
Environmental Pollution and the Risk of Developing Metabolic Disorders: Obesity and Diabetes


Abstract
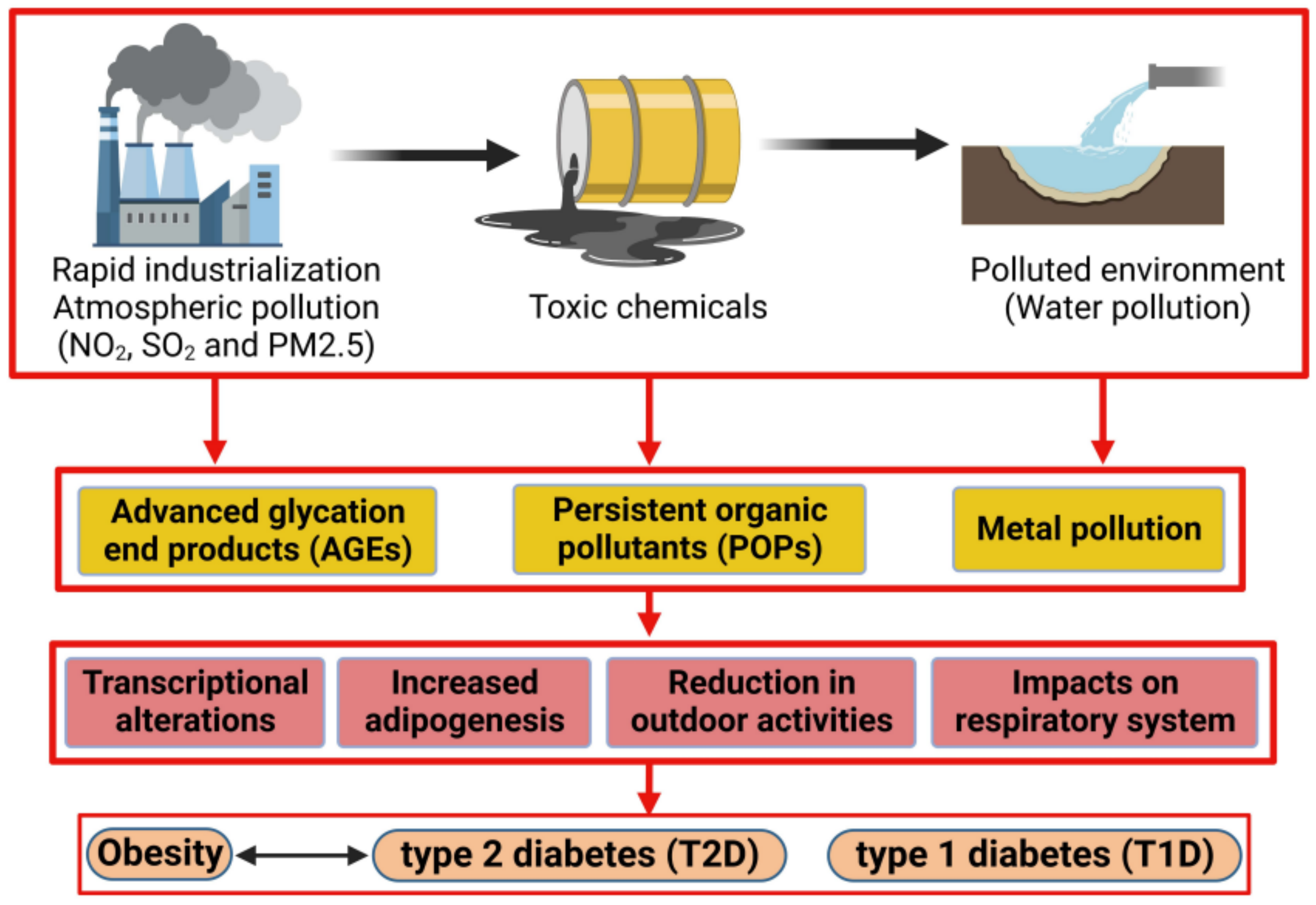
1. Introduction
2. Search Strategy
3. Endocrine Disruptor Compounds: Ambient Air Pollution, Persistent Organic Pollutants (POPs), Metals
3.1. Ambient Air Pollution
3.2. Persistent Organic Pollutants
3.2.1. Polychlorinated Biphenyls
3.2.2. Pesticides
3.2.3. Polycyclic Aromatic Hydrocarbons (PAHs)
3.2.4. Bisphenol A
3.2.5. Phthalates
3.2.6. Polybrominated Diphenyl Ethers
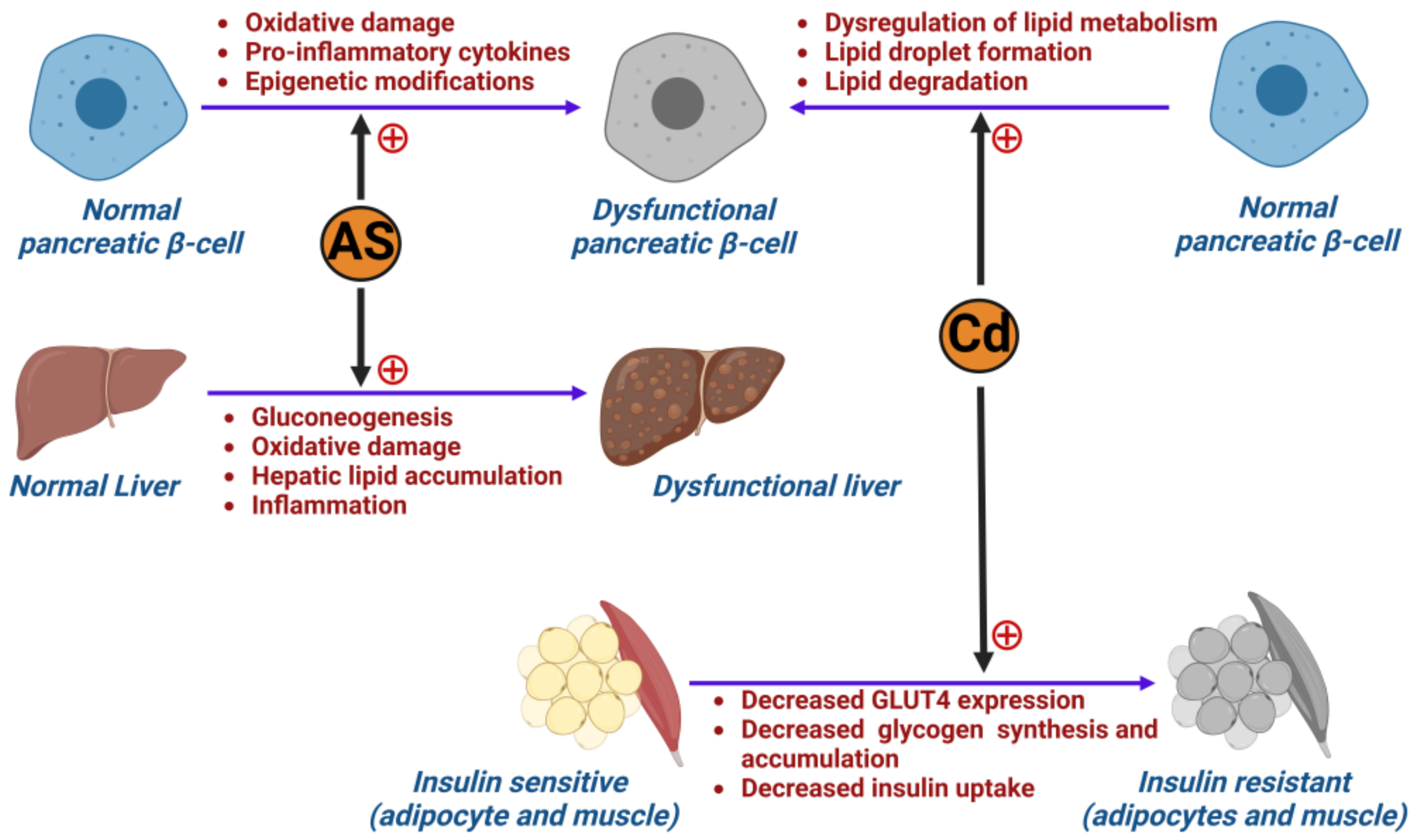
3.3. Metals
4. The Molecular and Cellular Mechanisms of Diabetes and Obesity
4.1. Molecular Mechanisms
4.2. Cellular Mechanisms
5. The Impact of Pollution on Obesity
5.1. The Development of Obesity
5.2. Advanced Glycation End Product (AGE) Impact on Obesity Prevalence
5.3. Impact of Persistent Organic Pollutants on Obesity
5.4. Pollution and the Impact on Childhood Obesity
6. Impact of Pollution on Type 2 Diabetes Mellitus (T2D)
6.1. The Development of T2D
6.2. The Role of Metals in Inducing Type 2 Diabetes Mellitus
6.3. Air Pollution and Its Impact on Developing Type 2 Diabetes (T2D)
7. The Impact of Pollution on Type 1 Diabetes Mellitus (T1D)
7.1. Impact of Pollution on Pancreatic β-Islet Cells
7.2. Impact of Pollution on Childhood Type 1 Diabetes Mellitus
8. Impact of Pollution on Gestational Diabetes (GDM)
8.1. The Development of GDM
8.2. Association between Particulate Matter (PM) and Gestational Diabetes (GDM)
9. Metabolic Syndrome
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Metabolic Disorders: Types, Causes, and Symptoms. Available online: https://www.medicalnewstoday.com/articles/metabolic-disorders (accessed on 17 September 2022).
- Common Metabolic Diseases. National Institutes of Health (NIH). 2021. Available online: https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/common-metabolic-diseases (accessed on 17 September 2022).
- Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 15 September 2022).
- CDC Obesity is a Common, Serious, and Costly Disease. Centers for Disease Control and Prevention. 2022. Available online: https://www.cdc.gov/obesity/data/adult.html (accessed on 17 September 2022).
