
The Role of IL-33/ST2 in COPD and Its Future as an Antibody Therapy

 , ,
, , 
Abstract
1. Introduction
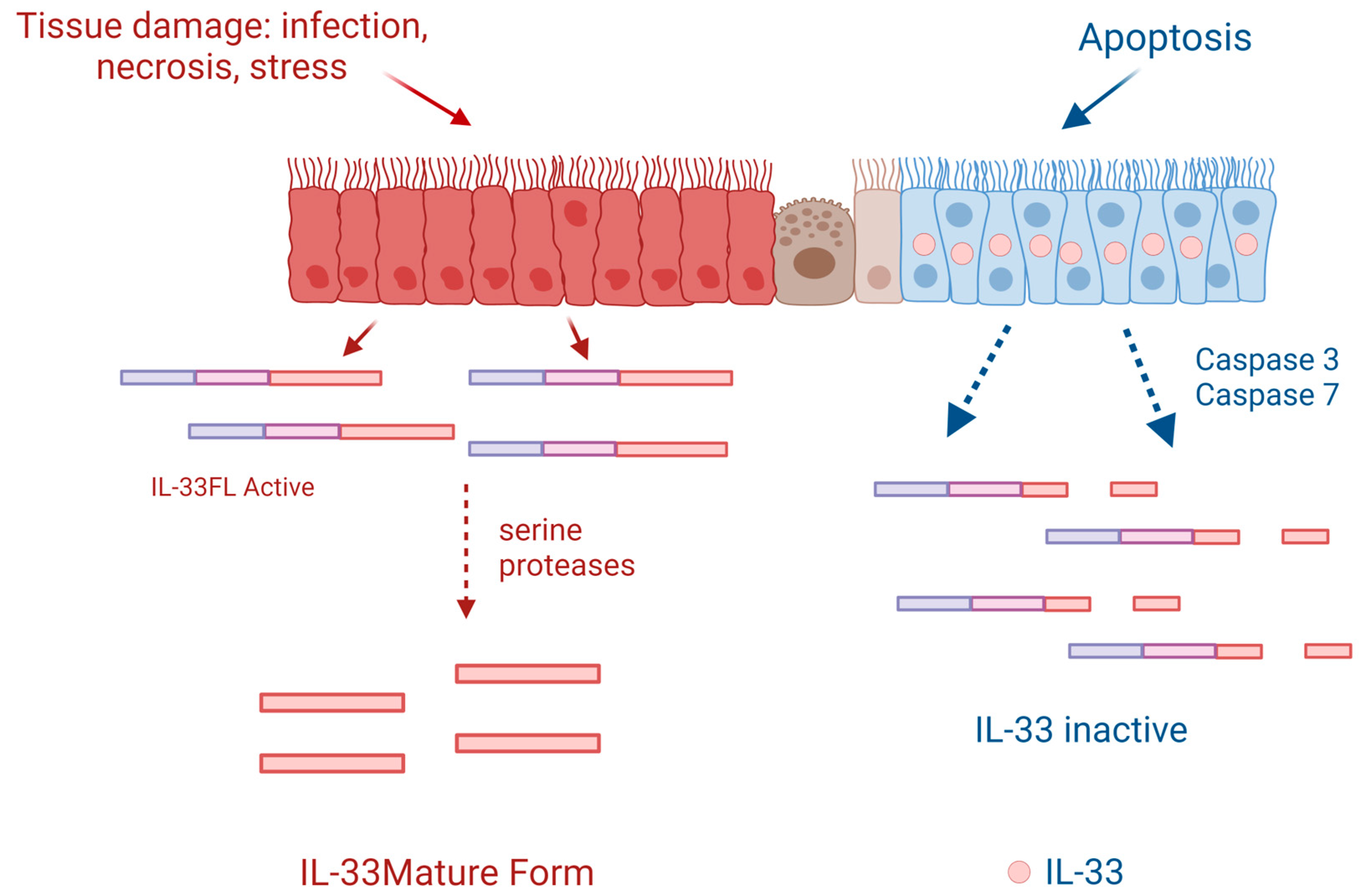
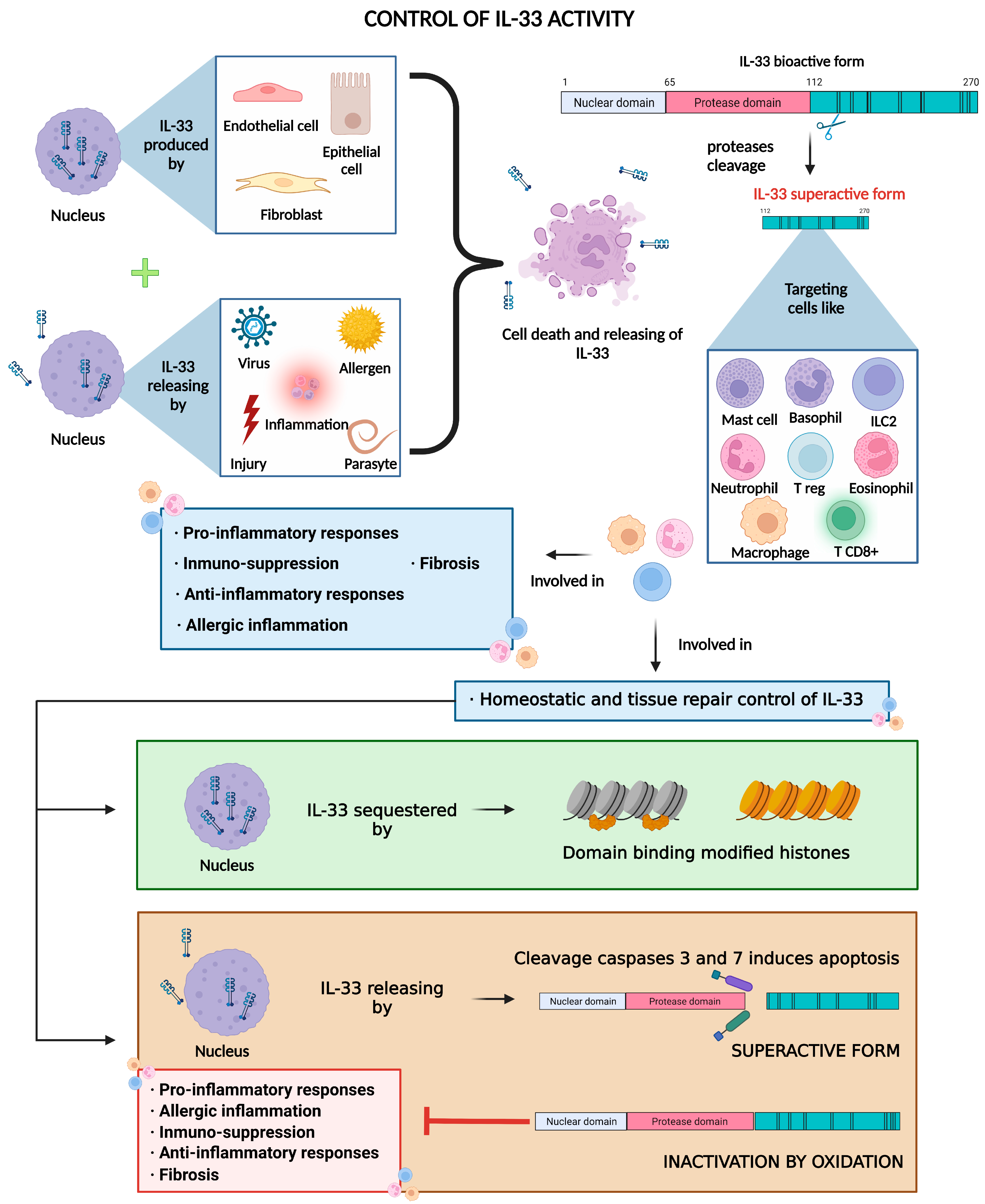
