
Multiscale Model of CVD Growth of Graphene on Cu(111) Surface

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
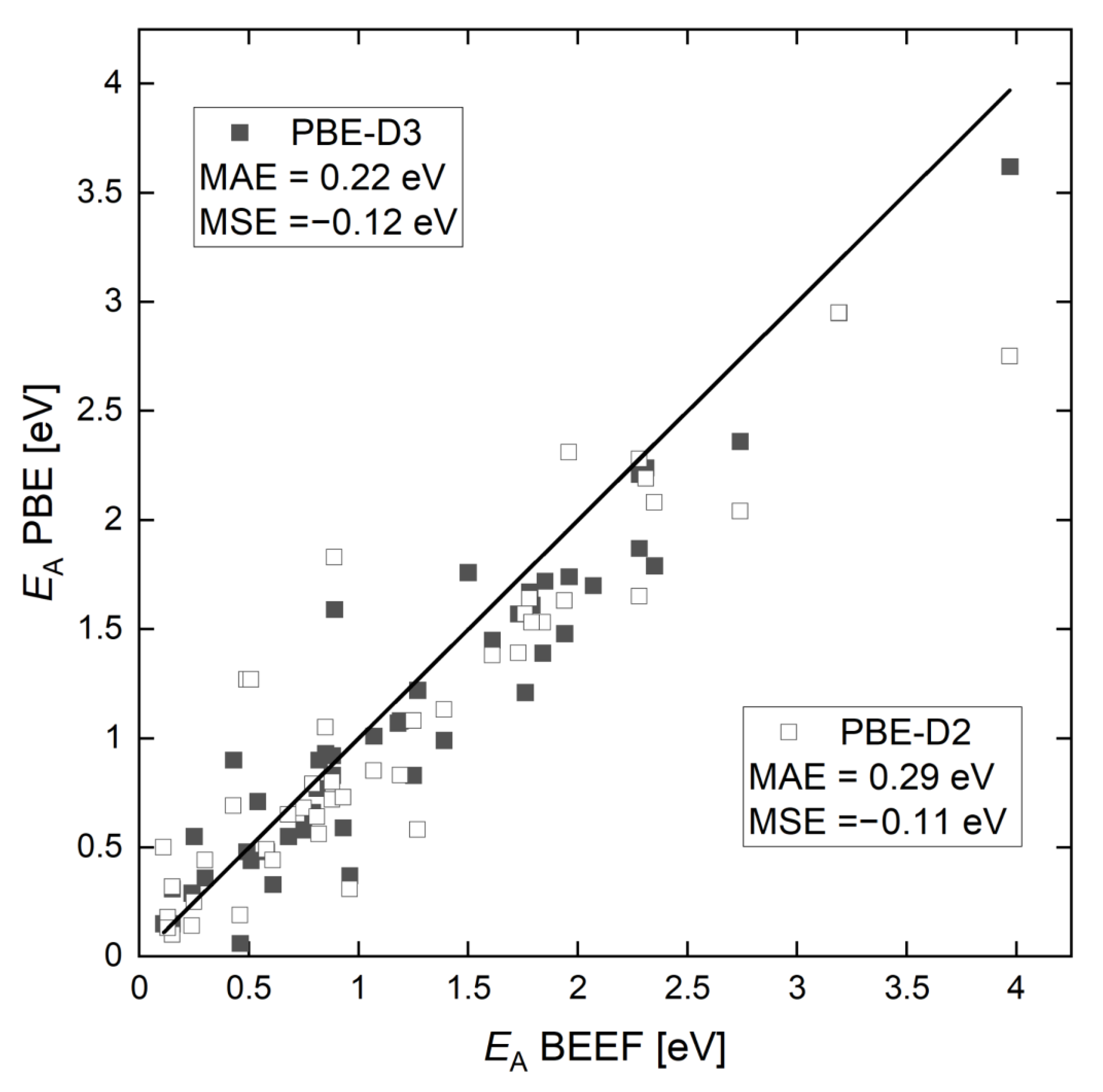
2.1. Activation Energy Barriers
2.2. Modulation of Graphene Growth by CH Partial Pressure
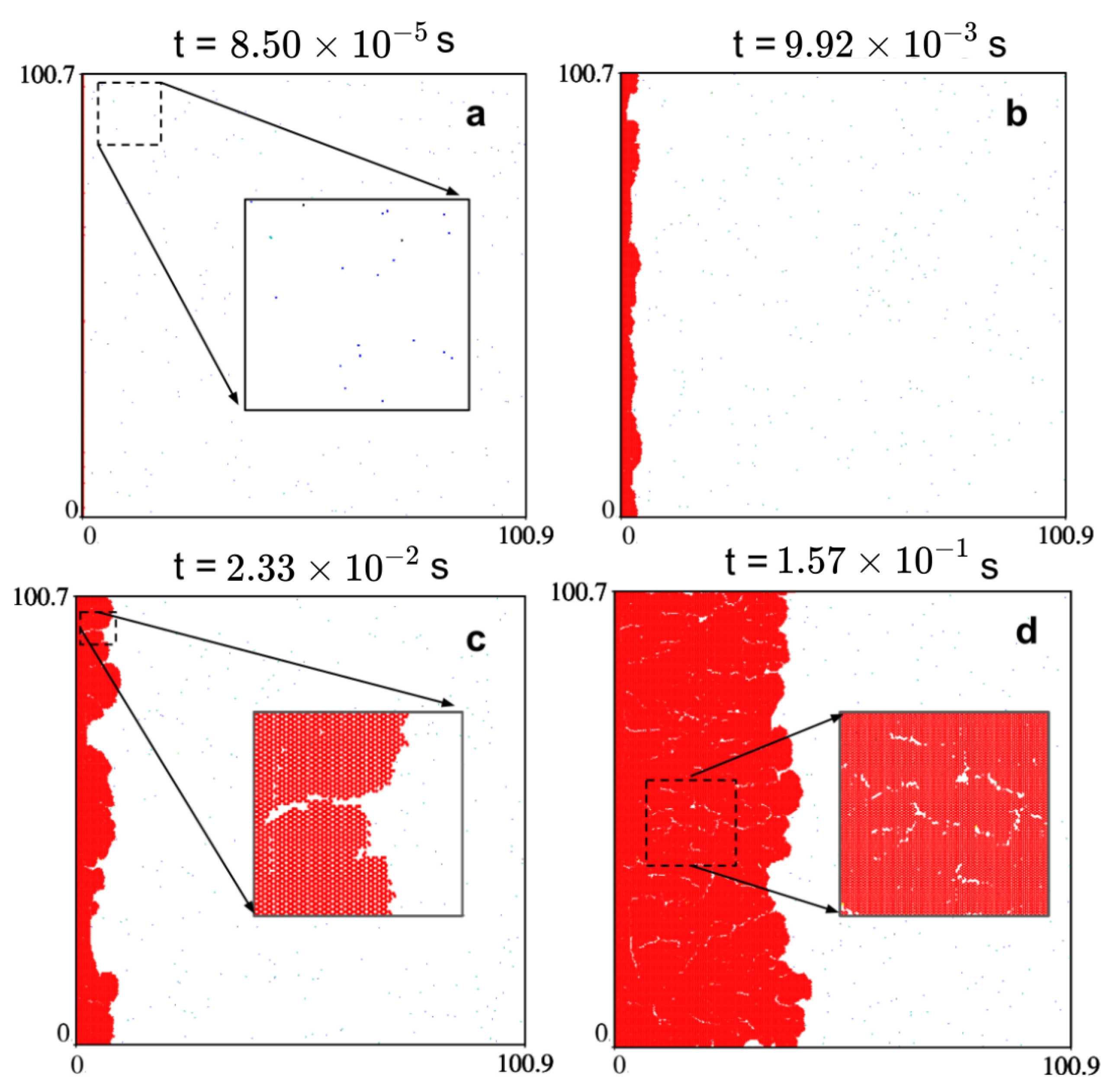
2.3. The Growth Mechanism
2.4. Hydrogenation Reactions
3. Materials and Methods
3.1. DFT Calculations and Reaction Rates
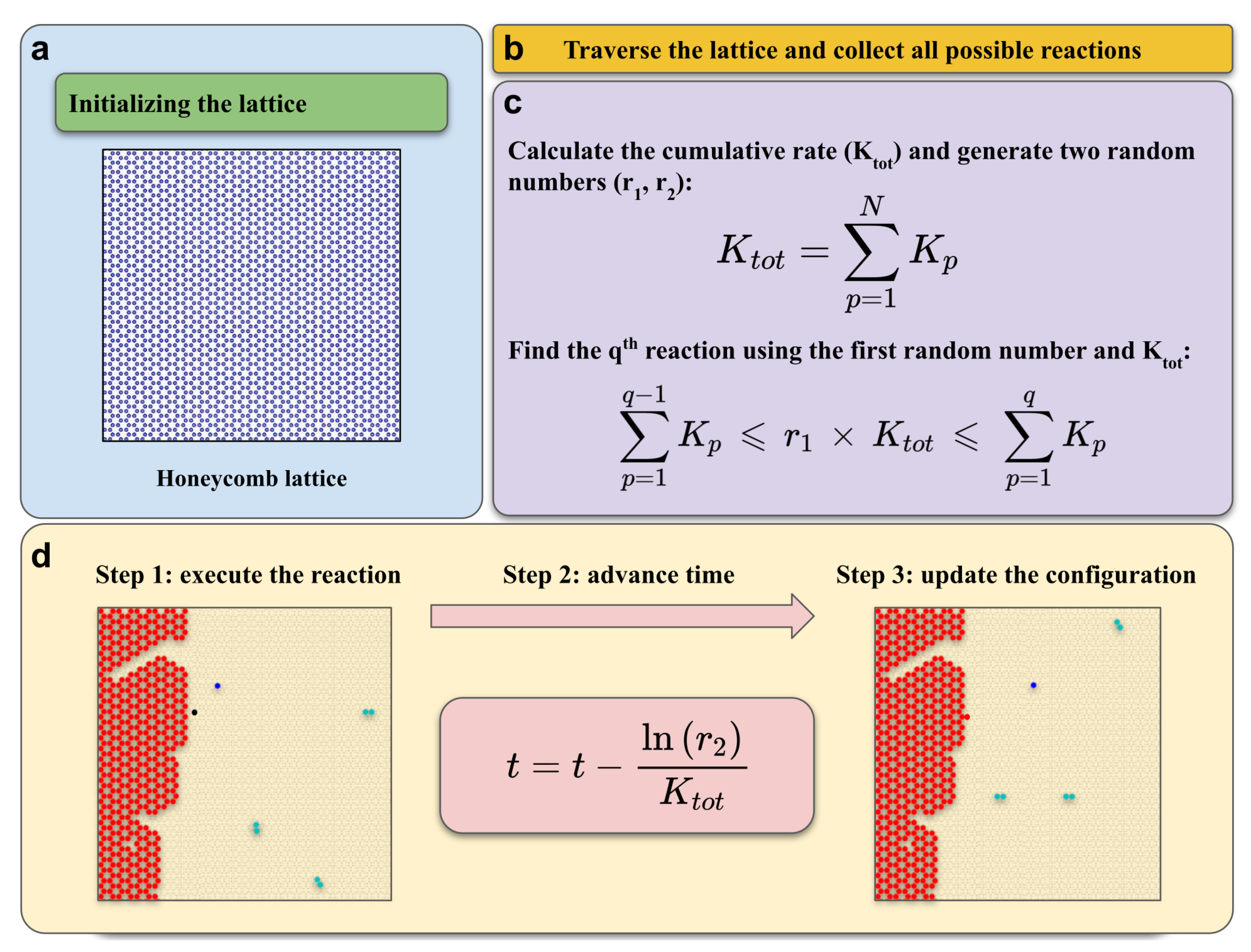
3.2. KMC Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-Dimensional |
| CVD | Chemical Vapor Deposition |
| PECVD | Plasma-Enhanced Chemical Vapor Deposition |
| KMC | Kinetic Monte Carlo |
| DFT | Density Functional Theory |
| VASP | Vienna Ab Initio Simulation Package |
| BKL | Bortz–Kalos–Lebowitz algorithm |
| PBE-D3 | Perdew–Burke–Ernzerhof Functional with Grimme D3 Dispersion Correction |
| BEEF-vdW | Bayesian Error Estimation Functional with van der Waals Correlation |
| PAW | Projector-Augmented Wave Method |
| NEB | Nudge Elastic Band Method |
| TST | Transition State Theory |
| MAE | Mean Absolute Error |
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, K.; Sikes, K.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.R.; Geim, A.K. Fine Structure Constant Defines Visual Transparency of Graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef]
- Lin, Y.M.; Jenkins, K.A.; Valdes-Garcia, A.; Small, J.P.; Farmer, D.B.; Avouris, P. Operation of graphene transistors at gigahertz frequencies. Nano Lett. 2009, 9, 422–426. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, Y.m.; Bol, A.A.; Jenkins, K.A.; Xia, F.; Farmer, D.B.; Zhu, Y.; Avouris, P. High-frequency, scaled graphene transistors on diamond-like carbon. Nature 2011, 472, 74–78. [Google Scholar] [CrossRef]
- Lin, Y.M.; Dimitrakopoulos, C.; Jenkins, K.A.; Farmer, D.B.; Chiu, H.Y.; Grill, A.; Avouris, P. 100-GHz transistors from wafer-scale epitaxial graphene. Science 2010, 327, 662. [Google Scholar] [CrossRef]
- Jang, H.; Park, Y.J.; Chen, X.; Das, T.; Kim, M.S.; Ahn, J.H. Graphene-based flexible and stretchable electronics. Adv. Mater. 2016, 28, 4184–4202. [Google Scholar] [CrossRef]
- Kim, K.S.; Zhao, Y.; Jang, H.; Lee, S.Y.; Kim, J.M.; Kim, K.S.; Ahn, J.H.; Kim, P.; Choi, J.Y.; Hong, B.H. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 2009, 457, 706–710. [Google Scholar] [CrossRef]