- ALNohair, S. Obesity in Gulf Countries. Int. J. Health Sci. 2014, 8, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Over Half of Adults in the EU Are Overweight. Available online: https://ec.europa.eu/eurostat/web/products-eurostat-news/-/ddn-20210721-2 (accessed on 21 April 2023).
- IDF Diabetes Atlas | Tenth Edition. Available online: https://diabetesatlas.org/ (accessed on 21 April 2023).
- Diabetes Symptoms, Causes, & Treatment | ADA. Available online: https://diabetes.org/diabetes (accessed on 15 September 2022).
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Alam, U.; Asghar, O.; Azmi, S.; Malik, R.A. Chapter 15—General aspects of diabetes mellitus. In Handbook of Clinical Neurology; Zochodne, D.W., Malik, R.A., Eds.; Diabetes and the Nervous System; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126, pp. 211–222. Available online: https://www.sciencedirect.com/science/article/pii/B9780444534804000151 (accessed on 15 September 2022).
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Increasing Environmental Pollution (GMT 10)—European Environment Agency. Available online: https://www.eea.europa.eu/soer/2015/global/pollution (accessed on 17 September 2022).
- Augustin, M.A.; Riley, M.; Stockmann, R.; Bennett, L.; Kahl, A.; Lockett, T.; Osmond, M.; Sanguansri, P.; Stonehouse, W.; Zajac, I.; et al. Role of food processing in food and nutrition security. Trends Food Sci. Technol. 2016, 56, 115–125. [Google Scholar] [CrossRef]
- Riera-Crichton, D.; Tefft, N. Macronutrients and obesity: Revisiting the calories in, calories out framework. Econ. Hum. Biol. 2014, 14, 33–49. [Google Scholar] [CrossRef]
- The exposome and health: Where chemistry meets biology | Science. Available online: https://www.science.org/doi/abs/10.1126/science.aay3164 (accessed on 15 September 2022).
- Brunekreef, B.; Holgate, S.T. Air pollution and health. Lancet 2002, 360, 1233–1242. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Endocrine-Disrupting Chemicals (EDCs). Available online: https://www.endocrine.org/patient-engagement/endocrine-library/edcs (accessed on 10 September 2022).