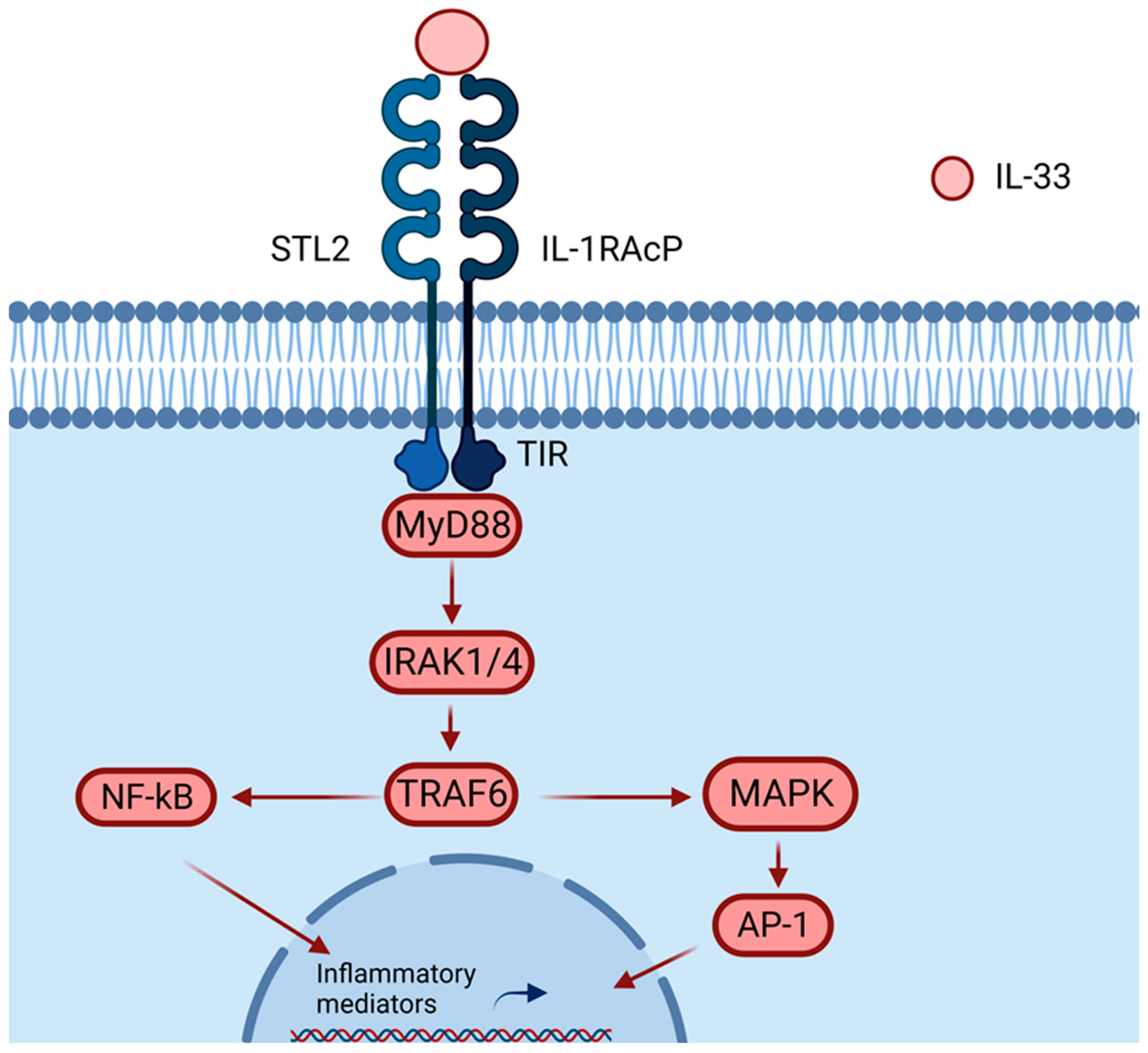
2. IL-33 and Inflammation
3. IL33 and the Lung
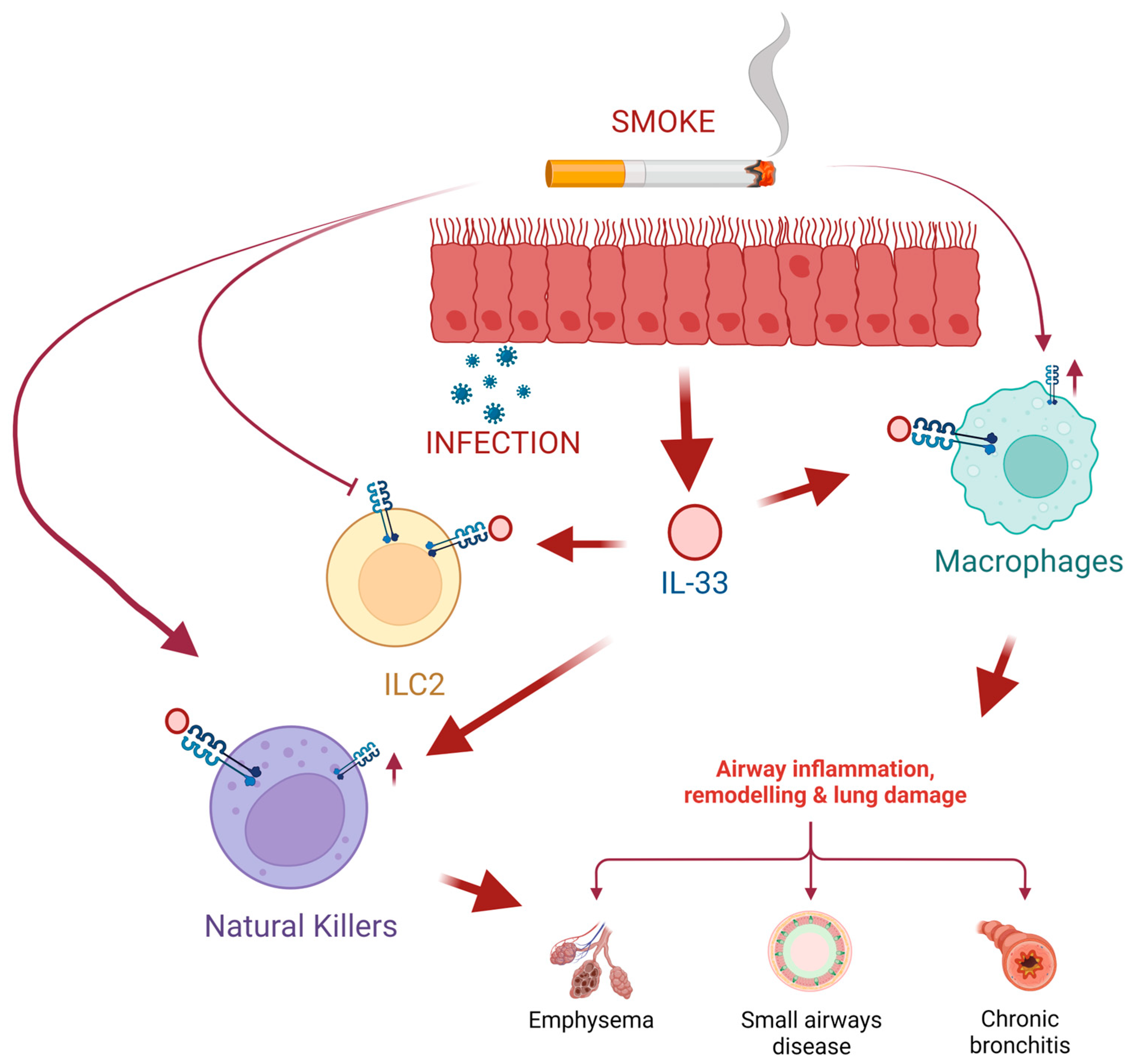
4. IL-33 in COPD
5. Targeting IL-33 in COPD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global Strategy for Prevention, Diagnosis and Management of Copd: 2021. Available online: https://goldcopd.org/2022-gold-reports-2/ (accessed on 23 March 2022).