- Furchi, M.; Urich, A.; Pospischil, A.; Lilley, G.; Unterrainer, K.; Detz, H.; Klang, P.; Andrews, A.M.; Schrenk, W.; Strasser, G.; et al. Microcavity-integrated graphene photodetector. Nano Lett. 2012, 12, 2773–2777. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Shiue, R.J.; Gao, Y.; Meric, I.; Heinz, T.F.; Shepard, K.; Hone, J.; Assefa, S.; Englund, D. Chip-integrated ultrafast graphene photodetector with high responsivity. Nat. Photonics 2013, 7, 883–887. [Google Scholar] [CrossRef]
- Kim, H.; Park, K.Y.; Hong, J.; Kang, K. All-graphene-battery: Bridging the gap between supercapacitors and lithium ion batteries. Sci. Rep. 2014, 4, 5278. [Google Scholar] [CrossRef] [PubMed]
- El-Kady, M.F.; Strong, V.; Dubin, S.; Kaner, R.B. Laser scribing of high-performance and flexible graphene-based electrochemical capacitors. Science 2012, 335, 1326–1330. [Google Scholar] [CrossRef]
- Robinson, J.T.; Perkins, F.K.; Snow, E.S.; Wei, Z.; Sheehan, P.E. Reduced graphene oxide molecular sensors. Nano Lett. 2008, 8, 3137–3140. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.D.; Allen, M.J.; Tung, V.C.; Yang, Y.; Kaner, R.B.; Weiller, B.H. Practical Chemical Sensors from Chemically Derived Graphene. ACS Nano 2009, 3, 301–306. [Google Scholar] [CrossRef]
- Traversi, F.; Raillon, C.; Benameur, S.; Liu, K.; Khlybov, S.; Tosun, M.; Krasnozhon, D.; Kis, A.; Radenovic, A. Detecting the translocation of DNA through a nanopore using graphene nanoribbons. Nat. Nanotechnol. 2013, 8, 939–945. [Google Scholar] [CrossRef]
- Schneider, G.F.; Kowalczyk, S.W.; Calado, V.E.; Pandraud, G.; Zandbergen, H.W.; Vandersypen, L.M.; Dekker, C. DNA translocation through graphene nanopores. Nano Lett. 2010, 10, 3163–3167. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cruz, A.; Ruiz-Hernández, A.R.; Vega-Clemente, J.F.; Luna-Gazcón, D.G.; Campos-Delgado, J. A review of top-down and bottom-up synthesis methods for the production of graphene, graphene oxide and reduced graphene oxide. J. Mater. Sci. 2022, 57, 14543–14578. [Google Scholar] [CrossRef]
- Olatomiwa, A.L.; Adam, T.; Gopinath, S.C.B.; Kolawole, S.Y.; Olayinka, O.H.; Hashim, U. Graphene synthesis, fabrication, characterization based on bottom-up and top-down approaches: An overview. J. Semicond. 2022, 43, 061101. [Google Scholar] [CrossRef]
- Zhang, Z.; Fraser, A.; Ye, S.; Merle, G.; Barralet, J. Top-down bottom-up graphene synthesis. Nano Futur. 2019, 3, 042003. [Google Scholar] [CrossRef]
- Yoon, K.Y.; Dong, G. Liquid-phase bottom-up synthesis of graphene nanoribbons. Mater. Chem. Front. 2020, 4, 29–45. [Google Scholar] [CrossRef]