- Darbre, P.D. Overview of air pollution and endocrine disorders. Int. J. Gen. Med. 2018, 11, 191–207. [Google Scholar] [CrossRef]
- Combarnous, Y.; Nguyen, T.M.D. Comparative Overview of the Mechanisms of Action of Hormones and Endocrine Disruptor Compounds. Toxics 2019, 7, 5. [Google Scholar] [CrossRef]
- Sever, R.; Glass, C.K. Signaling by Nuclear Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef]
- Endocrine-Disrupting Chemicals—UpToDate. Available online: https://www.uptodate.com/contents/endocrine-disrupting-chemicals?search=EDCs%20and%20reproduction&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 1 February 2023).
- Occupational and Environmental Risks to Reproduction in Females: Specific Exposures and Impact—UpToDate. Available online: https://www.uptodate.com/contents/occupational-and-environmental-risks-to-reproduction-in-females-specific-exposures-and-impact?search=EDCs%20and%20reproduction&topicRef=113968&source=see_link#H2180005789 (accessed on 2 February 2023).
- Ambient (Outdoor) Air Pollution. Available online: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health (accessed on 8 September 2022).
- Particulate Matter Overview. Utah Department of Environmental Quality. 2019. Available online: https://deq.utah.gov/air-quality/particulate-matter-overview (accessed on 4 February 2023).
- Poursafa, P.; Kamali, Z.; Fraszczyk, E.; Boezen, H.M.; Vaez, A.; Snieder, H. DNA methylation: A potential mediator between air pollution and metabolic syndrome. Clin. Epigenetics 2022, 14, 82. [Google Scholar] [CrossRef]
- Idowu, S.O.; Capaldi, N.; Zu, L.; Gupta, A.D. (Eds.) Stockholm Convention on Persistent Organic Pollutants (POPs). In Encyclopedia of Corporate Social Responsibility; Springer: Berlin/Heidelberg, Germany, 2013; p. 2336. ISBN 978-3-642-28035-1. Available online: http://link.springer.com/10.1007/978-3-642-28036-8_101506 (accessed on 10 September 2022).
- Yang, C.; Kong, A.P.S.; Cai, Z.; Chung, A.C.K. Persistent Organic Pollutants as Risk Factors for Obesity and Diabetes. Curr. Diab. Rep. 2017, 17, 132. [Google Scholar] [CrossRef]
- Rahman, M.L.; Zhang, C.; Smarr, M.M.; Lee, S.; Honda, M.; Kannan, K.; Tekola-Ayele, F.; Buck Louis, G.M. Persistent organic pollutants and gestational diabetes: A multi-center prospective cohort study of healthy US women. Environ. Int. 2019, 124, 249–258. [Google Scholar] [CrossRef]
- The 12 Initial POPs. Available online: http://www.pops.int/TheConvention/ThePOPs/The12InitialPOPs/tabid/296/Default.aspx (accessed on 10 September 2022).
- Listing of POPs in the Stockholm Convention. Available online: http://chm.pops.int/TheConvention/ThePOPs/AllPOPs/tabid/2509/Default.aspx (accessed on 4 February 2023).
- Fitzgerald, L.; Wikoff, D.S. Persistent Organic Pollutants. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 820–825. Available online: https://www.sciencedirect.com/science/article/pii/B9780123864543002116 (accessed on 18 September 2022).
- Overview. Available online: http://www.pops.int/TheConvention/Overview/tabid/3351/Default.aspx (accessed on 29 August 2022).
- Safe, S.; Bandiera, S.; Sawyer, T.; Robertson, L.; Safe, L.; Parkinson, A.; Thomas, P.E.; Ryan, D.E.; Reik, L.M.; Levin, W.; et al. PCBs: Structure–function relationships and mechanism of action. Environ. Health Perspect. 1985, 60, 47–56. [Google Scholar]
- Aaseth, J.; Javorac, D.; Djordjevic, A.; Bulat, Z.; Skalny, A.; Zaitseva, I.; Aschner, M.; Tinkov, A. The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies. Toxics 2022, 10, 65. [Google Scholar] [CrossRef]
- Mnif, W.; Hassine, A.I.H.; Bouaziz, A.; Bartegi, A.; Thomas, O.; Roig, B. Effect of Endocrine Disruptor Pesticides: A Review. Int. J. Environ. Res. Public. Health 2011, 8, 2265–2303. [Google Scholar] [CrossRef]
- Turusov, V.; Rakitsky, V.; Tomatis, L. Dichlorodiphenyltrichloroethane (DDT): Ubiquity, persistence, and risks. Environ. Health Perspect. 2002, 110, 125–128. [Google Scholar] [CrossRef]
- Polycyclic Aromatic Hydrocarbons (PAHs) Factsheet | National Biomonitoring Program | CDC. Available online: https://www.cdc.gov/biomonitoring/PAHs_FactSheet.html (accessed on 18 September 2022).
- Nagel, S.C.; Bromfield, J.J. Bisphenol A: A Model Endocrine Disrupting Chemical With a New Potential Mechanism of Action. Endocrinology 2013, 154, 1962–1964. [Google Scholar] [CrossRef]
- Plastics, EDCs & Health: Authoritative Guide. Available online: https://www.endocrine.org/topics/edc/plastics-edcs-and-health (accessed on 15 September 2022).
- Costa, L.G.; Giordano, G. Developmental neurotoxicity of polybrominated diphenyl ether (pbde) flame retardants. Neurotoxicology 2007, 28, 1047–1067. [Google Scholar] [CrossRef]
- Dishaw, L.; Macaulay, L.; Roberts, S.C.; Stapleton, H.M. Exposures, Mechanisms, and Impacts of Endocrine-Active Flame Retardants. Curr. Opin. Pharmacol. 2014, 19, 125–133. [Google Scholar] [CrossRef]
- McGovern, V. PCBs Are Endocrine Disruptors: Mixture Affects Reproductive Development in Female Mice. Environ. Health Perspect. 2006, 114, A368–A369. [Google Scholar] [CrossRef]
- Bell, M.R. Endocrine-disrupting actions of PCBs on brain development and social and reproductive behaviors. Curr. Opin. Pharmacol. 2014, 19, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Everett, C.J.; Frithsen, I.; Player, M. Relationship of polychlorinated biphenyls with type 2 diabetes and hypertension. J. Environ. Monit. JEM 2011, 13, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Chemical Safety: Pesticides. Available online: https://www.who.int/news-room/questions-and-answers/item/chemical-safety-pesticides (accessed on 9 September 2022).