- Soler-Cataluña, J.J.; Piñera, P.; Trigueros, J.A.; Calle, M.; Casanova, C.; Cosío, B.G.; López-Campos, J.L.; Molina, J.; Almagro, P.; Gómez, J.-T.; et al. [Translated Article] Spanish COPD Guidelines (GesEPOC) 2021 Update. Diagnosis and Treatment of COPD Exacerbation Syndrome. Arch. Bronconeumol. 2022, 58, T159–T170. [Google Scholar] [CrossRef]
- Diab, N.; Gershon, A.S.; Sin, D.D.; Tan, W.C.; Bourbeau, J.; Boulet, L.-P.; Aaron, S.D. Underdiagnosis and Overdiagnosis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2018, 198, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Zhang, C.-T.; Zhu, F.-Y.; Zheng, G.; Liu, Y.-F.; Liu, K.; Zhang, C.-H.; Zhang, H. Potential Natural Small Molecular Compounds for the Treatment of Chronic Obstructive Pulmonary Disease: An Overview. Front. Pharmacol. 2022, 13, 821941. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.M.; Tiew, P.Y.; Mac Aogáin, M.; Budden, K.F.; Yong, V.F.L.; Thomas, S.S.; Pethe, K.; Hansbro, P.M.; Chotirmall, S.H. The Role of Acute and Chronic Respiratory Colonization and Infections in the Pathogenesis of COPD. Respirology 2017, 22, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Szucs, B.; Szucs, C.; Petrekanits, M.; Varga, J.T. Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. Int. J. Mol. Sci. 2019, 20, 4329. [Google Scholar] [CrossRef]
- Hallstrand, T.S.; Hackett, T.L.; Altemeier, W.A.; Matute-Bello, G.; Hansbro, P.M.; Knight, D.A. Airway Epithelial Regulation of Pulmonary Immune Homeostasis and Inflammation. Clin. Immunol. 2014, 151, 1–15. [Google Scholar] [CrossRef]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and Immune Response in COPD: Where Do We Stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef] [PubMed]
- Bradford, E.; Jacobson, S.; Varasteh, J.; Comellas, A.P.; Woodruff, P.; O’Neal, W.; DeMeo, D.L.; Li, X.; Kim, V.; Cho, M.; et al. The Value of Blood Cytokines and Chemokines in Assessing COPD. Respir. Res. 2017, 18, 180. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A Critical Review of Its Biology and the Mechanisms Involved in Its Release as a Potent Extracellular Cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef]
- Guo, P.; Li, R.; Piao, T.H.; Wang, C.L.; Wu, X.L.; Cai, H.Y. Pathological Mechanism and Targeted Drugs of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2022, 17, 1565–1575. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. The Interleukin-1 Receptor/Toll-like Receptor Superfamily: 10 Years of Progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 Family: Regulators of Immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.; Arend, W.; Sims, J.; Smith, D.; Blumberg, H.; O’Neill, L.; Goldbach-Mansky, R.; Pizarro, T.; Hoffman, H.; Bufler, P.; et al. IL-1 Family Nomenclature. Nat. Immunol. 2010, 11, 973. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8. [Google Scholar] [CrossRef]
- Lefrançais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 Is Processed into Mature Bioactive Forms by Neutrophil Elastase and Cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef]
- Talabot-Ayer, D.; Lamacchia, C.; Gabay, C.; Palmer, G. Interleukin-33 Is Biologically Active Independently of Caspase-1 Cleavage. J. Biol. Chem. 2009, 284, 19420–19426. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A Nuclear Cytokine from the IL-1 Family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Lefrançais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.-P. Central Domain of IL-33 Is Cleaved by Mast Cell Proteases for Potent Activation of Group-2 Innate Lymphoid Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells in Vivo: A Novel “Alarmin”? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef]
- Hardman, C.S.; Panova, V.; McKenzie, A.N.J. IL-33 Citrine Reporter Mice Reveal the Temporal and Spatial Expression of IL-33 during Allergic Lung Inflammation. Eur. J. Immunol. 2013, 43, 488–498. [Google Scholar] [CrossRef]
- Mirchandani, A.S.; Salmond, R.J.; Liew, F.Y. Interleukin-33 and the Function of Innate Lymphoid Cells. Trends Immunol. 2012, 33, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Oboki, K.; Kajiwara, N.; Morii, E.; Aozasa, K.; Flavell, R.A.; Okumura, K.; Saito, H.; Nakae, S. Caspase-1, Caspase-8, and Calpain Are Dispensable for IL-33 Release by Macrophages. J. Immunol. 2009, 183, 7890–7897. [Google Scholar] [CrossRef] [PubMed]
- Sarrand, J.; Soyfoo, M. Involvement of IL-33 in the Pathophysiology of Systemic Lupus Erythematosus: Review. Int. J. Mol. Sci. 2022, 23, 3138. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, G. Role of IL-33 and Its Receptor in T Cell-Mediated Autoimmune Diseases. BioMed Res. Int. 2014, 2014, e587376. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Moritz, D.R.; Rodewald, H.R.; Gheyselinck, J.; Klemenz, R. The IL-1 Receptor-Related T1 Antigen Is Expressed on Immature and Mature Mast Cells and on Fetal Blood Mast Cell Progenitors. J. Immunol. 1998, 161, 4866–4874. [Google Scholar] [CrossRef]
