Abstract
Despite the remarkable progress in cancer treatment up to now, we are still far from conquering the disease. The most substantial change after the malignant transformation of normal cells into cancer cells is the alteration in their metabolism. Cancer cells reprogram their metabolism to support the elevated energy demand as well as the acquisition and maintenance of their malignancy, even in nutrient-poor environments. The metabolic alterations, even under aerobic conditions, such as the upregulation of the glucose uptake and glycolysis (the Warburg effect), increase the ROS (reactive oxygen species) and glutamine dependence, which are the prominent features of cancer metabolism. Among these metabolic alterations, high glutamine dependency has attracted serious attention in the cancer research community. In addition, the oncogenic signaling pathways of the well-known important genetic mutations play important regulatory roles, either directly or indirectly, in the central carbon metabolism. The identification of the convergent metabolic phenotypes is crucial to the targeting of cancer cells. In this review, we investigate the relationship between cancer metabolism and the signal transduction pathways, and we highlight the recent developments in anti-cancer therapy that target metabolism.
Keywords:
cancer; metabolism; aerobic glycolysis; glutamine; redox; signal transduction; anti-cancer drug; ROS 1. Introduction
Cancer is a complex disease that is caused by somatic mutations in key genes, such as those stimulating oncogenes, inactivating tumor suppressor genes, or inactivating stability genes, which are involved in proliferative cell division [1]. The characteristics of cancer include the limitless replicative potential, avoidance of cell death, insensitivity to growth suppressors, induction of angiogenesis, and activation of invasion and metastases [2]. Researchers have conducted extensive studies on these characteristics so far; however, they have mainly conducted them from the genetic viewpoint. Recently, researchers have recognized the importance of the alteration in metabolism after the transformation into cancer. Cancer cells enable continuous growth and division by reprogramming the metabolic signal transduction pathways related to cancer metabolism to overcome environmental limitations such as limited resources and a lack of oxygen under the control of oncogenic mutations. Normal differentiated cells depend on mitochondrial oxidative phosphorylation (OXPHOS) for the energy and biomass required for cellular processes under a precisely controlled system to prevent abnormal growth, whereas cancer cells mainly depend on aerobic glycolysis, which is called “the Warburg effect” [3].
Early on, Otto Warburg argued that cancer cells do not utilize OXPHOS due to damaged mitochondria, and that they are highly dependent on aerobic glycolysis (the conversion of glucose to lactate even in oxygen-abundant environments) [4]. However, since then, there has been increasing evidence that cancer cells show flexibility in metabolism by reversibly or simultaneously using two energy metabolism methods, depending on a wide variety of tumor microenvironments: glycolysis and respiration [5,6,7]. The metabolic reprograming of cancer cells is not limited to a high rate of aerobic glycolysis. In addition to the Warburg effect, a high reliance on glutamine is another unique hallmark of cancer cells [8]. The metabolic reprograming of cancer cells is accompanied by an increased redox homeostasis, or vice versa. In this review, we aimed to enrich the overall understanding of the complexity of cancer metabolism, as well as the role of that the signaling transduction pathway plays in it. Furthermore, we intended the review to help increase the effectiveness of cancer treatment by predicting the convergent metabolic phenotypes.
2. Glucose Metabolism: The Warburg Effect
Researchers have recently recognized an altered metabolism as one of the hallmarks of cancer [2]. Because glucose usually exists in abundance, mammalian cells mainly use it in carbon metabolism to provide energy sources and metabolites for various anabolic pathways [3]. Glucose is metabolized to the final product, pyruvate, through glycolysis. In normal resting cells, the metabolism is regulated to meet the energy needed to maintain cellular homeostasis through ATP production [9]. The glycolysis-derived pyruvate mainly enters the tri-carboxylic acid (TCA) cycle and is oxidized to produce high-efficiency ATP energy [10]. However, proliferating cancer cells must not only generate sufficient energy to support cellular replication, but they must also meet the anabolic requirements of biosynthetic macromolecules to build new cells and maintain an adequate redox balance in response to the increased production of ROS [11]. As such, cancer cells inevitably change their glucose metabolism as part of their survival and proliferation (Figure 1). Cancer cells have two biochemical characteristics: a markedly increased glucose uptake and aerobic glycolysis. Enhanced glycolysis provides metabolic intermediates and precursors for the biosynthesis of macromolecules, such as NADPH, amino acids, nucleotides, and lipids, which are required by proliferating cancer cells [12]. Furthermore, cancer cells prefer to convert most pyruvate to lactate rather than oxidize it through the TCA cycle, despite adequate oxygenation [13]. Mitochondrial OXPHOS produces 36 mol ATP/mol glucose; however, glycolysis only produces 2 mol ATP/mol glucose, which is a much smaller amount [1]. The reasons why cancer cells choose the less efficient glycolysis are as follows. First, the rate of glycolysis and the turnover of glucose into lactate is much faster than OXPHOS, which results in faster and greater ATP production [14]. Moreover, this high-rate but low-yield ATP production is an aspect of evolution, allowing cells to occupy advantageous positions when they are competing for shared energy resources [15]. High levels of glycolytic intermediates are interconnected with various metabolic pathways, particularly those that are associated with the synthesis of building blocks, and serve as substrates to support cellular anabolic reactions [16].
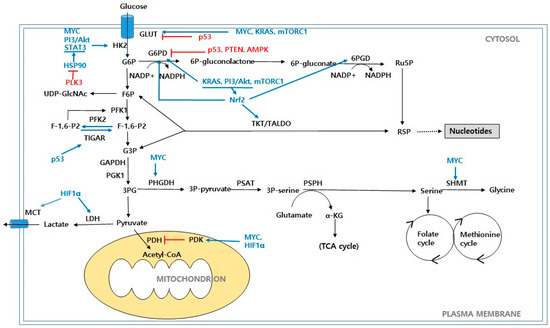
Figure 1.