- Fallon Nevada: FAQs: Organophosphates | CDC HSB. Available online: https://www.cdc.gov/nceh/clusters/fallon/organophosfaq.htm (accessed on 9 September 2022).
- Evangelou, E.; Ntritsos, G.; Chondrogiorgi, M.; Kavvoura, F.K.; Hernández, A.F.; Ntzani, E.E.; Tzoulaki, I. Exposure to pesticides and diabetes: A systematic review and meta-analysis. Environ. Int. 2016, 91, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Kaur, A.; Thind, S.S.; Singh, B.; Raina, S. Advanced glycation End-products (AGEs): An emerging concern for processed food industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Henao-Mejia, J.; Simmons, R.A. Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health. Endocrinology 2017, 159, 32–45. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, H. Phthalates and Their Impacts on Human Health. Healthcare 2021, 9, 603. [Google Scholar] [CrossRef]
- James-Todd, T.M.; Huang, T.; Seely, E.W.; Saxena, A.R. The association between phthalates and metabolic syndrome: The National Health and Nutrition Examination Survey 2001–2010. Environ. Health 2016, 15, 52. [Google Scholar] [CrossRef]
- Scoville, D.K.; Li, C.Y.; Wang, D.; Dempsey, J.L.; Raftery, D.; Mani, S.; Gu, H.; Cui, J.Y. Polybrominated Diphenyl Ethers and Gut Microbiome Modulate Metabolic Syndrome–Related Aqueous Metabolites in Mice. Drug Metab. Dispos. 2019, 47, 928–940. [Google Scholar] [CrossRef]
- Arsenic Toxicity: What Is the Biologic Fate of Arsenic in the Body? | Environmental Medicine | ATSDR. Available online: https://www.atsdr.cdc.gov/csem/arsenic/biologic_fate.html (accessed on 14 September 2022).
- Arsenic Toxicity: Where Is Arsenic Found? | Environmental Medicine | ATSDR. Available online: https://www.atsdr.cdc.gov/csem/arsenic/where_arsenic.html (accessed on 14 September 2022).
- Navas-Acien, A.; Silbergeld, E.K.; Pastor-Barriuso, R.; Guallar, E. Arsenic Exposure and Prevalence of Type 2 Diabetes in US Adults. JAMA 2008, 300, 814–822. [Google Scholar] [CrossRef]
- Arsenic Toxicity: What are the Physiologic Effects of Arsenic Exposure? | Environmental Medicine | ATSDR. Available online: https://www.atsdr.cdc.gov/csem/arsenic/physiologic_effects.html (accessed on 14 September 2022).
- Alonso-Magdalena, P.; Tudurí, E.; Marroquí, L.; Quesada, I.; Sargis, R.M.; Nadal, A. Toxic Effects of Common Environmental Pollutants in Pancreatic β-Cells and the Onset of Diabetes Mellitus. In Encyclopedia of Endocrine Diseases; Elsevier: Amsterdam, The Netherlands, 2019; pp. 764–775. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780128012383643258 (accessed on 23 August 2022).
- Bimonte, V.M.; Besharat, Z.M.; Antonioni, A.; Cella, V.; Lenzi, A.; Ferretti, E.; Migliaccio, S. The endocrine disruptor cadmium: A new player in the pathophysiology of metabolic diseases. J. Endocrinol. Investig. 2021, 44, 1363–1377. [Google Scholar] [CrossRef]
- Seo, M.Y.; Kim, S.-H.; Park, M.J. Air pollution and childhood obesity. Clin. Exp. Pediatr. 2020, 63, 382–388. [Google Scholar] [CrossRef]
- Xu, X.; Yavar, Z.; Verdin, M.; Ying, Z.; Mihai, G.; Kampfrath, T.; Wang, A.; Zhong, M.; Lippmann, M.; Chen, L.-C.; et al. Effect of Early Particulate Air Pollution Exposure on Obesity in Mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2518–2527. [Google Scholar] [CrossRef]
- Mirmiran, P.; Hadavi, H.; Mottaghi, A.; Azizi, F. Advanced glycation end products and risk of general and abdominal obesity in Iranian adults: Tehran lipid and glucose study. Med. J. Islam. Repub. Iran 2019, 33, 21. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Hong, S.-H.; Sung, Y.-A.; Hong, Y.S.; Ha, E.; Jeong, K.; Chung, H.; Lee, H. Urinary bisphenol A is associated with insulin resistance and obesity in reproductive-aged women. Clin. Endocrinol. 2017, 86, 506–512. [Google Scholar] [CrossRef]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. 2013, 37, 999–1005. [Google Scholar] [CrossRef]
- Amin, M.M.; Ebrahimpour, K.; Parastar, S.; Shoshtari-Yeganeh, B.; Hashemi, M.; Mansourian, M.; Poursafa, P.; Fallah, Z.; Rafiei, N.; Kelishadi, R. Association of urinary concentrations of phthalate metabolites with cardiometabolic risk factors and obesity in children and adolescents. Chemosphere 2018, 211, 547–556. [Google Scholar] [CrossRef]
- Chiu, C.-Y.; Sun, S.-C.; Chiang, C.-K.; Wang, C.-C.; Chan, D.-C.; Chen, H.-J.; Liu, S.-H.; Yang, R.-S. Plasticizer di(2-ethylhexyl)phthalate interferes with osteoblastogenesis and adipogenesis in a mouse model. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2018, 36, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Hurst, C.H.; Waxman, D.J. Activation of PPARalpha and PPARgamma by environmental phthalate monoesters. Toxicol. Sci. Off. J. Soc. Toxicol. 2003, 74, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Hernández, L.A.; Luzardo, O.P.; Valerón, P.F.; Zumbado, M.; Serra-Majem, L.; Camacho, M.; González-Antuña, A.; Boada, L.D. Persistent organic pollutants and risk of diabetes and obesity on healthy adults: Results from a cross-sectional study in Spain. Sci. Total Environ. 2017, 607–608, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Pesta, M.; Cedikova, M.; Dvorak, P.; Dvorakova, J.; Kulda, V.; Srbecka, K.; Muller, L.; Bouchalova, V.; Kralickova, M.; Babuska, V.; et al. Trends in gene expression changes during adipogenesis in human adipose derived mesenchymal stem cells under dichlorodiphenyldichloroethylene exposure. Mol. Cell. Toxicol. 2018, 14, 369–379. [Google Scholar] [CrossRef]
