

Understanding Carbohydrate Metabolism and Insulin Resistance in Acute Intermittent Porphyria

 , ,
, ,  and
and 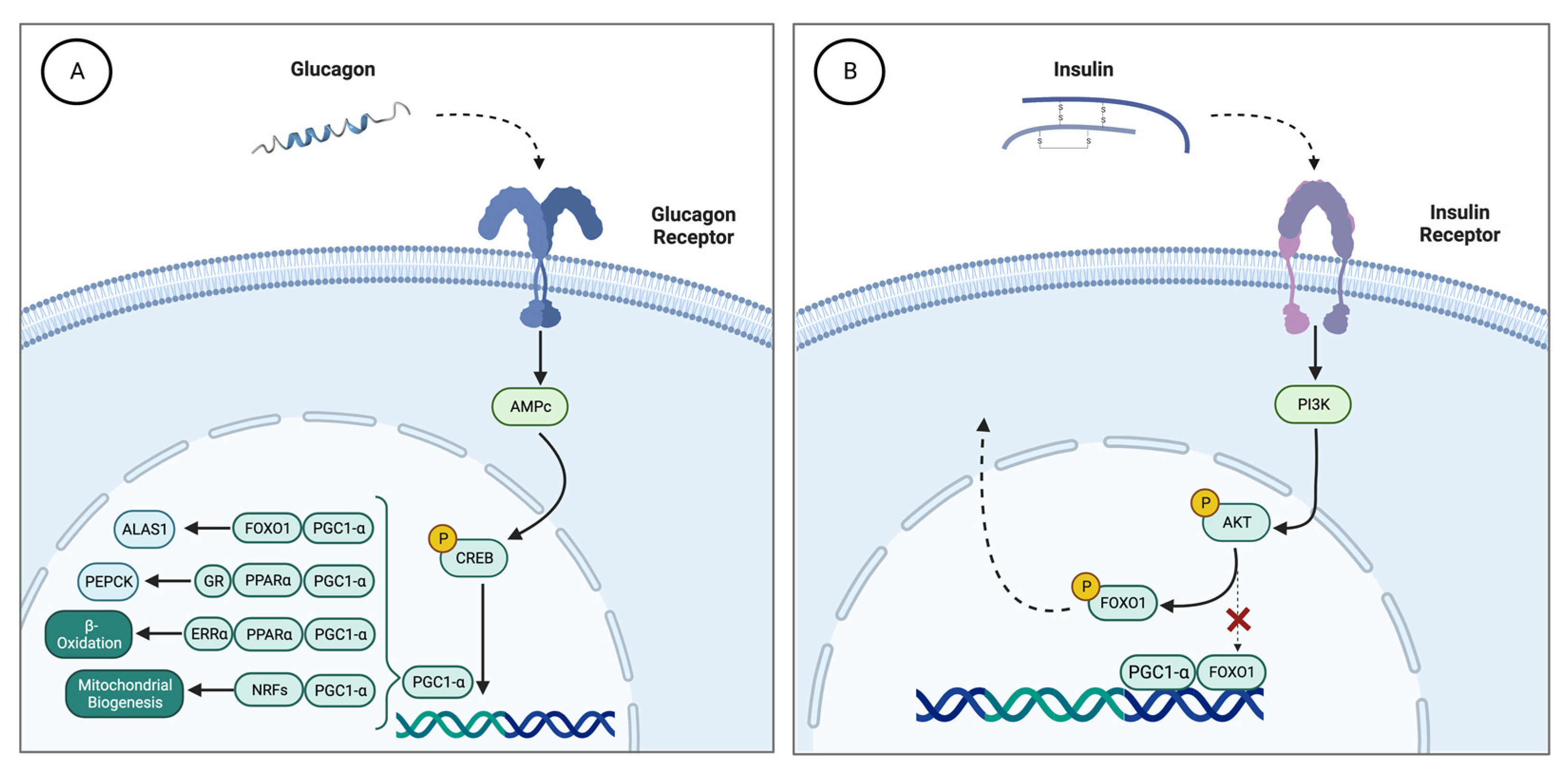{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Carbohydrate Metabolism in AIP
3. Role of Insulin in the Heme Synthesis Pathway
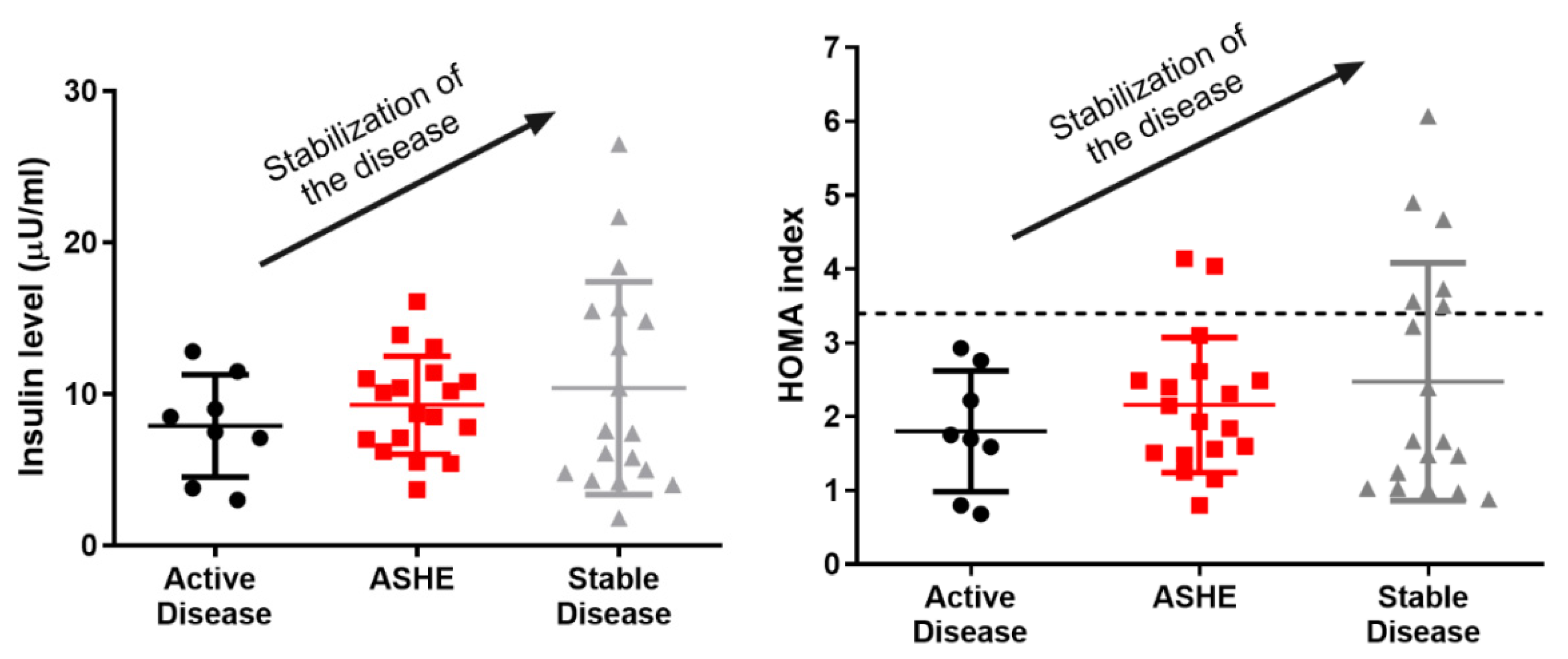
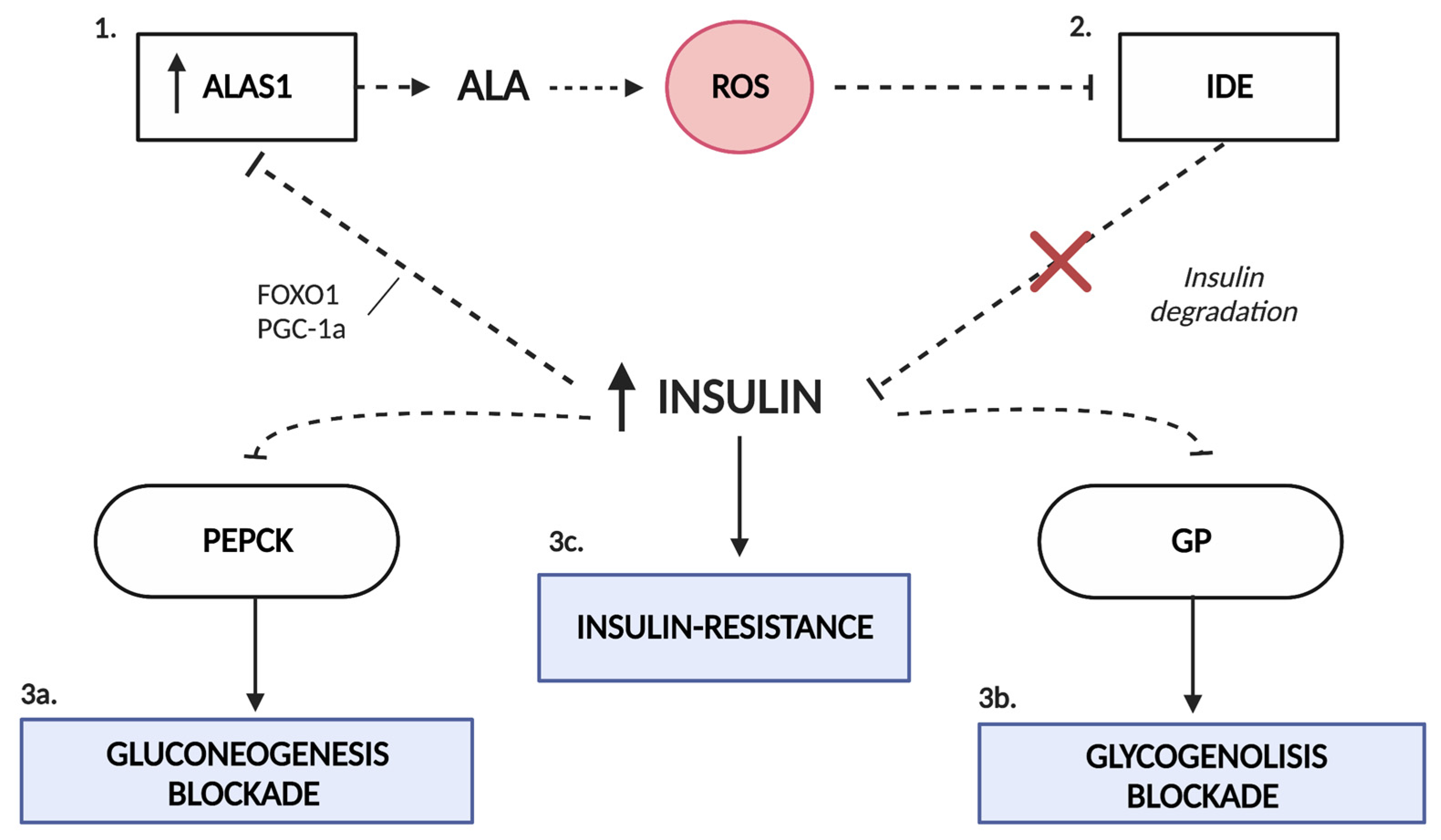
4. Insulin Resistance in AIP
5. Insulin as a Therapeutic Weapon
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, K.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the Diagnosis and Treatment of the Acute Porphyrias. Ann. Intern. Med. 2005, 142, 439–450, Erratum in: Ann Intern Med. 2005, 143, 316. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.; Badminton, M.; Barth, J.; Rees, D.; Stewart, M.F. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2013, 50, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Castelbón Fernández, F.J.; Solares Fernandez, I.; Arranz Canales, E.; Enríquez de Salamanca Lorente, R.; Morales Conejo, M. Protocol for Patients with Suspected Acute Porphyria [published online ahead of print, 2020 Mar 3]. Protocolo de actuación en pacientes con sospecha de porfiria aguda [published online ahead of print, 2020 Mar 3]. Rev. Clin. Esp. 2020, 220, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood 2012, 120, 4496–4504, Erratum in: Blood 2013, 122, 3090. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Li, H.; Du, Y.; Han, B. Congenital sideroblastic anemia: Advances in gene mutations and pathophysiology. Gene 2018, 668, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.-C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2012, 36, 849–857. [Google Scholar] [CrossRef]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.-C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef]
- Gomez-Gomez, A.; Aguilera, P.; Langohr, K.; Casals, G.; Pavon, C.; Marcos, J.; To-Figueras, J.; Pozo, O.J. Evaluation of Metabolic Changes in Acute Intermittent Porphyria Patients by Targeted Metabolomics. Int. J. Mol. Sci. 2022, 23, 3219. [Google Scholar] [CrossRef]