Glucose metabolism in cancer cells. Schematic representation of main metabolic pathways that contribute to production of biomass precursors, including glycolysis, PPP, HBP, and serine biosynthesis pathway. Abbreviations: GLUT: glucose transporter; HK2: hexokinase 2; G6P: glucose-6-phosphate; F6P: fructose-6-phosphate; PFK1: phosphofructokinase 1; PFK2: phosphofructokinase 2; F-1,6-P2: fructose-1,6-biphosphate; F-2,6-P2: fructose-2,6-biphosphate; TIGAR: TP53-induced glycolysis and apoptosis regulator; G3P: glyceraldehyde-3-phosphate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; PGK1: phosphoglycerate kinase 1; 3PG: 3-phosphoglycerate; PDH: pyruvate dehydrogenase; PDK: pyruvate dehydrogenase kinase; LDH: lactate dehydrogenase; MCT: monocarboxylate transporter; G6PD: glucose-6-phosphate dehydrogenase; 6P-gluconolactone: 6-phosphogluconolactone; 6P-gluconate: 6-phosphogluconate; 6PGD: 6-phosphogluconate dehydrogenase; Ru5P: ribulose-5-phosphate; R5P: ribose-5-phosphate; TKT: transketolase; TALDO: transaldolase; PHGDH: phosphoglycerate dehydrogenase; PAST: phosphoserine aminotransferase; 3P-serine: 3-phosphoserine; PSPH: phosphoserine phosphatase; α-KG: α-ketoglutarate; SHMT: serine hydroxymethyl transferase; UDP-GlcNAc: uridine diphospho-N-acetylglucosamine; PI3K/Akt: phosphoinositide 3-kinase/protein kinase B; mTORC1: mammalian target of rapamycin complex 1; Myc: c-Myc; KRAS: Kirsten Ras GTPase; HIF1α: hypoxia-inducible factors 1α; AMPK: AMP-activated protein kinase; PTEN: phosphate and tension homolog; Nrf2: nuclear factor erythroid-2-related factor 2; PLK3: polo-like kinase 3; HSP90: Heat shock protein 90; STAT3: signal transducer and activator of transcription 3.
The metabolic reprogramming of cancer cells starts with a mutagenic emergence of oncogenes from proto-oncogenes, which induce metabolic alteration by activating the proliferative metabolic pathways and/or by mediating the metabolic gene expressions that are involved in cell proliferation. The epidermal growth factor receptor (EGFR) signaling pathway is one of the main regulators for the cell cycle that regulates growth, survival, proliferation, and differentiation in normal cells [17,18,19]. Since the EGFR signaling pathway plays a critical role in regulating the cell cycle, a mutation for constitutive activation of the genes such as EGFR, RAS family (HRAS, KRAS, and NRAS), Myc, etc., in the EGFR signaling pathway is the main driver for malignant transformation [20,21,22]. Among these oncogenes, KRAS is the best-recognized oncogene with the highest mutation rate in all cancers including highly fatal cancers such as pancreatic ductal adenocarcinoma, non-small-cell lung cancer, and colorectal cancer [23]. The oncogenic mutations of KRAS result in mitochondrial dysfunction by disruption of complex I of the electron transport system (ETS) [24]. Despite a reduced oxygen consumption as a result of mitochondrial dysfunction, the KRAS mutations increase glucose transport into cancer cells by the over-expression of GLUT1 and hexokinase 2 [25,26,27]. The inevitable consequence of elevated ATP generation after high glucose influx under mitochondrial dysfunction would be aerobic glycolysis. In fact, decreased OXPHOS, elevated glycolysis, and increased generation of reactive oxygen species have been observed as the main features in the cancer cells with oncogenic mutations of KRAS [24,28,29].
Other than genetic mutations in the oncogenes, the signaling pathway is constitutively activated in most cancers through subsequent genetic modification [21,22]. The oncogenes of the EGFR signaling pathway generally activate the cancer metabolism including anaerobic glycolysis as a result of the cell cycle activation in which Myc exerts the final role [30]. Myc, which is the final effector of the EGFR signaling pathway, promotes glycolysis by inducing the expression of glucose transporter1 (GLUT1) and lactate dehydrogenase A (LDHA) and by upregulating the glycolytic enzymes, including hexokinase 2 (HK2), phosphofructokinase (PFK), and pyruvate dehydrogenase kinase 1 (PDK1) [31,32]. Moreover, most cancers are exposed to hypoxia [33]. In hypoxia, hypoxia-inducible factor 1α (HIF-1α) dimerizes with HIF-1β and accumulates in the nucleus. The HIF-1 dimer, which is the complex of HIF-1α and HIF-1β, then binds to the hypoxia response elements (HREs) and activates the regulatory genes involved in glycolysis [34,35]. These genes include the glucose transporter GLUT1, glycolytic enzyme HK2, phosphofructokinase-1 (PFK1), fructose-bisphosphate aldolase A (ALDOA), phosphoglycerate kinase 1 (PGK1), pyruvate, pyruvate kinase (PK), and the enzymes related to lactate production, extrusion LDHA, and monocarboxylate transporter 4 (MCT-4) [36,37]. HIF-1 inactivates pyruvate dehydrogenase (PDH) via the upregulation of PDH kinases (PDKs), which inhibit the transformation of pyruvate into acetyl-CoA [38]. Researchers have observed crosstalk between Myc and HIF, and this interplay promotes cancer cell survival and growth [39]. A key tumor suppressor, p53, also plays an important role in the regulation of cellular metabolism [40]. p53 regulates the glucose absorption by repressing GLUT1 and GLUT4, as well as GLUT3 through the inhibition of nuclear factor kB (NF-ĸB) signaling [41]. p53 suppresses the MCT1 expression to inhibit the transportation of lactate [42]. In addition, p53 inhibits glycolysis through the transcriptional activation of Parkin, which degrades HIF-1α, or by downregulating HK2 [43,44]. The phosphoinositide 3-kinase (PI3K)/Akt pathway, which is one of the most frequently dysregulated pathways in cancer, is activated in response to growth factors, insulin, and cytokines and it controls key metabolic processes [45]. PI3K/Akt promotes glucose uptake and glycolysis by increasing the glucose transporter expressions such as those of HK and PFK1 [46]. HIF-1α regulates glycolysis via the PI3K/Akt/mTOR signaling pathway [47]. Tumor suppressors, such as phosphate and tension homolog (PTEN) and p53, suppress the metabolic adaptation through the negative activation of the PI3K/Akt and MAPK/ERK pathways [48,49]. Recent studies provide evidence for associations with polo-like kinases (PLKs) in regulating cancer progression, apoptosis, and metabolic processes [50,51]. The PLKs, which belong to a family of highly conserved serine/threonine kinases, comprise all five members (PLK1-5) [50]. PLKs and signal transducer and activator of transcription 3 (STAT3) exhibit cross-regulatory interactions in human cancers. PLK1 promotes the migration of lung cancer cells via STAT3 signaling [52]. Furthermore, PLK3 binds to the heat shock protein 90 (HSP90) and facilitates its degradation, which leads to a decrease of phosphorylated STATS. The STAT3 dephosphorylation further inhibits the transcriptional activation of HK2, resulting in a lower glycolytic rate in colorectal cancer [53].