- Löhning, M.; Stroehmann, A.; Coyle, A.J.; Grogan, J.L.; Lin, S.; Gutierrez-Ramos, J.C.; Levinson, D.; Radbruch, A.; Kamradt, T. T1/ST2 Is Preferentially Expressed on Murine Th2 Cells, Independent of Interleukin 4, Interleukin 5, and Interleukin 10, and Important for Th2 Effector Function. Proc. Natl. Acad. Sci. USA 1998, 95, 6930–6935. [Google Scholar] [CrossRef]
- Suzukawa, M.; Iikura, M.; Koketsu, R.; Nagase, H.; Tamura, C.; Komiya, A.; Nakae, S.; Matsushima, K.; Ohta, K.; Yamamoto, K.; et al. An IL-1 Cytokine Member, IL-33, Induces Human Basophil Activation via Its ST2 Receptor. J. Immunol. 2008, 181, 5981–5989. [Google Scholar] [CrossRef]
- Cherry, W.B.; Yoon, J.; Bartemes, K.R.; Iijima, K.; Kita, H. A Novel IL-1 Family Cytokine, IL-33, Potently Activates Human Eosinophils. J. Allergy Clin. Immunol. 2008, 121, 1484–1490. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in Health and Disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Tanabe, T.; Shimokawaji, T.; Kanoh, S.; Rubin, B.K. IL-33 Stimulates CXCL8/IL-8 Secretion in Goblet Cells but Not Normally Differentiated Airway Cells. Clin. Exp. Allergy 2014, 44, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Kawaguchi, M.; Kokubu, F.; Ohara, G.; Ota, K.; Huang, S.-K.; Morishima, Y.; Ishii, Y.; Satoh, H.; Sakamoto, T.; et al. Interleukin-33 Induces Interleukin-17F in Bronchial Epithelial Cells. Allergy 2012, 67, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 Receptor Accessory Protein and ST2 Comprise the IL-33 Receptor Complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Huber, M.; Kollewe, C.; Bischoff, S.C.; Falk, W.; Martin, M.U. IL-1 Receptor Accessory Protein Is Essential for IL-33-Induced Activation of T Lymphocytes and Mast Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18660–18665. [Google Scholar] [CrossRef]
- Palmer, G.; Lipsky, B.P.; Smithgall, M.D.; Meininger, D.; Siu, S.; Talabot-Ayer, D.; Gabay, C.; Smith, D.E. The IL-1 Receptor Accessory Protein (AcP) Is Required for IL-33 Signaling and Soluble AcP Enhances the Ability of Soluble ST2 to Inhibit IL-33. Cytokine 2008, 42, 358–364. [Google Scholar] [CrossRef]
- Miller, A.M. Role of IL-33 in Inflammation and Disease. J. Inflamm. 2011, 8, 22. [Google Scholar] [CrossRef]
- Li, Q.; Hu, Y.; Chen, Y.; Lv, Z.; Wang, J.; An, G.; Du, X.; Wang, H.; Corrigan, C.J.; Wang, W.; et al. IL-33 Induces Production of Autoantibody against Autologous Respiratory Epithelial Cells: A Potential Mechanism for the Pathogenesis of COPD. Immunology 2019, 157, 137–150. [Google Scholar] [CrossRef]
- Wang, H.-H.; Cheng, S.-L. From Biomarkers to Novel Therapeutic Approaches in Chronic Obstructive Pulmonary Disease. Biomedicines 2021, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Kobayashi, T.; Hara, K.; Kephart, G.M.; Ziegler, S.F.; McKenzie, A.N.; Kita, H. IL-33 and Thymic Stromal Lymphopoietin Mediate Immune Pathology in Response to Chronic Airborne Allergen Exposure. J. Immunol. 2014, 193, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Christianson, C.A.; Goplen, N.P.; Zafar, I.; Irvin, C.; Good, J.T.; Rollins, D.R.; Gorentla, B.; Liu, W.; Gorska, M.M.; Chu, H.; et al. Persistence of Asthma Requires Multiple Feedback Circuits Involving Type 2 Innate Lymphoid Cells and IL-33. J. Allergy Clin. Immunol. 2015, 136, 59–68.e14. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-Yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 Induces Airway Hyperresponsiveness and Goblet Cell Hyperplasia in the Lungs in the Absence of Adaptive Immune System. Int. Immunol. 2008, 20, 791–800. [Google Scholar] [CrossRef]
- Calderon, A.A.; Dimond, C.; Choy, D.F.; Pappu, R.; Grimbaldeston, M.A.; Mohan, D.; Chung, K.F. Targeting Interleukin-33 and Thymic Stromal Lymphopoietin Pathways for Novel Pulmonary Therapeutics in Asthma and COPD. Eur. Respir. Rev. 2023, 32, 220144. [Google Scholar] [CrossRef] [PubMed]
- Tworek, D.; Majewski, S.; Szewczyk, K.; Kiszałkiewicz, J.; Kurmanowska, Z.; Górski, P.; Brzeziańska-Lasota, E.; Kuna, P.; Antczak, A. The Association between Airway Eosinophilic Inflammation and IL-33 in Stable Non-Atopic COPD. Respir. Res. 2018, 19, 108. [Google Scholar] [CrossRef]
- IL-33 Promotes ST2-Dependent Lung Fibrosis by the Induction of Alternatively Activated Macrophages and Innate Lymphoid Cells in Mice—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/24985397/ (accessed on 2 February 2023).