- Delaby, C.; To-Figueras, J.; Deybach, J.C.; Casamitjana, R.; Puy, H.; Herrero, C. Role of two nutritional hepatic markers (insulin-like growth factor 1 and transthyretin) in the clinical assessment and follow-up of acute intermittent porphyria patients. J. Intern. Med. 2009, 266, 277–285. [Google Scholar] [CrossRef]
- Bylesjö, I.; Wikberg, A.; Andersson, C. Clinical aspects of acute intermittent porphyria in northern Sweden: A population-based study. Scand. J. Clin. Lab. Investig. 2009, 69, 612–618. [Google Scholar] [CrossRef]
- Perez-Martinez, P.; Alcala-Diaz, J.F.; Delgado-Lista, J.; Garcia-Rios, A.; Gomez-Delgado, F.; Marin-Hinojosa, C.; Rodriguez-Cantalejo, F.; Delgado-Casado, N.; Perez-Caballero, A.I.; Fuentes-Jimenez, F.J.; et al. Metabolic phenotypes of obesity influence triglyceride and inflammation homoeostasis. Eur. J. Clin. Investig. 2014, 44, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.A.; Hellman, E.S.; Tschudy, D.P. Effect of diet on induction of experimental porphyria. Metabolism 1961, 10, 514–521. [Google Scholar]
- Tschudy, D.P.; Welland, F.H.; Collins, A.; Hunter, G.W. The effect of carbohydrate feeding on the induction of δ-aminolevulinic acid synthetase. Metabolism 1964, 13, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Bonkowsky, H.L.; Sinclair, P.R.; Sinclair, J.F. Hepatic heme metabolism and its control. Yale J. Biol. Med. 1979, 52, 13–37. [Google Scholar]
- Welland, F.H.; Hellman, E.S.; Gaddis, E.M.; Collins, A.; Hunter, G.W.; Tschudy, D.P. Factors affecting the excretion of porphyrin precursors by patients with acute intermittent porphyria I. The effect of diet. Metabolism 1964, 13, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.-K.; Chin, S.; Wu, P.-H.; Meyer, U.A.; Spiegelman, B.M. Nutritional Regulation of Hepatic Heme Biosynthesis and Porphyria through PGC-1α. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef]
- Newgard, C.B.; Hwang, P.K.; Fletterick, R.J. The Family of Glycogen Phosphorylases: Structure and Functio. Crit. Rev. Biochem. Mol. Biol. 1989, 24, 69–99. [Google Scholar] [CrossRef]