The pentose phosphate pathway (PPP), which is also called the hexose monophosphate shunt, is a major glucose catabolic pathway that regulates cancer cell survival and growth by supplying ribose-5-phosphate (R5P) [54]. Furthermore, the PPP plays an important role in the cellular redox balance as a major source of NADPH [55]. Glucose is phosphorylated by HK to form glucose-6-phosphate (G6P), which is further metabolized by the PPP. The PPP has two biochemical branches: oxidative and non-oxidative. In the oxidative phase, G6P is irreversibly converted into ribulose-5-phosphate (Ru5P) by a series of G6PD and 6-phosphogluconate dehydrogenase (6PGD). In addition, NADPH produced during the oxidative phase plays a role in reductive biosynthesis and the maintenance of the redox state of the cell [56]. Ru5P yielded by 6PGD enters the non-oxidative branch and is converted to R5P and erythrose 4-phosphate, which are required for the nucleic acid synthesis and are sugar phosphate precursors for amino acid biosynthesis, respectively [57]. The non-oxidative branch recruits glycolytic intermediates, including fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (G3P) by a series of reversible reactions, and it enables reciprocal coordination of the nutrient uptake and stress response in cancer cells [58]. Transketolase (TKL) and transaldolase (TALDO) are key enzymes that are responsible for a series of reversible reactions in the non-oxidative branch of the PPP [55]. The central role of glucose-6-phosphate dehydrogenase (G6PD),which is the first and rate-limiting enzyme in the PPP, is the generation of ribose and NADPH, which are vital for the biosynthesis of the building blocks and the maintenance of the antioxidant defenses [59]. G6PD acts as a “gatekeeper” to determine the glucose flux, acting as a partition between the glycolysis and PPP [60]. Increased NADP+ levels during oxidative stress increases the PPP flux by upregulating G6PD for the generation of NADPH and the antioxidant defense [1]. The pro-oncogenic signaling pathways that are related to the PPP are usually accelerated through G6PD [55]. Because p53 directly represses the glucose transporter genes GLUT1 and GLUT4, the loss of p53 increases both glycolysis and the PPP [61,62]. p53 induces the expression of TIGAR (TP53-induced glycolysis and apoptosis regulator) repressing the glycolysis by lowering the level of fructose-2,6-bisphosphate (F-2,6-P2). F-2,6-P2 is an allosteric activator of PFK1, and its reduction inhibits the PFK1 activity and results in a decreased glycolytic flux [63]. While p53, the p53 target gene PTEN, and AMP-activated protein kinase (AMPK) negatively regulate the PPP by suppressing the G6PD activity, the oncogenic pathways, such as KRAS, PI3/Akt, and mTORC1, increase the flux through the PPP by the upregulation of G6PD [60]. The oncogene KRASG120 stimulates glycolysis and substantially increases the nonoxidative PPP activity in pancreatic tumors [64]. mTORC1 increases both the flux through glycolysis and the oxidative PPP through the activation of SREBP1 (a sterol regulatory element-binding protein), which is involved in the induction of the G6PD’s expression [65]. Nrf2 (nuclear factor erythroid 2-related factor 2), through its binding to antioxidant response elements (AREs) under oxidative stress or increased ROS levels, activates both the oxidative and nonoxidative PPPs by upregulating the expressions of the PPP enzymes (G6PD, 6PGD, TKT, and TALDO) [66].
After the glucose that is taken up by the cell is converted to fructose-6-phosphate, most of the intermediates are metabolized through glycolysis, and from approximately 2 to 3% are shunted to the hexosamine biosynthetic pathway (HBP), which is a metabolic branch pathway of glycolysis [67]. The nucleotide–sugar uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), which is an end product of the HBP, serves as the sugar donor for O-linked-β-N-acetylglucosamine (O-GlcNAc) [68]. GlcNAcylation, which is similar to phosphorylation, is the post-translational cycling of a single O-GlcNAc on the hydroxyl groups of serine and/or threonine residues of the target proteins [69]. O-GlcNAc is added to or removed from the target proteins by two enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), respectively [70]. O-GlcNAcylation elevation is implicated in a variety of cancers, including breast cancer [71], prostate cancer [72], lung cancer [73], colorectal cancer [74], and chronic lymphatic leukemia [75]. The loss of p53 increases the HBP flux and leads to the enhancement of O-GlcNAcylation [76]. Reducing the O-GlcNAcylation in prostate cancer leads to a decrease in the level of c-Myc protein O-GlcNAcylation and decreased c-Myc stability, which researchers have associated with decreased cancer cell growth [77]. In breast cancer, the OGT and O-GlcNAcylation levels are upregulated via the PI3K/mTOR/Myc pathway for tumorigenesis [78].