- Cayrol, C. IL-33, an Alarmin of the IL-1 Family Involved in Allergic and Non Allergic Inflammation: Focus on the Mechanisms of Regulation of Its Activity. Cells 2021, 11, 107. [Google Scholar] [CrossRef]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate Lymphoid Cells Promote Lung-Tissue Homeostasis after Infection with Influenza Virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Li, D.; Guabiraba, R.; Besnard, A.-G.; Komai-Koma, M.; Jabir, M.S.; Zhang, L.; Graham, G.J.; Kurowska-Stolarska, M.; Liew, F.Y.; McSharry, C.; et al. IL-33 Promotes ST2-Dependent Lung Fibrosis by the Induction of Alternatively Activated Macrophages and Innate Lymphoid Cells in Mice. J. Allergy Clin. Immunol. 2014, 134, 1422–1432.e11. [Google Scholar] [CrossRef]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.-H.; Ward, C.K.; Criner, G.J.; et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef]
- Saglani, S.; Lui, S.; Ullmann, N.; Campbell, G.A.; Sherburn, R.T.; Mathie, S.A.; Denney, L.; Bossley, C.J.; Oates, T.; Walker, S.A.; et al. IL-33 Promotes Airway Remodeling in Pediatric Patients with Severe Steroid-Resistant Asthma. J. Allergy Clin. Immunol. 2013, 132, 676–685.e13. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhao, J.; Shang, J.; Li, M.; Zeng, Z.; Zhao, J.; Wang, J.; Xu, Y.; Xie, J. Increased IL-33 Expression in Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L619–L627. [Google Scholar] [CrossRef] [PubMed]
- Gabryelska, A.; Kuna, P.; Antczak, A.; Białasiewicz, P.; Panek, M. IL-33 Mediated Inflammation in Chronic Respiratory Diseases-Understanding the Role of the Member of IL-1 Superfamily. Front. Immunol. 2019, 10, 692. [Google Scholar] [CrossRef] [PubMed]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.-P.; et al. Long-Term IL-33-Producing Epithelial Progenitor Cells in Chronic Obstructive Lung Disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Choi, H.-J.; Min, J.-K.; Pyun, B.-J.; Maeng, Y.-S.; Park, H.; Kim, J.; Kim, Y.-M.; Kwon, Y.-G. Interleukin-33 Induces Angiogenesis and Vascular Permeability through ST2/TRAF6-Mediated Endothelial Nitric Oxide Production. Blood 2009, 114, 3117–3126. [Google Scholar] [CrossRef]
- Cerón-Pisa, N.; Shafiek, H.; Martín-Medina, A.; Verdú, J.; Jordana-Lluch, E.; Escobar-Salom, M.; Barceló, I.M.; López-Causapé, C.; Oliver, A.; Juan, C.; et al. Effects of Inhaled Corticosteroids on the Innate Immunological Response to Pseudomonas Aeruginosa Infection in Patients with COPD. Int. J. Mol. Sci. 2022, 23, 8127. [Google Scholar] [CrossRef]
- Lange, P.; Ahmed, E.; Lahmar, Z.M.; Martinez, F.J.; Bourdin, A. Natural History and Mechanisms of COPD. Respirology 2021, 26, 298–321. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of Inflammatory Cells in Airway Remodeling in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef]
- Shang, J.; Zhao, J.; Wu, X.; Xu, Y.; Xie, J.; Zhao, J. Interleukin-33 Promotes Inflammatory Cytokine Production in Chronic Airway Inflammation. Biochem. Cell Biol. 2015, 93, 359–366. [Google Scholar] [CrossRef]
- Tang, Y.; Guan, Y.; Liu, Y.; Sun, J.; Xu, L.; Jiang, Y. The Role of the Serum IL-33/SST2 Axis and Inflammatory Cytokines in Chronic Obstructive Pulmonary Disease. J. Interferon Cytokine Res. 2014, 34, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.K.; Hanania, N.A. Targeting IL-5 in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1045–1051. [Google Scholar] [CrossRef]
- Wu, H.; Yang, S.; Wu, X.; Zhao, J.; Zhao, J.; Ning, Q.; Xu, Y.; Xie, J. Interleukin-33/ST2 Signaling Promotes Production of Interleukin-6 and Interleukin-8 in Systemic Inflammation in Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease Mice. Biochem. Biophys. Res. Commun. 2014, 450, 110–116. [Google Scholar] [CrossRef] [PubMed]
- De Falco, G.; Colarusso, C.; Terlizzi, M.; Popolo, A.; Pecoraro, M.; Commodo, M.; Minutolo, P.; Sirignano, M.; D’Anna, A.; Aquino, R.P.; et al. Chronic Obstructive Pulmonary Disease-Derived Circulating Cells Release IL-18 and IL-33 under Ultrafine Particulate Matter Exposure in a Caspase-1/8-Independent Manner. Front. Immunol. 2017, 8, 1415. [Google Scholar] [CrossRef]
- Huang, Q.; Li, C.D.; Yang, Y.R.; Qin, X.F.; Wang, J.J.; Zhang, X.; Du, X.N.; Yang, X.; Wang, Y.; Li, L.; et al. Role of the IL-33/ST2 Axis in Cigarette Smoke-Induced Airways Remodelling in Chronic Obstructive Pulmonary Disease. Thorax 2021, 76, 750–762. [Google Scholar] [CrossRef]
- Joo, H.; Park, S.J.; Min, K.H.; Rhee, C.K. Association between Plasma Interleukin-33 Level and Acute Exacerbation of Chronic Obstructive Pulmonary Disease. BMC Pulm. Med. 2021, 21, 86. [Google Scholar] [CrossRef]
- Hoenderdos, K.; Condliffe, A. The Neutrophil in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef]
- Zhao, J.; Wei, J.; Mialki, R.K.; Mallampalli, D.F.; Chen, B.B.; Coon, T.; Zou, C.; Mallampalli, R.K.; Zhao, Y. F-Box Protein FBXL19-Mediated Ubiquitination and Degradation of the Receptor for IL-33 Limits Pulmonary Inflammation. Nat. Immunol. 2012, 13, 651–658. [Google Scholar] [CrossRef]
- Bhowmik, A.; Seemungal, T.A.; Sapsford, R.J.; Wedzicha, J.A. Relation of Sputum Inflammatory Markers to Symptoms and Lung Function Changes in COPD Exacerbations. Thorax 2000, 55, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Bucchioni, E.; Kharitonov, S.A.; Allegra, L.; Barnes, P.J. High Levels of Interleukin-6 in the Exhaled Breath Condensate of Patients with COPD. Respir. Med. 2003, 97, 1299–1302. [Google Scholar] [CrossRef] [PubMed]
- Hacievliyagil, S.S.; Gunen, H.; Mutlu, L.C.; Karabulut, A.B.; Temel, I. Association between Cytokines in Induced Sputum and Severity of Chronic Obstructive Pulmonary Disease. Respir. Med. 2006, 100, 846–854. [Google Scholar] [CrossRef] [PubMed]
- IL-33 in COPD: The Hunt for Responder Subgroups—ClinicalKey. Available online: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S2213260022000054?returnurl=https:%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS2213260022000054%3Fshowall%3Dtrue&referrer= (accessed on 2 February 2023).