- Lim, J.-S.; Lee, D.-H.; Jacobs, D.R. Association of brominated flame retardants with diabetes and metabolic syndrome in the U.S. population, 2003–2004. Diabetes Care 2008, 31, 1802–1807. [Google Scholar] [CrossRef]
- Helaleh, M.; Diboun, I.; Al-Tamimi, N.; Al-Sulaiti, H.; Al-Emadi, M.; Madani, A.; Mazloum, N.A.; Latiff, A.; Elrayess, M.A. Association of polybrominated diphenyl ethers in two fat compartments with increased risk of insulin resistance in obese individuals. Chemosphere 2018, 209, 268–276. [Google Scholar] [CrossRef]
- Yang, C.; Wong, C.-M.; Wei, J.; Chung, A.C.K.; Cai, Z. The brominated flame retardant BDE 47 upregulates purine metabolism and mitochondrial respiration to promote adipocyte differentiation. Sci. Total Environ. 2018, 644, 1312–1322. [Google Scholar] [CrossRef]
- Inoue, K.; Goto, A.; Sugiyama, T.; Ramlau-Hansen, C.H.; Liew, Z. The Confounder-Mediator Dilemma: Should We Control for Obesity to Estimate the Effect of Perfluoroalkyl Substances on Health Outcomes? Toxics 2020, 8, 125. [Google Scholar] [CrossRef]
- Lee, D.-H.; Lee, I.-K.; Porta, M.; Steffes, M.; Jacobs, D.R. Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 2007, 50, 1841–1851. [Google Scholar] [CrossRef]
- Scinicariello, F.; Buser, M.C. Urinary polycyclic aromatic hydrocarbons and childhood obesity: NHANES (2001–2006). Environ. Health Perspect. 2014, 122, 299–303. [Google Scholar] [CrossRef]
- Fleisch, A.F.; Luttmann-Gibson, H.; Perng, W.; Rifas-Shiman, S.L.; Coull, B.A.; Kloog, I.; Koutrakis, P.; Schwartz, J.D.; Zanobetti, A.; Mantzoros, C.S.; et al. Prenatal and early life exposure to traffic pollution and cardiometabolic health in childhood. Pediatr. Obes. 2017, 12, 48–57. [Google Scholar] [CrossRef]
- Fleisch, A.F.; Rifas-Shiman, S.L.; Koutrakis, P.; Schwartz, J.D.; Kloog, I.; Melly, S.; Coull, B.A.; Zanobetti, A.; Gillman, M.W.; Gold, D.R.; et al. Prenatal exposure to traffic pollution: Associations with reduced fetal growth and rapid infant weight gain. Epidemiol. Camb. Mass 2015, 26, 43–50. [Google Scholar] [CrossRef]
- Fleisch, A.F.; Aris, I.M.; Rifas-Shiman, S.L.; Coull, B.A.; Luttmann-Gibson, H.; Koutrakis, P.; Schwartz, J.D.; Kloog, I.; Gold, D.R.; Oken, E. Prenatal Exposure to Traffic Pollution and Childhood Body Mass Index Trajectory. Front. Endocrinol. 2018, 9, 771. [Google Scholar] [CrossRef]
- Wen, X.; Shenassa, E.D.; Paradis, A.D. Maternal smoking, breastfeeding, and risk of childhood overweight: Findings from a national cohort. Matern. Child Health J. 2013, 17, 746–755. [Google Scholar] [CrossRef]
- Hawkins, S.S.; Cole, T.J.; Law, C. Millennium Cohort Study Child Health Group An ecological systems approach to examining risk factors for early childhood overweight: Findings from the UK Millennium Cohort Study. J. Epidemiol. Community Health 2009, 63, 147–155. [Google Scholar] [CrossRef]
- Rundle, A.G.; Gallagher, D.; Herbstman, J.B.; Goldsmith, J.; Holmes, D.; Hassoun, A.; Oberfield, S.; Miller, R.L.; Andrews, H.; Widen, E.M.; et al. Prenatal exposure to airborne polycyclic aromatic hydrocarbons and childhood growth trajectories from age 5-14 years. Environ. Res. 2019, 177, 108595. [Google Scholar] [CrossRef]
- Rundle, A.; Hoepner, L.; Hassoun, A.; Oberfield, S.; Freyer, G.; Holmes, D.; Reyes, M.; Quinn, J.; Camann, D.; Perera, F.; et al. Association of childhood obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am. J. Epidemiol. 2012, 175, 1163–1172. [Google Scholar] [CrossRef]
- Jerrett, M.; McConnell, R.; Wolch, J.; Chang, R.; Lam, C.; Dunton, G.; Gilliland, F.; Lurmann, F.; Islam, T.; Berhane, K. Traffic-related air pollution and obesity formation in children: A longitudinal, multilevel analysis. Environ. Health 2014, 13, 49. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Jacobs, D.R., Jr.; Lee, D.-H. Persistent Organic Pollutants and Type 2 Diabetes: A Critical Review of Review Articles. Front. Endocrinol. 2018, 9, 712. [Google Scholar] [CrossRef]
- El-Sikaily, A.; Helal, M. Environmental pollution and diabetes mellitus. World J. Meta-Anal. 2021, 9, 220–326. [Google Scholar] [CrossRef]
- Liu, S.; Guo, X.; Wu, B.; Yu, H.; Zhang, X.; Li, M. Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci. Rep. 2014, 4, 6894. [Google Scholar] [CrossRef] [PubMed]
- Rajak, S.; Raza, S.; Tewari, A.; Sinha, R.A. Environmental Toxicants and NAFLD: A Neglected yet Significant Relationship. Dig. Dis. Sci. 2022, 67, 3497–3507. [Google Scholar] [CrossRef] [PubMed]
- Kalo, M.B.; Rezaei, M. In vitro toxic interaction of arsenic and hyperglycemia in mitochondria: An important implication of increased vulnerability in pre-diabetics. Environ. Sci. Pollut. Res. Vol. 2022, 29, 28375–28385. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.J. Impact of the exposome on the development and function of pancreatic β-cells. Mol. Aspects Med. 2022, 87, 100965. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Hodjat, M.; Rahimifard, M.; Nigjeh, M.N.; Azizi, M.; Baeeri, M.; Bayrami, Z.; Gholami, M.; Hassani, S.; Abdollahi, M. Assessment of arsenic-induced modifications in the DNA methylation of insulin-related genes in rat pancreatic islets. Ecotoxicol. Environ. Saf. 2020, 201, 110802. [Google Scholar] [CrossRef]
- WHO. Preventing Disease Through Healthy Environments. Exposure to Cadmium: A Major Public Health Concern; WHO: Geneva, Switzerland, 2019; Available online: https://apps.who.int/iris/bitstream/handle/10665/329480/WHO-CED-PHE-EPE-19.4.3-eng.pdf (accessed on 20 September 2022).