Researchers have observed glucose-derived metabolic fluxes into the serine biosynthesis pathway in cancer cells [79]. The glycolytic intermediate, 3-phosphoglycerate (3PG) can be diverted to produce serine, which is a major donor of carbon to the folate pool [80]. Phosphoglycerate dehydrogenase (PHGDH) is the rate-limiting enzyme that converts 3PG to serine [81]. During this serine synthesis process, phosphoserine aminotransferase (PAST) converts glutamate to α-KG, which serves as refuels for the TCA cycle; thus, the serine synthesis pathway supports the energy metabolism and anabolic processes [82]. Serine donates a one-carbon unit to tetrahydrofolate (THF) by serine hydroxymethyltransferase (SHMT1/2), which is then used for de novo purine synthesis or thymidylate synthesis [83]. Serine also transfers a one-carbon unit to homocysteine, which forms methionine [84]. Methionine is the precursor of cysteine, which is essential for the formation of glutathione, which plays an important role in redox buffering [85].
Researchers have observed the flux into this serine biosynthetic pathway through the amplification or upregulation of the PHGDH expression in many cancers [86,87]. The PHGDH expression levels in specific subtypes of cancer, such as triple-negative breast cancer [67] or estrogen-receptor-negative cancers [87], are upregulated. c-Myc is involved in the regulation of serine biosynthesis and the PHGDH expression is transcriptionally upregulated by activating c-Myc [88]. c-Myc also upregulates SHMT2 under hypoxia, and both the SHMT2 and PHGDH enzymes are positively correlated with each other in cancers such as breast cancer and neuroblastoma [89]. p53 allows cancer cells to overcome cellular stress, such as serine starvation, which preserves the cellular antioxidant capacity [90]. Glycine metabolism is also critical for tumorigenesis [91,92]. Glycine decarboxylase (GLDC) is commonly upregulated in cancers, and the mTORC1 activity regulates the posttranslational modifications of GLDC, which contribute to the modulation of glycine metabolism and tumorigenesis [93].
3. Tryptophan and Glutamine Metabolism
Amino acid metabolism is also significantly altered in cancer cells [94]. Although most amino acids are metabolized differently in cancer cells, the metabolisms of tryptophan and glutamine deviate most significantly in cancer cells [95]. Cancer cells unusually upregulate the expression of indoleamine 2,3-dioxygenase 1 (IDO1) that catalyzes the oxidation of L-tryptophan to N-formyl-kynurenine [96]. Overexpressed IDO1 in cancer cells makes cancer cells catabolize tryptophan leading to depletion of tryptophan. Since immune cells heavily rely on tryptophan for their immunological reaction, depletion of tryptophan by cancer cells causes the activation of apoptosis in the neighboring immune cells to cancer cells [97,98]. At the same time, the concomitant tryptophan metabolites by IDO1 such as kynurenine in cancer cells further induce T-lymphocytes to undergo apoptosis [97,98], as well as promote activation of immunosuppressive regulatory T-cells [98,99]. Although cancer cells prevent the immunological attacks in hosts by removing tryptophan, cancer cells in the same microenvironment are not affected by tryptophan depletion resulting from the accelerated tryptophan catabolism [95,96,97]. The mechanism for how cancer cells overcome IDO1-mediated tryptophan deprivation is not elucidated and is a current interesting scientific speculation.
Highly proliferating cells, such as cancer cells, have an explosively increased demand for amino acids, which are an important class of major nutrients, to support their growth rate. Thus, cancer cells try to meet these demands by increasing the glutamine metabolism (Figure 2). Glutamine is the most abundant amino acid, accounting for over 20% of the free amino acids in plasma and for 40% in muscle [100]. Most tissues synthesize glutamine, which is a nonessential amino acid (NEAA); however, in periods of rapid growth or stress, the cell’s glutamine requirements increase beyond their ability to rapidly synthesize macromolecules, which makes glutamine essential [101]. Many cancer cells are addicted to glutamine, and most of them show oncogene-dependent glutamine addictions in culture [102,103]. Glutamine is emerging as an important nutrient for cancer cell proliferation and survival through the supply of carbon and nitrogen or the production of macromolecules, energy generation, and cellular homeostasis [104]. The amide group (γ-nitrogen) from glutamine contributes to both purine and pyrimidine nucleotide biosynthesis in proliferating cells [105]. Moreover, glutamine’s α-nitrogen through transaminases is transferred into various pools of nonessential amino acids (NEAAs), promoting the generation of alanine, asparagine, and serine [106]. In addition to its role as a nitrogen donor, glutamine provides a source of carbon to fuel the TCA cycle for bioenergetics and the biosynthetic requirements of cancer cells [106]. Proliferating cancer cells that display aerobic glycolysis convert glucose to lactate, which shunts glucose carbon away from the TCA cycle and fatty acid (FA) synthesis [107]. Eventually, glutamine serves as the carbon donor to maintain the TCA cycle in proliferating cells by replenishing the intermediates that act as precursors in the biosynthetic and NAPDH-producing pathways [16]. Glutamine is imported into cells through the SLC1A5 (also called ASCT2) glutamine transporter [108], and it is deaminated to glutamate by glutaminases (GLS1/2) and transported into the mitochondria [109]. Glutamate is converted into α-ketoglutarate (α-KG) in the mitochondria through oxidative deamination by glutamate dehydrogenase (GLUD) to fuel the TCA cycle [110]. Glutamine-derived aspartate is transported to the cytosol, where it is converted into oxaloacetate (OAA) by transaminase, which is subsequently converted to malate and pyruvate. Alternatively, glutamate is converted to α-KG by transaminase generated NEAAs in the cytosol or mitochondria [104]. Glutamate is also a precursor of glutathione, which serves as a redox buffer against oxidative stress, and its formation is highly dependent on glutamine [101]. Under normoxic conditions and with an adequate glucose supply, proliferating cancer cells shunt α-KG into OXPHOS to produce energy, or they shunt it into the TCA cycle to produce citrate through the condensation with glutamine-derived OAA, which is produced through a series of oxidation processes, and with glucose-derived acetyl-CoA [111,112]. Citrate is transported to the cytosol and contributes to de novo lipid synthesis. The cytosolic OAA is converted into malate through NADP-dependent malate dehydrogenase (MDH1), and the malate is converted into pyruvate by malic enzyme 1 (ME1), producing CO2 and NADPH [105]. TCA cycle-derived OAA can be transferred to aspartate to contribute a carbon source for nucleotide biosynthesis [16]. Glutamine anaplerosis in the TCA cycle plays an important role in the provision of the critical precursors that are used to fuel the nucleotides, NEAAs, and lipids. Under conditions of hypoxia or glucose deprivation, α-KG produces citrate and supports lipid synthesis through a reductive carboxylation by NADP-dependent isocitrate dehydrogenase 2 (IDH2) [113,114]. In addition, α-KG exported to the cytosol is reductively carboxylated into citrate via NADPH-dependent isoforms of isocitrate dehydrogenase 1 (IDH1), which in turn helps maintain lipogenesis [115]. In this way, the NADPH-producing pathways from glutamine metabolism provide the reducing power for lipid synthesis as well as the redox balance.