- Walzl, G.; Matthews, S.; Kendall, S.; Gutierrez-Ramos, J.C.; Coyle, A.J.; Openshaw, P.J.; Hussell, T. Inhibition of T1/ST2 during Respiratory Syncytial Virus Infection Prevents T Helper Cell Type 2 (Th2)- but Not Th1-Driven Immunopathology. J. Exp. Med. 2001, 193, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Kim, H.Y.; Albacker, L.A.; Baumgarth, N.; McKenzie, A.N.J.; Smith, D.E.; Dekruyff, R.H.; Umetsu, D.T. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat. Immunol. 2011, 12, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Hansbro, P.M.; Kaiko, G.E.; Foster, P.S. Cytokine/Anti-Cytokine Therapy—Novel Treatments for Asthma? Br. J. Pharmacol. 2011, 163, 81–95. [Google Scholar] [CrossRef]
- Hansbro, P.M.; Scott, G.V.; Essilfie, A.-T.; Kim, R.Y.; Starkey, M.R.; Nguyen, D.H.; Allen, P.D.; Kaiko, G.E.; Yang, M.; Horvat, J.C.; et al. Th2 Cytokine Antagonists: Potential Treatments for Severe Asthma. Expert Opin. Investig. Drugs 2013, 22, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Hansbro, P.M.; Kim, R.Y.; Starkey, M.R.; Donovan, C.; Dua, K.; Mayall, J.R.; Liu, G.; Hansbro, N.G.; Simpson, J.L.; Wood, L.G.; et al. Mechanisms and Treatments for Severe, Steroid-Resistant Allergic Airway Disease and Asthma. Immunol. Rev. 2017, 278, 41–62. [Google Scholar] [CrossRef]
- Kosloski, M.P.; Kalliolias, G.D.; Xu, C.R.; Harel, S.; Lai, C.-H.; Zheng, W.; Davis, J.D.; Kamal, M.A. Pharmacokinetics and Pharmacodynamics of Itepekimab in Healthy Adults and Patients with Asthma: Phase I First-in-Human and First-in-Patient Trials. Clin. Transl. Sci. 2022, 15, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Allinne, J.; Scott, G.; Lim, W.K.; Birchard, D.; Erjefält, J.S.; Sandén, C.; Ben, L.-H.; Agrawal, A.; Kaur, N.; Kim, J.H.; et al. IL-33 Blockade Affects Mediators of Persistence and Exacerbation in a Model of Chronic Airway Inflammation. J. Allergy Clin. Immunol. 2019, 144, 1624–1637.e10. [Google Scholar] [CrossRef]
- Rabe, K.F.; Celli, B.R.; Wechsler, M.E.; Abdulai, R.M.; Luo, X.; Boomsma, M.M.; Staudinger, H.; Horowitz, J.E.; Baras, A.; Ferreira, M.A.; et al. Safety and Efficacy of Itepekimab in Patients with Moderate-to-Severe COPD: A Genetic Association Study and Randomised, Double-Blind, Phase 2a Trial. Lancet Respir. Med. 2021, 9, 1288–1298. [Google Scholar] [CrossRef]
- Sanofi. A Phase 2a, Open-Label, Two-Part Study to Evaluate the Mechanism of Action of Itepekimab (Anti-IL-33 MAb) on Airway Inflammation in Patients with Chronic Obstructive Pulmonary Disease (COPD); Sanofi: Paris, France, 2023. [Google Scholar]
- Donovan, C.; Hansbro, P.M. IL-33 in Chronic Respiratory Disease: From Preclinical to Clinical Studies. ACS Pharmacol. Transl. Sci. 2020, 3, 56–62. [Google Scholar] [CrossRef]
- Hoffmann-La Roche. A Phase III, Randomized, Double-Blind, Placebo-Controlled, Multicenter Study to Evaluate the Efficacy and Safety of Astegolimab in Patients with Chronic Obstructive Pulmonary Disease; Hoffmann-La Roche: Basel, Switzerland, 2023. [Google Scholar]
- Genentech, Inc. A Phase IIb, Randomized, Double-Blind, Placebo-Controlled, Multicenter Study to Evaluate the Efficacy and Safety of Astegolimab in Patients with Chronic Obstructive Pulmonary Disease; Genentech, Inc.: South San Francisco, CA, USA, 2023. [Google Scholar]
- Scott, I.C.; England, E.; Rees, D.G.; Erngren, T.; Huntington, C.C.; Houslay, K.F.; Sims, D.A.; Hollins, C.; Hinchy, E.C.; Colley, C.; et al. Tozorakimab: A Dual-Pharmacology Anti-IL-33 Antibody That Inhibits IL-33 Signalling via ST2 and RAGE/EGFR to Reduce Inflammation and Epithelial Dysfunction. Eur. Respir. J. 2022, 60, 2467. [Google Scholar] [CrossRef]
- MedImmune LLC. Safety, Tolerability, Pharmacokinetics and Immunogenicity of MEDI3506 Administered as Single Ascending Doses in Healthy Adult Subjects, as Multiple Ascending Doses in COPD Subjects and Single Dose in Healthy Japanese Subjects; MedImmune LLC: Gaithersburg, MD, USA, 2020. [Google Scholar]