- Xie, Y.; Liu, J.; Benbrahim-Tallaa, L.; Ward, J.M.; Logsdon, D.; Diwan, B.A.; Waalkes, M.P. Aberrant DNA methylation and gene expression in livers of newborn mice transplacentally exposed to a hepatocarcinogenic dose of inorganic arsenic. Toxicology 2007, 236, 7–15. [Google Scholar] [CrossRef]
- Jacquet, A.; Arnaud, J.; Hininger-Favier, I.; Hazane-Puch, F.; Couturier, K.; Lénon, M.; Lamarche, F.; Ounnas, F.; Fontaine, E.; Moulis, J.-M.; et al. Impact of chronic and low cadmium exposure of rats: Sex specific disruption of glucose metabolism. Chemosphere 2018, 207, 764–773. [Google Scholar] [CrossRef]
- Hong, H.; Xu, Y.; Xu, J.; Zhang, J.; Xi, Y.; Pi, H.; Yang, L.; Yu, Z.; Wu, Q.; Meng, Z.; et al. Cadmium exposure impairs pancreatic β-cell function and exaggerates diabetes by disrupting lipid metabolism. Environ. Int. 2021, 149, 106406. [Google Scholar] [CrossRef]
- Buha, A.; Đukić-Ćosić, D.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Moulis, J.-M.; Goumenou, M.; Wallace, D. Emerging Links between Cadmium Exposure and Insulin Resistance: Human, Animal, and Cell Study Data. Toxics 2020, 8, 63. [Google Scholar] [CrossRef]
- Little, B.B.; Reilly, R.; Walsh, B.; Vu, G.T. Cadmium Is Associated with Type 2 Diabetes in a Superfund Site Lead Smelter Community in Dallas, Texas. Int. J. Environ. Res. Public. Health 2020, 17, E4558. [Google Scholar] [CrossRef]
- Han, J.C.; Park, S.Y.; Hah, B.G.; Choi, G.H.; Kim, Y.K.; Kwon, T.H.; Kim, E.K.; Lachaal, M.; Jung, C.Y.; Lee, W. Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Arch. Biochem. Biophys. 2003, 413, 213–220. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yang, C.Y.; Huang, C.F.; Hung, D.Z.; Leung, Y.M.; Liu, S.H. Heavy metals, islet function and diabetes development. Islets 2009, 1, 169–176. [Google Scholar] [CrossRef]
- Tinkov, A.A.; Filippini, T.; Ajsuvakova, O.P.; Aaseth, J.; Gluhcheva, Y.G.; Ivanova, J.M.; Bjørklund, G.; Skalnaya, M.G.; Gatiatulina, E.R.; Popova, E.V.; et al. The role of cadmium in obesity and diabetes. Sci. Total Environ. 2017, 601–602, 741–755. [Google Scholar] [CrossRef]
- Nie, X.; Wang, N.; Chen, Y.; Chen, C.; Han, B.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Lu, M.; et al. Blood cadmium in Chinese adults and its relationships with diabetes and obesity | SpringerLink. Available online: https://link.springer.com/article/10.1007/s11356-016-7078-2 (accessed on 20 September 2022).
- Zhang, S.; Jin, Y.; Zeng, Z.; Liu, Z.; Fu, Z. Subchronic Exposure of Mice to Cadmium Perturbs Their Hepatic Energy Metabolism and Gut Microbiome. Chem. Res. Toxicol. 2015, 28, 2000–2009. [Google Scholar] [CrossRef]
- Treviño, S.; Waalkes, M.P.; Flores Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef]
- Sun, Q.; Yue, P.; Deiuliis, J.A.; Lumeng, C.N.; Kampfrath, T.; Mikolaj, M.B.; Cai, Y.; Ostrowski, M.C.; Lu, B.; Parthasarathy, S.; et al. Ambient Air Pollution Exaggerates Adipose Inflammation and Insulin Resistance in a Mouse Model of Diet-Induced Obesity. Circulation 2009, 119, 538–546. [Google Scholar] [CrossRef]
- Acharjee, S.; Ghosh, B.; Al-Dhubiab, B.E.; Nair, A.B. Understanding Type 1 Diabetes: Etiology and Models. Can. J. Diabetes 2013, 37, 269–276. [Google Scholar] [CrossRef]
- Hasham, A.; Tomer, Y. The recent rise in the frequency of Type 1 Diabetes: Who pulled the trigger? J. Autoimmun. 2011, 37, 1–2. [Google Scholar] [CrossRef]
- Krämer, U.; Herder, C.; Sugiri, D.; Strassburger, K.; Schikowski, T.; Ranft, U.; Rathmann, W. Traffic-related air pollution and incident type 2 diabetes: Results from the SALIA cohort study. Environ. Health Perspect. 2010, 118, 1273–1279. [Google Scholar] [CrossRef]
- Kelishadi, R.; Mirghaffari, N.; Poursafa, P.; Gidding, S.S. Lifestyle and environmental factors associated with inflammation, oxidative stress and insulin resistance in children. Atherosclerosis 2009, 203, 311–319. [Google Scholar] [CrossRef]
- Novelli, M.; Beffy, P.; Masini, M.; Vantaggiato, C.; Martino, L.; Marselli, L.; Marchetti, P.; De Tata, V. Selective beta-cell toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin on isolated pancreatic islets. Chemosphere 2021, 265, 129103. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Portincasa, P. Relationships between emissions of toxic airborne molecules and type 1 diabetes incidence in children: An ecologic study. World J. Diabetes 2021, 12, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Hathout, E.H.; Beeson, W.L.; Nahab, F.; Rabadi, A.; Thomas, W.; Mace, J.W. Role of exposure to air pollutants in the development of type 1 diabetes before and after 5 yr of age. Pediatr. Diabetes 2002, 3, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Bresson, S.E.; Isom, S.; Jensen, E.T.; Huber, S.; Oulhote, Y.; Rigdon, J.; Lovato, J.; Liese, A.D.; Pihoker, C.; Dabelea, D.; et al. Associations between persistent organic pollutants and type 1 diabetes in youth. Environ. Int. 2022, 163, 107175. [Google Scholar] [CrossRef] [PubMed]