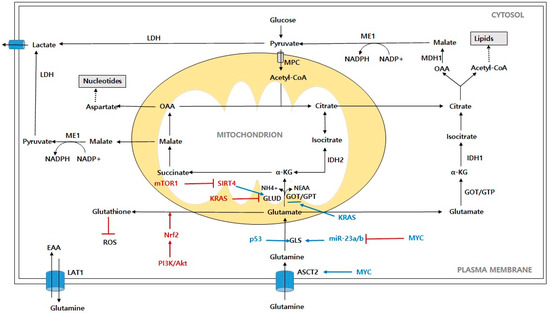
Figure 2.
Schematic representation of glutamine contribution to cancer cells metabolism. Abbreviations: ASCT2: alanine-serine-cysteine-transporter-2; LAT1: L-type amino acid transporter; GLS: glutaminase; GLUD: glutamate dehydrogenase; α-KG: α-ketoglutarate; OAA: oxaloacetate; IDH1: isocitrate dehydrogenase 1; IDH2: isocitrate dehydrogenase 2; ME1: malic enzyme; LDH: lactate dehydrogenase; ROS: reactive oxygen species; MPC: mitochondrial pyruvate carrier; MDH1: malate dehydrogenase; NEAA: nonessential amino acid; EAA: essential amino acid; NH4+: free ammonia; PI3K/Akt: phosphoinositide 3-kinase/protein kinase B; mTORC1: mammalian target of rapamycin complex 1; Myc: c-Myc; KRAS: Kirsten Ras GTPase; Nrf2: nuclear factor erythroid-2-related factor 2; SIRT4: Sirtuin 4.
Oncogenes reprogram glutamine metabolism to support cancer cell growth as in the case of glucose metabolism. Since cancer cells are heavily addicted to glutamine, it is natural to observe that oncogenes, especially KRAS, elevate the genes associated with glutamine catabolism such GLS, GLUD, SLC1A5, transaminase, etc., through Myc, which is the downstream molecule of the oncogenes [116,117,118]. Myc uses glutamine as a nitrogen source to synthesize purines and pyrimidines, and it directly regulates three (CTP synthetase, PRPP amidotranferase, and carbamoyl phosphate synthetase II) of the five enzymatic steps [119]. Myc also activates the expressions of the genes involved in glutamine uptake and glutaminolysis. Myc transcriptionally binds to the promoter regions of the high affinity glutamine transporters, SLC1A5 and SLC38A5, which facilitates the glutamine uptake [120]. The bidirectional transport of glutamine through SCL1A5 and SLC7A5 is required for the activation of mTORC1 signaling [121]. Furthermore, Myc indirectly stimulates GLS through the suppression of miR-23a and miR-23b, which results in the upregulation of the glutamine metabolism [122]. Myc leads to the diversion of glucose away from the mitochondria, which is its conversion to lactate [120]. As a result, Myc enhances the cellular dependence on glutamine. Glutamine depletion induces Myc-mediated cell death via the Bax and caspase activities, which means that glutamine is a carbon source for the maintenance of mitochondrial anaplerosis [107,123]. In addition, apoptosis that is caused by glutamine depletion is rescued by oxaloacetate or pyruvate, suggesting that glutamine depletion causes apoptosis by affecting the TCA cycle [104]. The oncogene KRAS also plays an important role in coordinating the shift in glutamine metabolism to support tumor growth and survival by increasing the GOT1 expression and decreasing the GLUD1 expression [111]. PI3K/Akt can activate Nrf2, in which case the activation of Nrf2 upregulates the expression of glutathione synthetase and glutamate–cysteine ligase to produce glutathione [124,125]. mTOR1 promotes glutamine anaplerosis by activating GLUD by repressing silent information regulator two 4 (SIRT4), which is the mitochondrial-localized sirtuin that inhibits GLUD [126]. p53-inducible GLS2 facilitates glutamine metabolism and lowers the intracellular ROS levels to protect against oxidative stress [127].
4. Lipid Metabolism
Lipids are the essential components of membranes, and they are the building blocks that constitute cells [128]. Lipid metabolism dysregulation is one of the major metabolic hallmarks of cancer [129]. Cancer cells optimize their requirements through lipidomic remodeling, which includes overall alterations in the FA transport, de novo synthesis, storage as lipid droplets, and β-oxidation to produce ATP (Figure 3).
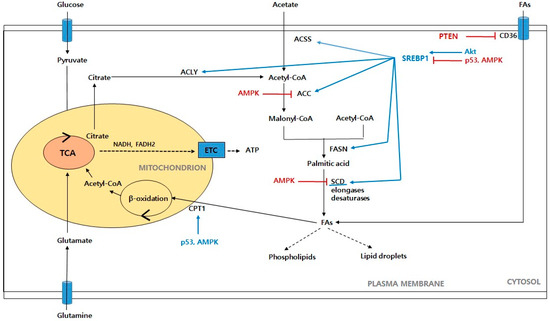
Figure 3.