- Jiang, L.; Niu, T.; Lu, X.; Dong, H.; Chen, W.; Liu, Y.; Hu, W.; Zhu, D. Low-temperature, bottom-up synthesis of graphene via a radical-coupling reaction. J. Am. Chem. Soc. 2013, 135, 9050–9054. [Google Scholar] [CrossRef]
- Kumar, N.; Salehiyan, R.; Chauke, V.; Botlhoko, O.J.; Setshedi, K.; Scriba, M.; Masukume, M.; Ray, S.S. Top-down synthesis of graphene: A comprehensive review. FlatChem 2021, 27, 100224. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.; Salvatierra, R.V.; McHugh, E.; Tour, J.M. Top-down synthesis of graphene nanoribbons using different sources of carbon nanotubes. Carbon 2020, 158, 615–623. [Google Scholar] [CrossRef]
- Muñoz, R.; Gómez-Aleixandre, C. Review of CVD Synthesis of Graphene. Chem. Vap. Depos. 2013, 19, 297–322. [Google Scholar] [CrossRef]
- Moreau, E.; Godey, S.; Ferrer, F.; Vignaud, D.; Wallart, X.; Avila, J.; Asensio, M.; Bournel, F.; Gallet, J.J. Graphene growth by molecular beam epitaxy on the carbon-face of SiC. Appl. Phys. Lett. 2010, 97, 241907. [Google Scholar] [CrossRef]
- Eigler, S.; Enzelberger-Heim, M.; Grimm, S.; Hofmann, P.; Kroener, W.; Geworski, A.; Dotzer, C.; Röckert, M.; Xiao, J.; Papp, C.; et al. Wet chemical synthesis of graphene. Adv. Mater. 2013, 25, 3583–3587. [Google Scholar] [CrossRef]
- Bhuyan, M.S.A.; Uddin, M.N.; Islam, M.M.; Bipasha, F.A.; Hossain, S.S. Synthesis of graphene. Int. Nano Lett. 2016, 6, 65–83. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; Huang, Y.; Ma, Y.; Wang, Y.; Chen, Y. Size-controlled synthesis of graphene oxide sheets on a large scale using chemical exfoliation. Carbon 2009, 47, 3365–3368. [Google Scholar] [CrossRef]
- Shah, J.; Lopez-Mercado, J.; Carreon, M.G.; Lopez-Miranda, A.; Carreon, M.L. Plasma synthesis of graphene from mango peel. ACS Omega 2018, 3, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Mertens, R. General Graphene Launched Its New High-Capacity CVD Graphene Roll-to-Roll Production Line. 2021. Available online: https://www.graphene-info.com/general-graphene-launched-its-new-high-capacity-cvd-graphene-roll-roll (accessed on 21 February 2022).
- Lee, B.; Chu, W.; Li, W. Effects of Process Parameters on Graphene Growth Via Low-Pressure Chemical Vapor Deposition. J. Micro-Nano-Manuf. 2020, 8, 031005. [Google Scholar] [CrossRef]
- Ramli, N.; Nayan, N.A.; Lee, H.W.; Embong, S.S. Analysis of the effect of growth parameters on graphene synthesized by chemical vapor deposition. J. Nanoelectron. Optoelectron. 2015, 10, 50–55. [Google Scholar] [CrossRef]