- Agius, L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol. Asp. Med. 2015, 46, 34–45. [Google Scholar] [CrossRef]
- She, P.; Shiota, M.; Shelton, K.D.; Chalkley, R.; Postic, C.; Magnuson, M.A. Phosphoenolpyruvate Carboxykinase Is Necessary for the Integration of Hepatic Energy Metabolism. Mol. Cell. Biol. 2000, 20, 6508–6517. [Google Scholar] [CrossRef]
- Hanson, R.W.; Reshef, L. Regulation of phosphoenolpyruvate carboxykinase (gtp) gene expression. Annu. Rev. Biochem. 1997, 66, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Cimbala, M.A.; Lamers, W.H.; Nelson, K.; Monahan, J.E.; Yoo-Warren, H.; Hanson, R.W. Rapid changes in the concentration of phosphoenolpyruvate carboxykinase mRNA in rat liver and kidney. Effects of insulin and cyclic AMP. J. Biol. Chem. 1982, 257, 7629–7636. [Google Scholar] [CrossRef] [PubMed]
- Lelli, S.M.; De Viale, L.C.S.M.; Mazzetti, M.B. Response of glucose metabolism enzymes in an acute porphyria model: Role of reactive oxygen species. Toxicology 2005, 216, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Collantes, M.; Serrano-Mendioroz, I.; Benito, M.; Molinet-Dronda, F.; Delgado, M.; Vinaixa, M.; Sampedro, A.; de Salamanca, R.E.; Prieto, E.; Pozo, M.A.; et al. Glucose metabolism during fasting is altered in experimental porphobilinogen deaminase deficiency. Hum. Mol. Genet. 2016, 25, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.W.; Stephens, J.K.; Marks, G.S. Effect of varying the insulin to glucagon ratio on porphyrin biosynthesis in chick embryo liver cells. Mol. Pharmacol. 1978, 14, 717–721. [Google Scholar]
- Marks, G.S.; Stephens, J.K.; Fischer, P.W.F.; Morgan, R.O. Hormonal effects on the regulation of hepatic heme biosynthesis. Mol. Cell. Biochem. 1979, 25, 111–123. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Chen, B.; Wang, M.; Gan, L.; Zhang, B.; Desnick, R.J.; Yasuda, M. Characterization of the hepatic transcriptome following phenobarbital induction in mice with AIP. Mol. Genet. Metab. 2019, 128, 382–390. [Google Scholar] [CrossRef]