Schematic representation of fatty acid metabolism in cancer cells. Glucose-derived pyruvate enters the mitochondria to form citrate, which then moves to the cytoplasm and contributes to the de novo synthesis of fatty acids. Acetate is also another source of lipogenesis. When cellular fatty acids are in excess, the fatty acids absorbed through CD36 are converted into lipid droplets and are made available when needed. Acetate also provides reducing equivalents, such as NADH and FADH2 through β-oxidation. Finally, ATP is generated from the ETC. Abbreviations: ACSS: acetyl-CoA synthetase; ACLY: ATP citrate lyase; ACC: acetyl-CoA carboxylase; FASN: fatty acid synthase; SCD: stearoyl-CoA desaturase; FAs: fatty acids; CPT1: carnitine palmitoyl transferase 1; ETC: electron transport chain. Akt: protein kinase B; AMPK: AMP-activated protein kinase; PTEN: phosphate and tension homolog; SREBP1: sterol regulatory element-binding proteins.
Fatty acids (FAs) are the building blocks of most lipids, and they are composed of a hydrocarbon chain with one terminal carboxyl group of varying carbon lengths and degrees of desaturation [130]. Consequently, FAs serve to regulate various biochemical processes, such as membrane biosynthesis, energy storage, and signaling pathways, in which they act as secondary messengers [131]. Tumor cells synthesize up to 95% of FAs de novo despite a sufficient dietary lipid supply, which suggests that the activity of the FA synthesis pathway is required for carcinogenesis [132,133]. A number of researchers have shown high levels of fatty acid synthesis in various human cancers and their precursor lesions, including prostate cancer [134], ovarian cancer [135], breast cancer [136], endometrium cancer [137], and colon cancer [138]. Thus, we focus on the FA metabolism in cancer cells and the oncogenic signaling pathways that support tumorigenesis and cancer progression.
Mammalian cells can obtain FAs through exogenous uptake or de novo synthesis [139]. The cellular uptake at the plasma membrane is the first step in FA utilization, and it is highly regulated through specific membrane transporters. These transporters include CD36 FA translocase, the FA transport proteins (FATPs; also known as SLC27), and plasma membrane FA-binding proteins (FABPs), all of which show increased gene and protein expression in tumors [140]. In particular, a high CD36 expression is associated with poor prognosis in cancers, such as breast, ovarian, gastric, and prostate cancers [141,142]. In PTEN-deficient prostate cancer, CD36 promotes an increased FA uptake and accelerated cancer progression, which suggests that it renders cancer cells dependent on the exogenous lipid uptake [143]. Cancer cells use the extracellular FA uptake to fill the lipid pool that is required for their rapid growth and proliferation under conditions of metabolic stress, such as hypoxia. Tumors under hypoxia exhibit an increased FA uptake through the upregulation of the FABP3/7 expression that is induced by HIF-1α, which accompanies repressed de novo FA synthesis [144]. In ovarian cancer, FABP4 is related to the supply of FAs from the surrounding adipocytes, and it is also a key determinant of the metastatic potential [145,146]. Hypoxia can downregulate miR-409-3p, thus removing its inhibitory effects on FABP4, which results in increased FABP4 levels. Therefore, the high expression of FABP4 can regulate various metabolites such as FA oxidation (FAO) and lysophospholipids and can lead to metastases in ovarian cancer.
Lipogenesis in most normal cells is usually confined to hepatocytes and adipocytes and is activated when necessary, whereas cancer cells often exhibit a shift toward de novo lipid synthesis, even in the presence of exogenous lipid sources [147]. FAs are synthesized from cytoplasmic acetyl-CoA, which is generated by ATP citrate lyase (ACLY), and acetyl-CoA synthetase (ACSS) from citrate and acetate, respectively [148]. In addition, the carbon sources from glucose and glutamine contribute to citrate production [128]. Pyruvate is converted to mitochondrial acetyl-CoA by PDH (pyruvate dehydrogenase), which is followed by citrate synthase and subsequent citrate generation [149]. As mentioned above, glutamine is converted to α-KG by GLS1 and GLS2, which is followed by IDH1 and IDH2, respectively, and subsequent citrate production [150]. Several steps are needed to convert carbons from citrate to FAs. Citrate is converted to cytoplasmic acetyl-CoA by ACLY, and then acetyl-CoA carboxylase (ACC), which is the rate-limiting enzyme of de novo lipogenesis, catalyzes the carboxylation of acetyl-CoA to malonyl-CoA [151]. Fatty acid synthase (FASN), which is a key step in the FA synthetic pathway, catalyzes the successive condensation of seven malonyl-CoA molecules and one acetyl-CoA molecule, producing 16-carbon palmitate [152]. Palmitate can be elongated and desaturated using FA elongases and stearoyl-CoA desaturases (SCD), generating additional FAs for the production of more complex lipids [130,153]. Lipogenesis is transcriptionally regulated by sterol regulatory element-binding proteins (SREBPs), which are membrane-bound transcription factors that consist of three isoforms: SREBP1a, SREBP1c, and SREBP2 [154]. SREBPs that are synthesized as inactive precursors bind to the SREBP cleavage-activating protein (SCAP) in the endoplasmic reticulum (ER) [155]. Insig-1 and Insig-2 are ER proteins that bind to the SCAP in a sterol-regulated fashion and prevent the proteolytic processing of SREBPs [155]. Under low-sterol-concentration conditions, the SCAP/SREBP complex is transported to the Golgi. In the Golgi, site-1 protease (S1P) and site-2 protease (S2P) subsequently cleave the SREBPs to release the active N terminus, which enters the nucleus and induces the transcription of the genes that are involved in lipogenesis [152,156]. As such, SREBP1 enhances lipogenesis by increasing the transcriptions of its target genes: ACLY, ACC, FASN, and SCD1. The activation of SREBPs is stimulated by the PI3K/Akt/mTOR pathway, which is the most frequently activated oncogenic signaling pathway in cancers [157]. The ACLY activity is regulated by the PI3K/Akt pathway, and Akt can directly activate ACLY [158,159]. The acetylation of ACLY increases the ACLY stability by blocking its ubiquitinylation and degradation, which leads to the promotion of de novo lipid synthesis and tumor growth [160]. Conversely, sirtuin 2 (SIRT2) deacetylates ACLY [140]. In addition, the increased phosphorylation of ACLY by Akt can enable sustained acetyl-CoA production, and thereby histone acetylation, even if nutrients are limited [161]. The activation of AMPK, which suppresses FA synthesis by phosphorylating ACC, also inhibits tumorigenesis [162,163]. The FASN activity is upregulated by the increase in the epidermal growth factor signaling via the mitogen-activated protein kinase (MAPK) and the PI3 kinase signaling cascades [164]. In addition, SCD1 contributes to the maintenance of the lipid metabolism in cancer cells, which favors cell proliferation and survival by the stimulation of the ACC activity through the inactivation of the AMPK pathway and the activation of Akt signals [165]. AMPK phosphorylates SREBPs to retain them in the ER, eventually inhibiting their transcriptional activities and suppressing lipogenesis in liver cancer cells [166]. Akt activation upregulates SREBP1 gene expression, and it also inhibits the ubiquitin-proteasome pathway degradation of SREBP1 through protein arginine methyltransferase 5 (PRMT5), inducing SREBP1 hyperactivity, which results in de novo lipogenesis and tumor growth [167]. Furthermore, mTORC2 positively regulates the lipid synthesis via the stabilization of SREBP1 by suppressing the GSK3/FBXW7-mediated degradation in cancer cells [153].