- Li, X.; Magnuson, C.W.; Venugopal, A.; Tromp, R.M.; Hannon, J.B.; Vogel, E.M.; Colombo, L.; Ruoff, R.S. Large-Area Graphene Single Crystals Grown by Low-Pressure Chemical Vapor Deposition of Methane on Copper. J. Am. Chem. Soc. 2011, 133, 2816–2819. [Google Scholar] [CrossRef] [PubMed]
- Bhaviripudi, S.; Jia, X.; Dresselhaus, M.S.; Kong, J. Role of Kinetic Factors in Chemical Vapor Deposition Synthesis of Uniform Large Area Graphene Using Copper Catalyst. Nano Lett. 2010, 10, 4128–4133. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Lu, Y.; Singer, D.W.; Berck, M.E.; Somers, L.A.; Goldsmith, B.R.; Johnson, A.T.C. Effect of Substrate Roughness and Feedstock Concentration on Growth of Wafer-Scale Graphene at Atmospheric Pressure. Chem. Mater. 2011, 23, 1441–1447. [Google Scholar] [CrossRef]
- Wu, W.; Yu, Q.; Peng, P.; Liu, Z.; Bao, J.; Pei, S.S. Control of thickness uniformity and grain size in graphene films for transparent conductive electrodes. Nanotechnology 2011, 23, 035603. [Google Scholar] [CrossRef]
- Li, X.; Magnuson, C.W.; Venugopal, A.; An, J.; Suk, J.W.; Han, B.; Borysiak, M.; Cai, W.; Velamakanni, A.; Zhu, Y.; et al. Graphene Films with Large Domain Size by a Two-Step Chemical Vapor Deposition Process. Nano Lett. 2010, 10, 4328–4334. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Geng, D.; Guo, Y.; Huang, L.; Xue, Y.; Zheng, J.; Chen, J.; Yu, G.; Liu, Y.; Jiang, L.; et al. Equiangular Hexagon-Shape-Controlled Synthesis of Graphene on Copper Surface. Adv. Mater. 2011, 23, 3522–3525. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, L.; Wang, B.; Ruoff, R.S. Chemical vapor deposition of graphene on thin-metal films. Cell Rep. Phys. Sci. 2021, 2, 100372. [Google Scholar] [CrossRef]
- Lyding, J.; Wood, J.; Pop, E. Growing better graphene by finding the best copper surface. SPIE Newsroom 2012, 10, 004110. [Google Scholar] [CrossRef]
- Zhao, L.; Rim, K.; Zhou, H.; He, R.; Heinz, T.; Pinczuk, A.; Flynn, G.; Pasupathy, A. Influence of copper crystal surface on the CVD growth of large area monolayer graphene. Solid State Commun. 2011, 151, 509–513. [Google Scholar] [CrossRef]
- Momeni, K.; Ji, Y.; Zhang, K.; Robinson, J.A.; Chen, L.Q. Multiscale framework for simulation-guided growth of 2D materials. NPJ 2D Mater. Appl. 2018, 2, 27. [Google Scholar] [CrossRef]
- Saeed, M.; Alshammari, Y.; Majeed, S.A.; Al-Nasrallah, E. Chemical Vapour Deposition of Graphene-Synthesis, Characterisation, and Applications: A Review. Molecules 2020, 25, 3856. [Google Scholar] [CrossRef]
- Seah, C.M.; Chai, S.P.; Mohamed, A.R. Mechanisms of graphene growth by chemical vapour deposition on transition metals. Carbon 2014, 70, 1–21. [Google Scholar] [CrossRef]
