of Resveratrol against Traumatic Brain Injury in Rats: Involvement of Synaptic Proteins and Neuronal Autophagy. Mol. Med. Rep. 2016, 13, 5248–5254. [Google Scholar] [CrossRef]
- Feng, Y.; Cui, Y.; Gao, J.-L.; Li, M.-H.; Li, R.; Jiang, X.-H.; Tian, Y.-X.; Wang, K.-J.; Cui, C.-M.; Cui, J.-Z. Resveratrol Attenuates Neuronal Autophagy and Inflammatory Injury by Inhibiting the TLR4/NF-ΚB Signaling Pathway in Experimental Traumatic Brain Injury. Int. J. Mol. Med. 2016, 37, 921–930. [Google Scholar] [CrossRef]
- Gatson, J.W.; Liu, M.-M.; Abdelfattah, K.; Wigginton, J.G.; Smith, S.; Wolf, S.; Minei, J.P. Resveratrol Decreases Inflammation in the Brain of Mice with Mild Traumatic Brain Injury. J. Trauma Acute Care Surg. 2013, 74, 470–475. [Google Scholar] [CrossRef]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by Resveratrol against Traumatic Brain Injury in Rats. Mol. Cell. Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef]
- Atalay, T.; Gulsen, I.; Colcimen, N.; Alp, H.H.; Sosuncu, E.; Alaca, I.; Ak, H.; Ragbetli, M.C. Resveratrol Treatment Prevents Hippocamal Neurodegeneration in a Rodent Model of Traumatic Brain Injury. Turk. Neurosurg. 2016, 27, 924–930. [Google Scholar] [CrossRef]
- Shi, Z.; Qiu, W.; Xiao, G.; Cheng, J.; Zhang, N. Resveratrol Attenuates Cognitive Deficits of Traumatic Brain Injury by Activating P38 Signaling in the Brain. Med. Sci. Monit. 2018, 24, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Singleton, R.H.; Yan, H.Q.; Fellows-Mayle, W.; Dixon, C.E. Resveratrol Attenuates Behavioral Impairments and Reduces Cortical and Hippocampal Loss in a Rat Controlled Cortical Impact Model of Traumatic Brain Injury. J. Neurotrauma 2010, 27, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Sönmez, Ü.; Sönmez, A.; Erbil, G.; Tekmen, I.; Baykara, B. Neuroprotective Effects of Resveratrol against Traumatic Brain Injury in Immature Rats. Neurosci. Lett. 2007, 420, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.Y.; Lee, S.H.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. Int. J. Mol. Sci. 2017, 18, 2510. [Google Scholar] [CrossRef]
- Erbil, G.; Sacik, U.; Yilmaz, F.; Kisaoglu, H.; Erbayraktar, Z.; Pekcetin, C.; Ozogul, C. The Effect of Ferulic Acid on Experimental Traumatic Brain Damage in Rats. Bratisl. Med. J. 2019, 120, 372–379. [Google Scholar] [CrossRef]
- Nasution, R.A.; Islam, A.A.; Hatta, M.; Prihantono; Turchan, A.; Nasrullah; Faruk, M. Role of CAPE in Reducing Oxidative Stress in Animal Models with Traumatic Brain Injury. Ann. Med. Surg. 2020, 57, 118–122. [Google Scholar] [CrossRef]
- Kerman, M.; Kanter, M.; Coşkun, K.K.; Erboga, M.; Gurel, A. Neuroprotective Effects of Caffeic Acid Phenethyl Ester on Experimental Traumatic Brain Injury in Rats. J. Mol. Histol. 2012, 43, 49–57. [Google Scholar] [CrossRef]
- Guo, S.; Zhen, Y.; Zhu, Z.; Zhou, G.; Zheng, X. Cinnamic Acid Rescues Behavioral Deficits in a Mouse Model of Traumatic Brain Injury by Targeting MiR-455-3p/HDAC2. Life Sci. 2019, 235, 116819. [Google Scholar] [CrossRef]
- Zhao, J.; Pati, S.; Redell, J.B.; Zhang, M.; Moore, A.N.; Dash, P.K. Caffeic Acid Phenethyl Ester Protects Blood–Brain Barrier Integrity and Reduces Contusion Volume in Rodent Models of Traumatic Brain Injury. J. Neurotrauma 2012, 29, 1209–1218. [Google Scholar] [CrossRef]
- Nasution, R.A.; Islam, A.A.; Hatta, M.; Prihantono; Warsinggih; Ludong, D.H.; Ismail; Wangi, H.; Massi, M.N.; Nasution, K.I. Effects of Caffeic Acid Phenethyl Ester in Reducing Cerebral Edema in Rat Subjects Experiencing Brain Injury: An In Vivo Study. Ann. Med. Surg. 2020, 57, 328–333. [Google Scholar] [CrossRef]