- Scassa, M.E.; Guberman, A.S.; Varone, C.L.; Cánepa, E.T. Phosphatidylinositol 3-Kinase and Ras/Mitogen-Activated Protein Kinase Signaling Pathways Are Required for the Regulation of 5-Aminolevulinate Synthase Gene Expression by Insulin. Exp. Cell Res. 2001, 271, 201–213. [Google Scholar] [CrossRef]
- Storjord, E.; Dahl, A.J.; Landsem, A.; Fure, H.; Ludviksen, J.K.; Goldbeck-Wood, S.; Karlsen, O.B.; Berg, K.S.; Mollnes, E.T.; Nielsen, E.W.; et al. Systemic inflammation in acute intermittent porphyria: A case–control study. Clin. Exp. Immunol. 2016, 187, 466–479. [Google Scholar] [CrossRef][Green Version]
- Storjord, E.; Dahl, J.A.; Landsem, A.; Ludviksen, J.K.; Karlsen, M.B.; Karlsen, B.O.; Brekke, O.-L. Lifestyle factors including diet and biochemical biomarkers in acute intermittent porphyria: Results from a case-control study in northern Norway. Mol. Genet. Metab. 2018, 128, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Hedger, R.W.; Wehrmacher, W.H.; French, A.V. Porphyria syndrome associated with diabetic nephrosclerosis and erythropoietin. Compr. Ther. 2006, 32, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sixel-Dietrich, F.; Verspohl, F.; Doss, M. Hyperinsulinemia in Acute Intermittent Porphyria. Horm. Metab. Res. 1985, 17, 375–376. [Google Scholar] [CrossRef]
- Yalouris, A.G.; A Raptis, S. Effect of diabetes on porphyric attacks. BMJ 1987, 295, 1237–1238. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andersson, C.; Bylesjo, I.; Lithner, F. Effects of diabetes mellitus on patients with acute intermittent porphyria. J. Intern. Med. 1999, 245, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.; Lithner, F. Diabetisk metabolism skydd vid svår akut intermittent porfyri [Diabetic metabolism protective in severe acute intermittent porphyria]. Lakartidningen 2001, 98, 5874–5876. [Google Scholar] [PubMed]
- Bitar, M.; Weiner, M. Diabetes-induced Metabolic Alterations in Heme Synthesis and Degradation and Various Heme-containing Enzymes in Female Rats. Diabetes 1984, 33, 37–44. [Google Scholar] [CrossRef]