- Alnuaimi, A.; Almansouri, I.; Saadat, I.; Nayfeh, A. Toward fast growth of large area high quality graphene using a cold-wall CVD reactor. RSC Adv. 2017, 7, 51951–51957. [Google Scholar] [CrossRef]
- Andersen, M.; Cingolani, J.S.; Reuter, K. Ab Initio Thermodynamics of Hydrocarbons Relevant to Graphene Growth at Solid and Liquid Cu Surfaces. J. Phys. Chem. C 2019, 123, 22299–22310. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, Y.; Teng, W.; Xia, T.; Rong, Y.; Li, N.; Ma, H. A molecular dynamics simulation of the graphene growth on Cu(111) surface. Comput. Mater. Sci. 2017, 130, 10–15. [Google Scholar] [CrossRef]
- Li, P.; Li, Z.; Yang, J. Dominant Kinetic Pathways of Graphene Growth in Chemical Vapor Deposition: The Role of Hydrogen. J. Phys. Chem. C 2017, 121, 25949–25955. [Google Scholar] [CrossRef]
- Qiu, Z.; Li, P.; Li, Z.; Yang, J. Atomistic Simulations of Graphene Growth: From Kinetics to Mechanism. Accounts Chem. Res. 2018, 51, 728–735. [Google Scholar] [CrossRef]
- Chen, S.; Gao, J.; Srinivasan, B.M.; Zhang, G.; Sorkin, V.; Hariharaputran, R.; Zhang, Y.W. An all-atom kinetic Monte Carlo model for chemical vapor deposition growth of graphene on Cu(111) substrate. J. Phys. Condens. Matter 2020, 32, 155401. [Google Scholar] [CrossRef]
- Gaillard, P.; Chanier, T.; Henrard, L.; Moskovkin, P.; Lucas, S. Multiscale simulations of the early stages of the growth of graphene on copper. Surf. Sci. 2015, 637–638, 11–18. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, L.; Ding, F. Kinetics of Graphene and 2D Materials Growth. Adv. Mater. 2019, 31, 1801583. [Google Scholar] [CrossRef]
- Popov, I.; Bügel, P.; Kozlowska, M.; Fink, K.; Studt, F.; Sharapa, D.I. Analytical Model of CVD Growth of Graphene on Cu(111) Surface. Nanomaterials 2022, 12, 2963. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, Y.; Cui, P.; Li, Z.; Yang, J.; Zhang, Z. Carbon Dimers as the Dominant Feeding Species in Epitaxial Growth and Morphological Phase Transition of Graphene on Different Cu Substrates. Phys. Rev. Lett. 2005, 114, 216102. [Google Scholar] [CrossRef]
- Taioli, S. Computational study of graphene growth on copper by first-principles and kinetic Monte Carlo calculations. J. Mol. Model. 2014, 20, 1–13. [Google Scholar] [CrossRef]
- Jiang, H.; Hou, Z. Large-scale epitaxial growth kinetics of graphene: A kinetic Monte Carlo study. J. Chem. Phys. 2015, 143, 084109. [Google Scholar] [CrossRef]