Cells convert excess lipids into triglycerides (TGs) and cholesterol esters in the ER, and they store them as lipid droplets (LDs) for use as energy generation and membrane synthesis in times of starvation [168]. Cancer cells exhibit the presence of abundant LDs, which suggests that the storage of lipids may be a common feature of malignancies [169]. TG serves as the main form of energy storage and diglyceride acyltransferase 1/2 (DGAT1/2) synthesizes TG and acyl-CoA [170]. Mitochondrial fatty acid oxidation (FAO, also known as β-oxidation) plays an important role in maintaining energy homeostasis by generating energy in the form of ATP [171]. The FAO pathway converts long-chain FAs into acetyl-CoA, which in turn generates NADPH and FADH2 through the TCA cycle, which enter the electron transport chain to produce ATP [152]. Cancers exhibit the overexpression of FAO pathway proteins [172,173]. Carnitine palmitoyl transferase 1 (CPT1), which is the rate-limiting enzyme in FAO, catalyzes the acylation of long-chain FAs and their entry into the mitochondria for FAO [174]. One of the isoforms, CPT1C, is upregulated in human lung cancer, depending on a mechanism that involves activated AMPK, and it increases the FAO and ATP generation [175]. Peroxisome proliferator activated receptor α (PPARα) is a major transcriptional regulator of FAO and extended PPARα activation causes hepatocellular carcinoma by involving the perturbation of the cell cycle and the production of ROS [152,176]. AMPK enables cancer cells to survive under metabolic stress by maintaining NADPH as well as the abovementioned function of ATP homeostasis, and this metabolic adaptation is achieved through the liver kinase B1 (LKB1)-AMPK pathway [177]. The phosphorylation and inactivation of the acetyl-CoA carboxylases ACC1 and ACC2 by AMPK maintains the NADPH levels by inhibiting FA synthesis and activating FAO, respectively [177,178]. Emerging evidence supports an additional role for p53 in lipid metabolism. The CPT1C expression is activated through the AMPK/p53 signaling pathway under metabolic stress, which leads to tumor proliferation and survival [179]. Lpin1 is essential for fat metabolism, and the ROS-mediated p53 induction of Lpin1 regulates the FAO in response to nutritional stress [180].
5. Redox Metabolism
ROS are the ionized oxygens and derived unstable chemicals from ionized oxygens. ROS contain unstable bonds or unpaired valence electrons that make these molecules highly reactive. Hydroxyl radical (HO·), hydrogen peroxide (H2O2), and superoxide (O2−) are typical examples of the ROS found in mammalian cells. ROS are generally highly oxidative and are therefore innately toxic in normal cells. However, ROS do not only act as toxic molecules in normal cells, but they also function as signaling molecules so that the many biological processes and functions of normal cells rely on the ROS of the redox metabolism [181,182,183]. ROS play important roles in the modulation of cell survival, death, differentiation, proliferation, growth, and migration, cytoskeletal regulation, and the cell signaling, fertilization, and production of cytokines [184,185,186]. The roles of ROS are not limited to the modulation of these cellular functions, as ROS are directly involved in various biochemical pathways. ROS react with biomacromolecules, which play important roles in inflammatory diseases, neurodegenerative diseases, the aging process, etc., to oxidize them [183,187]. In blood, ROS react with nitric oxide (NO) to produce cytotoxic reactive nitrogen species such as NO2 and NO3, which cause oxidative injury to the endothelium, which causes cardiovascular diseases [188]. The iodination of the thyroid hormone also relies on hydrogen peroxide (H2O2) as an oxidative agent [189]. Because ROS play a dual role in oxidative damages and as either signaling molecules or enzyme substrates, normal cells tightly control the cellular redox balance to maintain the optimum ROS levels [187].