- Mashhadizadeh, S.; Farbood, Y.; Dianat, M.; Khodadadi, A.; Sarkaki, A. Therapeutic Effects of Ellagic Acid on Memory, Hippocampus Electrophysiology Deficits, and Elevated TNF-α Level in Brain Due to Experimental Traumatic Brain Injury. Iran J. Basic Med. Sci. 2017, 20, 399–407. [Google Scholar] [CrossRef]
- Krishna, G.; Ying, Z.; Gomez-Pinilla, F. Blueberry Supplementation Mitigates Altered Brain Plasticity and Behavior after Traumatic Brain Injury in Rats. Mol. Nutr. Food Res. 2019, 63, 1801055. [Google Scholar] [CrossRef]
- Zahedi, H.; Hosseinzadeh-Attar, M.J.; Shadnoush, M.; Sahebkar, A.; Barkhidarian, B.; Sadeghi, O.; Najafi, A.; Hosseini, S.; Qorbani, M.; Ahmadi, A.; et al. Effects of Curcuminoids on Inflammatory and Oxidative Stress Biomarkers and Clinical Outcomes in Critically Ill Patients: A Randomized double-blind Placebo-controlled Trial. Phytother. Res. 2021, 35, 4605–4615. [Google Scholar] [CrossRef]
- Campesi, I.; Romani, A.; Marino, M.; Franconi, F. Phenolic Compounds from a Sex-Gender Perspective. In Recent Advances in Polyphenol Research; John Wiley & Sons, Ltd: Chichester, UK, 2014; pp. 327–339. [Google Scholar]
- Gupte, R.P.; Brooks, W.M.; Vukas, R.R.; Pierce, J.D.; Harris, J.L. Sex Differences in Traumatic Brain Injury: What We Know and What We Should Know. J. Neurotrauma 2019, 36, 3063–3091. [Google Scholar] [CrossRef]
- De Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The Pharmacokinetics of Anthocyanins and Their Metabolites in Humans. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef]
- Hazeldine, J.; Lord, J.M.; Belli, A. Traumatic Brain Injury and Peripheral Immune Suppression: Primer and Prospectus. Front. Neurol. 2015, 6, 235. [Google Scholar] [CrossRef]
- Manso, T.; Lores, M.; de Miguel, T. Antimicrobial Activity of Polyphenols and Natural Polyphenolic Extracts on Clinical Isolates. Antibiotics 2021, 11, 46. [Google Scholar] [CrossRef]
- Raj, R.; Wennervirta, J.M.; Tjerkaski, J.; Luoto, T.M.; Posti, J.P.; Nelson, D.W.; Takala, R.; Bendel, S.; Thelin, E.P.; Luostarinen, T.; et al. Dynamic Prediction of Mortality after Traumatic Brain Injury Using a Machine Learning Algorithm. NPJ Digit. Med. 2022, 5, 96. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carecho, R.; Carregosa, D.; Ratilal, B.O.; Figueira, I.; Ávila-Gálvez, M.A.; dos Santos, C.N.; Loncarevic-Vasiljkovic, N. Dietary (Poly)phenols in Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 8908. https://doi.org/10.3390/ijms24108908
Carecho R, Carregosa D, Ratilal BO, Figueira I, Ávila-Gálvez MA, dos Santos CN, Loncarevic-Vasiljkovic N. Dietary (Poly)phenols in Traumatic Brain Injury. International Journal of Molecular Sciences. 2023; 24(10):8908. https://doi.org/10.3390/ijms24108908
Chicago/Turabian StyleCarecho, Rafael, Diogo Carregosa, Bernardo Oliveira Ratilal, Inês Figueira, Maria Angeles Ávila-Gálvez, Cláudia Nunes dos Santos, and Natasa Loncarevic-Vasiljkovic. 2023. "Dietary (Poly)phenols in Traumatic Brain Injury" International Journal of Molecular Sciences 24, no. 10: 8908. https://doi.org/10.3390/ijms24108908
APA StyleCarecho, R., Carregosa, D., Ratilal, B. O., Figueira, I., Ávila-Gálvez, M. A., dos Santos, C. N., & Loncarevic-Vasiljkovic, N. (2023). Dietary (Poly)phenols in Traumatic Brain Injury. International Journal of Molecular Sciences, 24(10), 8908. https://doi.org/10.3390/ijms24108908