- Chen, S.; Gao, J.; Srinivasan, B.M.; Zhang, G.; Sorkin, V.; Hariharaputran, R.; Zhang, Y.W. A kinetic Monte Carlo model for the growth and etching of graphene during chemical vapor deposition. Carbon 2019, 146, 399–405. [Google Scholar] [CrossRef]
- Kong, X.; Zhuang, J.; Zhu, L.; Ding, F. The complementary graphene growth and etching revealed by large-scale kinetic Monte Carlo simulation. NPJ Comput. Mater. 2021, 7, 14. [Google Scholar] [CrossRef]
- Li, X.; Cai, W.; Colombo, L.; Ruoff, R.S. Evolution of Graphene Growth on Ni and Cu by Carbon Isotope Labeling. Nano Lett. 2009, 9, 4268–4272. [Google Scholar] [CrossRef]
- Mallikarjun Sharada, S.; Bligaard, T.; Luntz, A.C.; Kroes, G.J.; Nørskov, J.K. SBH10: A Benchmark Database of Barrier Heights on Transition Metal Surfaces. J. Phys. Chem. C 2017, 121, 19807–19815. [Google Scholar] [CrossRef]
- Sung, C.M.; Tai, M.F. Reactivities of transition metals with carbon: Implications to the mechanism of diamond synthesis under high pressure. Int. J. Refract. Met. Hard Mater. 1997, 15, 237–256. [Google Scholar] [CrossRef]
- Wang, S.; Petzold, V.; Tripkovic, V.; Kleis, J.; Howalt, J.G.; Skúlason, E.; Fernández, E.M.; Hvolbæk, B.; Jones, G.; Toftelund, A.; et al. Universal transition state scaling relations for (de)hydrogenation over transition metals. Phys. Chem. Chem. Phys. 2011, 13, 20760–20765. [Google Scholar] [CrossRef]
- Wang, S.; Vorotnikov, V.; Sutton, J.E.; Vlachos, D.G. Brønsted–Evans–Polanyi and Transition State Scaling Relations of Furan Derivatives on Pd(111) and Their Relation to Those of Small Molecules. ACS Catal. 2014, 4, 604–612. [Google Scholar] [CrossRef]
- Falsig, H.; Shen, J.; Khan, T.S.; Guo, W.; Jones, G.; Dahl, S.; Bligaard, T. On the Structure Sensitivity of Direct NO Decomposition over Low-Index Transition Metal Facets. Top Catal. 2014, 57, 80–88. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Hvolbæk, B.; Abild-Pedersen, F.; Chorkendorff, I.; Christensen, C.H. The nature of the active site in heterogeneous metal catalysis. Chem. Soc. Rev. 2008, 37, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pintos, D.; Voss, J.; Jensen, A.D.; Studt, F. Hydrodeoxygenation of Phenol to Benzene and Cyclohexane on Rh(111) and Rh(211) Surfaces: Insights from Density Functional Theory. J. Phys. Chem. C 2016, 120, 18529–18537. [Google Scholar] [CrossRef]
- Sharma, I.; Papanai, G.S.; Paul, S.J.; Gupta, B.K. Partial Pressure Assisted Growth of Single-Layer Graphene Grown by Low-Pressure Chemical Vapor Deposition: Implications for High-Performance Graphene FET Devices. ACS Omega 2020, 5, 22109–22118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, P.; Li, Z.; Yang, J. First-Principles Thermodynamics of Graphene Growth on Cu Surfaces. J. Phys. Chem. C 2011, 115, 17782–17787. [Google Scholar] [CrossRef]