- Storjord, E.; Airila-Månsson, S.; Karlsen, K.; Madsen, M.; Dahl, J.A.; Landsem, A.; Fure, H.; Ludviksen, J.K.; Fjøse, J.; Dickey, A.K.; et al. Dental and Periodontal Health in Acute Intermittent Porphyria. Life 2022, 12, 1270. [Google Scholar] [CrossRef]
- Solares, I.; Izquierdo-Sánchez, L.; Morales-Conejo, M.; Jericó, D.; Castelbón, F.; Córdoba, K.; Sampedro, A.; Lumbreras, C.; Moreno-Aliaga, M.; de Salamanca, R.E.; et al. High Prevalence of Insulin Resistance in Asymptomatic Patients with Acute Intermittent Porphyria and Liver-Targeted Insulin as a Novel Therapeutic Approach. Biomedicines 2021, 9, 255. [Google Scholar] [CrossRef]
- Zaccardi, F.; Webb, D.R.; Yates, T.; Davies, M.J. Pathophysiology of type 1 and type 2 diabetes mellitus: A 90-year perspective. Postgrad. Med. J. 2016, 92, 63–69. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia Drives Diet-Induced Obesity Independently of Brain Insulin Production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal-State Hyperinsulinemia in Healthy Normoglycemic Adults Is Predictive of Type 2 Diabetes Over a 24-Year Follow-Up: A preliminary report. Diabetes Care 2009, 32, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Morita, I.; Tanimoto, K.; Akiyama, N.; Naya, N.; Fujieda, K.; Iwasaki, T.; Yukioka, H. Chronic hyperinsulinemia contributes to insulin resistance under dietary restriction in association with altered lipid metabolism in Zucker diabetic fatty rats. Am. J. Physiol. Metab. 2017, 312, E264–E272. [Google Scholar] [CrossRef] [PubMed]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef]
- Matkovic, L.B.; D’Andrea, F.; Fornes, D.; de Viale, L.C.S.M.; Mazzetti, M.B. How porphyrinogenic drugs modeling acute porphyria impair the hormonal status that regulates glucose metabolism. Their relevance in the onset of this disease. Toxicology 2011, 290, 22–30. [Google Scholar] [CrossRef]
- Monteiro, H.P.; Abdalla, D.S.; Augusto, O.; Bechara, E.J. Free radical generation during δ-Aminolevulinic acid autoxidation: Induction by hemoglobin and connections with porphyrinpathies. Arch. Biochem. Biophys. 1989, 271, 206–216. [Google Scholar] [CrossRef]