- Fabricio, G.; Malta, A.; Chango, A.; De Freitas Mathias, P.C. Environmental Contaminants and Pancreatic Beta-Cells. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 257–263. [Google Scholar] [CrossRef]
- Lin, Y.; Wei, J.; Li, Y.; Chen, J.; Zhou, Z.; Song, L.; Wei, Z.; Lv, Z.; Chen, X.; Xia, W.; et al. Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E527–E538. [Google Scholar] [CrossRef]
- Meier, J.J.; Köhler, C.U.; Alkhatib, B.; Sergi, C.; Junker, T.; Klein, H.H.; Schmidt, W.E.; Fritsch, H. Beta-cell development and turnover during prenatal life in humans. Eur. J. Endocrinol. 2010, 162, 559–568. [Google Scholar] [CrossRef]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef]
- Yahaya, T.O.; Salisu, T.; Abdulrahman, Y.B.; Umar, A.K. Update on the genetic and epigenetic etiology of gestational diabetes mellitus: A review. Egypt. J. Med. Hum. Genet. 2020, 21, 13. [Google Scholar] [CrossRef]
- Gestational Diabetes Mellitus (GDM). Available online: https://www.hopkinsmedicine.org/health/conditions-and-diseases/diabetes/gestational-diabetes (accessed on 25 August 2022).
- Butler, A.E.; Cao-Minh, L.; Galasso, R.; Rizza, R.A.; Corradin, A.; Cobelli, C.; Butler, P.C. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010, 53, 2167–2176. [Google Scholar] [CrossRef]
- Robledo, C.A.; Mendola, P.; Yeung, E.; Männistö, T.; Sundaram, R.; Liu, D.; Ying, Q.; Sherman, S.; Grantz, K.L. Preconception and early pregnancy air pollution exposures and risk of gestational diabetes Mellitus. Environ. Res. 2015, 137, 316–322. [Google Scholar] [CrossRef]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, X.; Yang, X.; Dong, T.; Hu, W.; Guan, Q.; Tun, H.M.; Chen, Y.; Chen, R.; Sun, Z.; et al. Increased risk of gestational diabetes mellitus in women with higher prepregnancy ambient PM2.5 exposure. Sci. Total Environ. 2020, 730, 138982. [Google Scholar] [CrossRef]
- Ramanjaneya, M.; Butler, A.E.; Alkasem, M.; Bashir, M.; Jerobin, J.; Godwin, A.; Moin, A.S.M.; Ahmed, L.; Elrayess, M.A.; Hunt, S.C.; et al. Association of Complement-Related Proteins in Subjects With and Without Second Trimester Gestational Diabetes. Front. Endocrinol. 2021, 12, 641361. Available online: https://www.frontiersin.org/articles/10.3389/fendo.2021.641361 (accessed on 29 September 2022). [CrossRef]
- Ramanjaneya, M.; Butler, A.E.; Bashir, M.; Bettahi, I.; Moin, A.S.M.; Ahmed, L.; Elrayess, M.A.; Hunt, S.C.; Atkin, S.L.; Abou-Samra, A.B. apoA2 correlates to gestational age with decreased apolipoproteins A2, C1, C3 and E in gestational diabetes. BMJ Open Diabetes Res. Care 2021, 9, e001925. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Q.; He, S.; Wu, K.; Ren, M.; Dong, H.; Di, J.; Yu, Z.; Huang, C. Ambient air pollution and gestational diabetes mellitus: A review of evidence from biological mechanisms to population epidemiology. Sci. Total Environ. 2020, 719, 137349. [Google Scholar] [CrossRef]
- Lawrence, R.L.; Wall, C.R.; Bloomfield, F.H. Prevalence of gestational diabetes according to commonly used data sources: An observational study. BMC Pregnancy Childbirth 2019, 19, 349. [Google Scholar] [CrossRef]
- Zhou, T.; Sun, D.; Li, X.; Heianza, Y.; Nisa, H.; Hu, G.; Pei, X.; Shang, X.; Qi, L. Prevalence and Trends in Gestational Diabetes Mellitus among Women in the United States, 2006–2016. Diabetes 2018, 67, 121-OR. [Google Scholar] [CrossRef]
- Gao, C.; Sun, X.; Lu, L.; Liu, F.; Yuan, J. Prevalence of gestational diabetes mellitus in mainland China: A systematic review and meta-analysis. J. Diabetes Investig. 2019, 10, 154–162. [Google Scholar] [CrossRef]
- Al-Rifai, R.H.; Abdo, N.M.; Paulo, M.S.; Saha, S.; Ahmed, L.A. Prevalence of Gestational Diabetes Mellitus in the Middle East and North Africa, 2000–2019: A Systematic Review, Meta-Analysis, and Meta-Regression. Front. Endocrinol. 2021, 12, 668447. Available online: https://www.frontiersin.org/articles/10.3389/fendo.2021.668447 (accessed on 16 September 2022). [CrossRef]
- Paulo, M.S.; Abdo, N.M.; Bettencourt-Silva, R.; Al-Rifai, R.H. Gestational Diabetes Mellitus in Europe: A Systematic Review and Meta-Analysis of Prevalence Studies. Front. Endocrinol. 2021, 12, 691033. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, C. Prevalence of Gestational Diabetes and Risk of Progression to Type 2 Diabetes: A Global Perspective. Curr. Diab. Rep. 2016, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, E.; Ashley-Martin, J.; Dodds, L.; Arbuckle, T.E.; Hystad, P.; Johnson, M.; Crouse, D.L.; Ettinger, A.S.; Shapiro, G.D.; Fisher, M.; et al. Air Pollution Exposure During Pregnancy and Fetal Markers of Metabolic function. Am. J. Epidemiol. 2016, 183, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.D.; Dodds, L.; Arbuckle, T.E.; Ashley-Martin, J.; Ettinger, A.S.; Fisher, M.; Taback, S.; Bouchard, M.F.; Monnier, P.; Dallaire, R.; et al. Exposure to organophosphorus and organochlorine pesticides, perfluoroalkyl substances, and polychlorinated biphenyls in pregnancy and the association with impaired glucose tolerance and gestational diabetes mellitus: The MIREC Study. Environ. Res. 2016, 147, 71–81. [Google Scholar] [CrossRef]