- Chen, S.; Gao, J.; Srinivasan, B.M.; Zhang, G.; Sorkin, V.; Hariharaputran, R.; Zhang, Y.W. Unveiling the competitive role of etching in graphene growth during chemical vapor deposition. 2D Mater. 2018, 6, 015031. [Google Scholar] [CrossRef]
- Tsakonas, C.; Manikas, A.; Andersen, M.; Dimitropoulos, M.; Reuter, K.; Galiotis, C. In situ kinetic studies of CVD graphene growth by reflection spectroscopy. Chem. Eng. J. 2021, 421, 129434. [Google Scholar] [CrossRef]
- Balerba, A.K.; Kotanidis, A.; Paraskeuas, A.; Gialampouki, M.; Moreno, J.J.G.; Papageorgiou, D.G.; Evangelakis, G.A.; Lekka, C.E. Chapter 9—Graphene nano-flakes on Cu low-index surfaces by density functional theory and molecular dynamics simulations. Front. Nanosci. 2020, 17, 141–159. [Google Scholar] [CrossRef]
- Wellendorff, J.; Lundgaard, K.T.; Møgelhøj, A.; Petzold, V.; Landis, D.D.; Nørskov, J.K.; Bligaard, T.; Jacobsen, K.W. Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation. Phys. Rev. B 2012, 85, 235149. [Google Scholar] [CrossRef]
- Dirac, P.A.M.; Fowler, R.H. Quantum mechanics of many-electron systems. Proc. R. Soc. Lond. A. Math. Phys. Sci. 1929, 123, 714–733. [Google Scholar] [CrossRef]
- Slater, J.C. A Simplification of the Hartree-Fock Method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Bortz, A.; Kalos, M.; Lebowitz, J. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Schulze, T.P. Efficient kinetic Monte Carlo simulation. J. Comput. Phys. 2008, 227, 2455–2462. [Google Scholar] [CrossRef]
- Bittner, E.R. Chemical Dynamics in the Condensed Phases: Relaxation, Transfer, and Reactions in Condensed Molecular Systems. J. Am. Chem. Soc. 2006, 128, 17156–17157. [Google Scholar] [CrossRef]
- Sun, L.; Chen, B.; Wang, W.; Li, Y.; Zeng, X.; Liu, H.; Liang, Y.; Zhao, Z.; Cai, A.; Zhang, R.; et al. Toward Epitaxial Growth of Misorientation-Free Graphene on Cu(111) Foils. ACS Nano 2021, 16, 285–294. [Google Scholar] [CrossRef]
- Chen, B.; Huang, H.; Ma, X.; Huang, L.; Zhang, Z.; Peng, L.M. How good can CVD-grown monolayer graphene be? Nanoscale 2014, 6, 15255–15261. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.; Schoenhalz, A.; Moskovkin, P.; Lucas, S.; Henrard, L. Growth of nitrogen-doped graphene on copper: Multiscale simulations. Surf. Sci. 2016, 644, 102–108. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938, 34, 11–24. [Google Scholar] [CrossRef]