- A Stein, J.; Tschudy, D.P. Acute intermittent porphyria. A clinical and biochemical study of 46 patients. Medicine 1970, 49, 1–16. [Google Scholar] [CrossRef]
- Oliveri, L.M.; Davio, C.; Batlle, A.M.D.C.; Gerez, E.N. ALAS1 gene expression is down-regulated by Akt-mediated phosphorylation and nuclear exclusion of FOXO1 by vanadate in diabetic mice. Biochem. J. 2012, 442, 303–310. [Google Scholar] [CrossRef]
- Rajpal, G.; Liu, M.; Zhang, Y.; Arvan, P. Single-Chain Insulins as Receptor Agonists. Mol. Endocrinol. 2009, 23, 679–688. [Google Scholar] [CrossRef]
- Drew, B.G.; Rye, K.-A.; Duffy, S.J.; Barter, P.; Kingwell, B.A. The emerging role of HDL in glucose metabolism. Nat. Rev. Endocrinol. 2012, 8, 237–245. [Google Scholar] [CrossRef]
- Kim, S.I.; Shin, D.; Choi, T.H.; Lee, J.C.; Cheon, G.-J.; Kim, K.-Y.; Park, M.; Kim, M. Systemic and Specific Delivery of Small Interfering RNAs to the Liver Mediated by Apolipoprotein A-I. Mol. Ther. 2007, 15, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.J.; Sun, Y.; Ong, K.L.; Li, Y.; Tang, S.; Barter, P.J.; Rye, K.-A. Apolipoprotein A-I Protects Against Pregnancy-Induced Insulin Resistance in Rats. Arter. Thromb. Vasc. Biol. 2019, 39, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Paolini, E.; Meroni, M.; Duca, L.; Motta, I.; Fracanzani, A.L.; Di Pierro, E.; Dongiovanni, P. α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics 2021, 11, 1628. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Kwon, Y.; Yea, K.; Moon, H.-Y.; Yoon, J.H.; Ghim, J.; Hyun, H.; Kim, D.; Koh, A.; Berggren, P.-O.; et al. Apolipoprotein a1 increases mitochondrial biogenesis through AMP-activated protein kinase. Cell. Signal. 2015, 27, 1873–1881. [Google Scholar] [CrossRef]