- Eslami, B.; Naddafi, K.; Rastkari, N.; Rashidi, B.H.; Djazayeri, A.; Malekafzali, H. Association between serum concentrations of persistent organic pollutants and gestational diabetes mellitus in primiparous women. Environ. Res. 2016, 151, 706–712. [Google Scholar] [CrossRef]
- Vafeiadi, M.; Roumeliotaki, T.; Chalkiadaki, G.; Rantakokko, P.; Kiviranta, H.; Fthenou, E.; Kyrtopoulos, S.A.; Kogevinas, M.; Chatzi, L. Persistent organic pollutants in early pregnancy and risk of gestational diabetes mellitus. Environ. Int. 2017, 98, 89–95. [Google Scholar] [CrossRef]
- Shmerling, R.H. Metabolic Syndrome is on the Rise: What It Is and Why It Matters. Harvard Health. 2020. Available online: https://www.health.harvard.edu/blog/metabolic-syndrome-is-on-the-rise-what-it-is-and-why-it-matters-2020071720621 (accessed on 25 April 2023).
- Moore, J.X. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev. Chronic. Dis. 2017, 14, E24. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Mathangasinghe, Y.; Jayawardena, R.; Hills, A.P.; Misra, A. Prevalence and trends of metabolic syndrome among adults in the asia-pacific region: A systematic review. BMC Public Health 2017, 17, 101. [Google Scholar] [CrossRef]
- Rossnerova, A.; Tulupova, E.; Tabashidze, N.; Schmuczerova, J.; Dostal, M.; Rossner, P.; Gmuender, H.; Sram, R.J. Factors affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat. Res. 2013, 741–742, 18–26. [Google Scholar] [CrossRef]
- Li, H.; Chen, R.; Cai, J.; Cui, X.; Huang, N.; Kan, H. Short-term exposure to fine particulate air pollution and genome-wide DNA methylation: A randomized, double-blind, crossover trial. Environ. Int. 2018, 120, 130–136. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pollutant | Source of Pollutant | Impact on Endocrine System |
|---|---|---|
| Polychlorinated biphenyls (PCBs) | Industrial chemicals (mainly electrical products) [19,34] |
|
| Dichlorodiphenyltrichloroethane (DDT) | Pesticide Currently used to rid vectors of disease in parts of the world [30] |
|
| Polycyclic aromatic hydrocarbons (PAHs) | Synthetic chemical used in industries, food preparation and cigarettes [19,38] | |
| Bisphenol A (BPA) | Industrial chemical used for plastic and resin [19,38] |
|
| Phthalates | Industrial chemical used in plastics [19,38] |
|
| Polybrominated diphenyl ethers (PBDEs) | Class of fire-retardant chemicals [41] |
|
| Annex A: Elimination | Annex B: Restriction | Annex C: Unintentional Production |
|---|---|---|
| Aldrin Chlordane Chlordecone Decabromodiphenyl ether Dicofol Dieldrin Endrin Heptachlor Hexabromobiphenyl Hexabromocyclododecane Hexabromodiphenyl ether and heptabromodiphenyl ether Hexachlorobenzene Hexachlorobutadiene Alpha hexachlorocyclohexane Beta hexachlorocyclohexane Lindane Mirex Pentachlorobenzene Pentachlorophenol and its salts and esters Polychlorinated biphenyls Polychlorinated naphthalenes Perfluorooctanoic acid, its salts and PFOA-related compounds Short-chain chlorinated paraffins Technical endosulfan and its related isomers Tetrabromodiphenyl ether and pentabromodiphenyl ether Toxaphene | DDT Perfluorooctane sulfonic acid, its salts and perfluorooctane sulfonyl fluoride | Hexachlorobenzene Hexachlorobutadiene Pentachlorobenzene Polychlorinated biphenyls Polychlorinated dibenzo-p-dioxins Polychlorinated dibenzofurans Polychlorinated naphthalenes |
| Types of Compounds | Examples |
|---|---|
| Organochlorine compounds |
|
| Organophosphate compounds |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, W.J.; Akeblersane, M.; Khan, A.S.; Moin, A.S.M.; Butler, A.E. Environmental Pollution and the Risk of Developing Metabolic Disorders: Obesity and Diabetes. Int. J. Mol. Sci. 2023, 24, 8870. https://doi.org/10.3390/ijms24108870
Khalil WJ, Akeblersane M, Khan AS, Moin ASM, Butler AE. Environmental Pollution and the Risk of Developing Metabolic Disorders: Obesity and Diabetes. International Journal of Molecular Sciences. 2023; 24(10):8870. https://doi.org/10.3390/ijms24108870
Chicago/Turabian StyleKhalil, William Junior, Meriem Akeblersane, Ana Saad Khan, Abu Saleh Md Moin, and Alexandra E. Butler. 2023. "Environmental Pollution and the Risk of Developing Metabolic Disorders: Obesity and Diabetes" International Journal of Molecular Sciences 24, no. 10: 8870. https://doi.org/10.3390/ijms24108870
APA StyleKhalil, W. J., Akeblersane, M., Khan, A. S., Moin, A. S. M., & Butler, A. E. (2023). Environmental Pollution and the Risk of Developing Metabolic Disorders: Obesity and Diabetes. International Journal of Molecular Sciences, 24(10), 8870. https://doi.org/10.3390/ijms24108870