- Bell, R.P.; Hinshelwood, C.N. The theory of reactions involving proton transfers. Proc. R. Soc. Lond. Ser. A-Math. Phys. Sci. 1936, 154, 414–429. [Google Scholar] [CrossRef]
- Anglada, J.M.; Besalú, E.; Bofill, J.M.; Crehuet, R. Prediction of approximate transition states by Bell–Evans–Polanyi principle: I. J. Comput. Chem. 1999, 20, 1112–1129. [Google Scholar] [CrossRef]
- Anand, M.; Nørskov, J.K. Scaling Relations in Homogeneous Catalysis: Analyzing the Buchwald–Hartwig Amination Reaction. ACS Catal. 2020, 10, 336–345. [Google Scholar] [CrossRef]
- Hoffmann, M.J.; Bligaard, T. A Lattice Kinetic Monte Carlo Solver for First-Principles Microkinetic Trend Studies. J. Chem. Theory Comput. 2018, 14, 1583–1593. [Google Scholar] [CrossRef]
- NASCAM (Nanoscale Modeling). Available online: https://www.unamur.be/sciences/physique/ur/larn/logiciels/nascam (accessed on 27 April 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | P | P |
|---|---|---|
| S1 | 100 | 0.01 |
| S2 | 10 | 0.001 |
| S3 | 60 | 0.01 |
| S4 | 30 | 0.01 |
| S5 | 10 | 0.005 |
| S6 | 10 | 0.01 |
| S7 | 10 | 0.05 |
| S8 | 10 | 0.08 |
| Sample | R (nm) | R (nm) | R (nm) |
|---|---|---|---|
| S6 | 3.88 | 1.24 | 1.51 |
| S4 | 13.14 | 1.3 | 1.63 |
| S3 | 25.14 | 1.31 | 1.69 |
| S1 | 38.51 | 1.73 | 2.22 |
| % | S1 | S1 | S2 | S2 |
|---|---|---|---|---|
| Hydrogenated edge | 0.44 | 1.39 | 0.15 | 1.38 |
| Vacancy defect | 5.7 | 7.8 | 6.8 | 10.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esmaeilpour, M.; Bügel, P.; Fink, K.; Studt, F.; Wenzel, W.; Kozlowska, M. Multiscale Model of CVD Growth of Graphene on Cu(111) Surface. Int. J. Mol. Sci. 2023, 24, 8563. https://doi.org/10.3390/ijms24108563
Esmaeilpour M, Bügel P, Fink K, Studt F, Wenzel W, Kozlowska M. Multiscale Model of CVD Growth of Graphene on Cu(111) Surface. International Journal of Molecular Sciences. 2023; 24(10):8563. https://doi.org/10.3390/ijms24108563
Chicago/Turabian StyleEsmaeilpour, Meysam, Patrick Bügel, Karin Fink, Felix Studt, Wolfgang Wenzel, and Mariana Kozlowska. 2023. "Multiscale Model of CVD Growth of Graphene on Cu(111) Surface" International Journal of Molecular Sciences 24, no. 10: 8563. https://doi.org/10.3390/ijms24108563
APA StyleEsmaeilpour, M., Bügel, P., Fink, K., Studt, F., Wenzel, W., & Kozlowska, M. (2023). Multiscale Model of CVD Growth of Graphene on Cu(111) Surface. International Journal of Molecular Sciences, 24(10), 8563. https://doi.org/10.3390/ijms24108563