- Higashikawa, F.; Noda, M.; Awaya, T.; Tanaka, T.; Sugiyama, M. 5-aminolevulinic acid, a precursor of heme, reduces both fasting and postprandial glucose levels in mildly hyperglycemic subjects. Nutrition 2013, 29, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, B.L.; Rodriguez, B.L.; Rodriguez, B.L.; Curb, J.D.; Curb, J.D.; Curb, J.D.; Davis, J.; Davis, J.; Davis, J.; Shintani, T.; et al. Use of the Dietary Supplement 5-Aminiolevulinic Acid (5-ALA) and Its Relationship with Glucose Levels and Hemoglobin A1C among Individuals with Prediabetes. Clin. Transl. Sci. 2012, 5, 314–320. [Google Scholar] [CrossRef]
- Saitoh, S.; Okano, S.; Nohara, H.; Nakano, H.; Shirasawa, N.; Naito, A.; Yamamoto, M.; Kelly, V.P.; Takahashi, K.; Tanaka, T.; et al. 5-aminolevulinic acid (ALA) deficiency causes impaired glucose tolerance and insulin resistance coincident with an attenuation of mitochondrial function in aged mice. PLoS ONE 2018, 13, e0189593. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solares, I.; Jericó, D.; Córdoba, K.M.; Morales-Conejo, M.; Ena, J.; Enríquez de Salamanca, R.; Fontanellas, A. Understanding Carbohydrate Metabolism and Insulin Resistance in Acute Intermittent Porphyria. Int. J. Mol. Sci. 2023, 24, 51. https://doi.org/10.3390/ijms24010051
Solares I, Jericó D, Córdoba KM, Morales-Conejo M, Ena J, Enríquez de Salamanca R, Fontanellas A. Understanding Carbohydrate Metabolism and Insulin Resistance in Acute Intermittent Porphyria. International Journal of Molecular Sciences. 2023; 24(1):51. https://doi.org/10.3390/ijms24010051
Chicago/Turabian StyleSolares, Isabel, Daniel Jericó, Karol M. Córdoba, Montserrat Morales-Conejo, Javier Ena, Rafael Enríquez de Salamanca, and Antonio Fontanellas. 2023. "Understanding Carbohydrate Metabolism and Insulin Resistance in Acute Intermittent Porphyria" International Journal of Molecular Sciences 24, no. 1: 51. https://doi.org/10.3390/ijms24010051
APA StyleSolares, I., Jericó, D., Córdoba, K. M., Morales-Conejo, M., Ena, J., Enríquez de Salamanca, R., & Fontanellas, A. (2023). Understanding Carbohydrate Metabolism and Insulin Resistance in Acute Intermittent Porphyria. International Journal of Molecular Sciences, 24(1), 51. https://doi.org/10.3390/ijms24010051