- AstraZeneca. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy, Safety and Tolerability of MEDI3506 in Participants with Moderate to Severe Chronic Obstructive Pulmonary Disease and Chronic Bronchitis (FRONTIER 4); AstraZeneca: Cambridge, UK, 2023. [Google Scholar]
- AstraZeneca. A Phase III, Multicentre, Randomised, Double-Blind, Chronic-Dosing, Parallel-Group, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Two Dose Regimens of Tozorakimab in Participants with Symptomatic Chronic Obstructive Pulmonary Disease (COPD) with a History of COPD Exacerbations (OBERON); AstraZeneca: Cambridge, UK, 2023. [Google Scholar]
- AstraZeneca. A Phase III, Multicentre, Randomised, Double-Blind, Chronic-Dosing, Parallel-Group, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Two Dose Regimens of Tozorakimab in Participants with Symptomatic Chronic Obstructive Pulmonary Disease (COPD) with a History of COPD Exacerbations (TITANIA); AstraZeneca: Cambridge, UK, 2023. [Google Scholar]
- AstraZeneca. A Phase III, Multicentre, Randomised, Double-Blind, Chronic-Dosing, Parallel-Group, Placebo-Controlled Extension Study to Evaluate the Long-Term Efficacy and Safety of Tozorakimab (MEDI3506) in Participants with Chronic Obstructive Pulmonary Disease (COPD) with a History of Exacerbations; AstraZeneca: Cambridge, UK, 2023. [Google Scholar]
- Yousuf, A.J.; Mohammed, S.; Carr, L.; Yavari Ramsheh, M.; Micieli, C.; Mistry, V.; Haldar, K.; Wright, A.; Novotny, P.; Parker, S.; et al. Astegolimab, an Anti-ST2, in Chronic Obstructive Pulmonary Disease (COPD-ST2OP): A Phase 2a, Placebo-Controlled Trial. Lancet Respir. Med. 2022, 10, 469–477. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Phase | Population | ClinicalTrials.gov |
|---|---|---|---|
| Anti-IL33 SAR440340 | Phase 2 | Moderate-to-severe acute exacerbations of Chronic Obstructive Pulmonary Disease | NCT03546907 |
| Anti-IL33 Itepekimab | Phase 2a | Chronic Obstructive Pulmonary Disease | NCT05326412 |
| Anti-IL33 Itepekimab/SAR440340 | Phase 3 | Chronic Obstructive Pulmonary Disease | NCT04701983 |
| SAR440340/REGN3500/Itepekimab | Phase 3 | Chronic Obstructive Pulmonary Disease | NCT04751487 |
| Anti-ST2 Astegolimab | Phase 2a | Moderate-to-very severe Chronic Obstructive Pulmonary Disease | NCT03615040 |
| Anti-ST2 Astegolimab | Phase 2b | Chronic Obstructive Pulmonary Disease | NCT05595642 |
| Anti-ST2 Astegolimab | Phase 2 | Chronic Obstructive Pulmonary Disease | NCT05037929 |
| MEDI-3506/Tozorakimab | Phase 1 | Chronic Obstructive Pulmonary Disease and Healthy Japanese Participants | NCT03096795 |
| MEDI-3506/Tozorakimab | Phase 2 | Chronic Obstructive Pulmonary Disease and Chronic Bronchitis. | NCT04631016 |
| Tozorakimab | Phase 3 | Chronic Obstructive Pulmonary Disease | NCT05166889 |
| Tozorakimab | Phase 3 | Chronic Obstructive Pulmonary Disease | NCT05158387 |
| Tozorakimab | Phase 3 | Chronic Obstructive Pulmonary Disease | NCT05742802 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riera-Martínez, L.; Cànaves-Gómez, L.; Iglesias, A.; Martin-Medina, A.; Cosío, B.G. The Role of IL-33/ST2 in COPD and Its Future as an Antibody Therapy. Int. J. Mol. Sci. 2023, 24, 8702. https://doi.org/10.3390/ijms24108702
Riera-Martínez L, Cànaves-Gómez L, Iglesias A, Martin-Medina A, Cosío BG. The Role of IL-33/ST2 in COPD and Its Future as an Antibody Therapy. International Journal of Molecular Sciences. 2023; 24(10):8702. https://doi.org/10.3390/ijms24108702
Chicago/Turabian StyleRiera-Martínez, Lluc, Laura Cànaves-Gómez, Amanda Iglesias, Aina Martin-Medina, and Borja G. Cosío. 2023. "The Role of IL-33/ST2 in COPD and Its Future as an Antibody Therapy" International Journal of Molecular Sciences 24, no. 10: 8702. https://doi.org/10.3390/ijms24108702
APA StyleRiera-Martínez, L., Cànaves-Gómez, L., Iglesias, A., Martin-Medina, A., & Cosío, B. G. (2023). The Role of IL-33/ST2 in COPD and Its Future as an Antibody Therapy. International Journal of Molecular Sciences, 24(10), 8702. https://doi.org/10.3390/ijms24108702