ROS molecules are generally generated from incompletely reduced oxygen, which accepts electrons from the ETCs of the mitochondria [190]. During the ETC process, some electrons escape and settle to O2, which become ROS. ROS generated by the leakage from the ETC is the most substantial source of cellular ROS (Figure 4). However, in addition to the ETC-generated ROS, the cytochrome P450 enzymes (CYPs) and NADPH oxidases (NOXs) also produce them. CYPs are a superfamily of enzymes that contain hemes as a cofactor and that function as monooxygenases, exhibiting a broad range of substrate specificities [191]. CYPs accidentally produce ROS as byproducts during their oxidation reactions, as in the case of the ETC [192]. Unlike the ROS from the ETC and CYPs, NOXs produce ROS as their intended enzymatic product rather than as a byproduct [193]. NOXs are six-transmembrane proteins with a conserved core element that contains four heme-binding histidines to transfer electrons to oxygen for the production of ROS [193]. Researchers have identified six members of the NOX family, including NOX1, NOX3, NOX4, NOX5, DUOX1, and DUOX2 [194]. These enzymes are expressed in a cell-specific manner and constitute one of the major sources of the intracellular ROS beside the mitochondria. Researchers initially identified the function of NOXs (NOX1) in the membrane of phagocytic cells, where the ROS production led to the destruction of pathogens by immune cells [195,196]. Later, researchers elucidated that NOXs play various functions in post-translational modifications, lipids, the calcium level, the oxygen sensor, and metabolic reprograming, as well as cell differentiation, transformation, growth, and death in normal cells depending on the cell type [197,198,199]. As described above for glucose and lipid metabolism, the oxidative pathways of G6PDH, 6PGLDH, ME1, and IDH1, which produce NADPH, are elevated. The increased concentration of NADPH drives the enzymatic activities of NOXs to increase the production of ROS. In this context, NOX-derived ROS are a logical contributor to the elevated concentrations of ROS in cancer cells. In fact, cancer cells ubiquitously up-regulate the expression of NOX4, and the up-regulated NOX4 enhances the production of ROS [200].
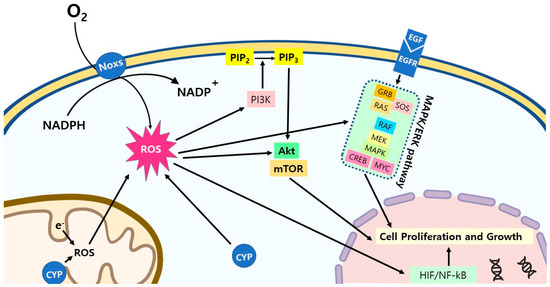
Figure 4.
ROS signaling pathways in cancer cells. ROS activate multiple signaling pathways in cancer cells for the stimulation of the cell proliferation and growth. Abbreviations: CYP: cytochrome P450; PI3K: phosphoinositide 3-kinases; PIP2: phosphatidylinositol 4,5-bisphosphate; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; AKT: protein kinase B; mTOR: mammalian target of rapamycin; EGF/EGFR: epidermal growth factor/epidermal growth factor receptor; GRB: growth factor receptor-bound protein; SOS: Son of Sevenless; RAS: Ras GTPase; RAF: RAF kinase; MEK: mitogen-activated protein kinase; MAPK: mitogen-activated protein kinase; CREB: cAMP response element-binding protein; MYC: c-MYC; HIF: hypoxia-inducible factors; NF-κB: nuclear factor kappa B.
The emergence of cancers starts with the mutagenic conversion of proto-oncogenes to oncogenes. The transformation of proto-oncogenes such as RAS, RAF, MEK, ERK, and AKT to oncogenes due to chemical, physical, or genetic mutations stimulates the cell proliferation in early cancer by activating the signaling pathways of the unbridled cell proliferation, and by inactivating the signaling pathways of the apoptosis suppression [201]. Cell proliferation demands that ATP is mostly generated from mitochondrial oxidative phosphorylation; thus, the oxidative phosphorylation pathway is upregulated in fast-dividing cells. Because of electron leakage, which is the source of ROS is an inevitable consequence of the OXPHOS pathway, the upregulation of mitochondrial OXPHOS in early cancer cells leads to an increased production of ROS [202,203]. The structure of cancer tissue also contributes to the increased production of ROS. The uncontrolled, rapid growth of cancer cells leads to the formation of unorganized cell masses, which make it impossible to develop functional neovascularization, as in the case of normal tissues. The lack of a proper vascular system in cancer gives rise to an inadequate blood supply, which results in chronic hypoxia [204]. The generation of ROS, as well as the NF-κB-dependent transcription, are facilitated in the hypoxic condition [205,206]. Moreover, the hypoxia-sensitive transcription factor HIF-1α activates the expression of NOX4 [207]. Conversely, H2O2 generated from ETS, CYPs, and NOX4 stabilizes HIF-1α for an even higher increase in the production of ROS [208,209,210]. The chain reaction between H2O2 and NOX4 that is initiated by hypoxia seems to make NOX4 a key player in ROS generation in cancer.
Both the increased OXPHOS pathway and the development of the chronic hypoxic condition inevitably contribute to the increased production of ROS in cancer. The increased ROS stimulate the proximal activation of the PI3K/Akt and MAPK/ERK signaling pathways, which stimulate the early cancer cells to more rapidly proliferate [211,212,213,214,215]. In addition to the activation of the signaling pathways, the increased ROS activate the HIF and NF-kB transcription factors [215]. These transcription factors also contribute to the stimulation of cell proliferation. Finally, the increased ROS apodictically mutate genomic DNA through their oxidative damaging capability, which contributes to genome instability [216]. Overall, ROS play the key role in the evolution of early cancer cells into more malignant cancerous cells as they progress into the later stage, while oncogenes drive the transformation of cancer cells from normal cells.
The high redox state endows cancer cells with another unique property. Most redox enzymes contain hemes with a central iron ion at their protoporphyrin cores as a cofactor [217]. Because of the high demand for redox enzymes, one of the key characteristics of cancer cells is that they actively absorb hemin, protoporphyrin, and iron [218,219,220]. Cancer cells increase the cellular concentrations of hemin and protoporphyrin by both the activate synthesis of the compounds as a result of the activated RAS/MEK pathway [221], and the upregulation of two ABC transporters: ABCG2 and ABCB6 [222,223]. Porphyrin compounds, such as hemin, selectively accumulate in cancer cells in vivo [224]. Because of the trophic feature of porphyrin compounds to cancer cells, researchers are actively developing a technology that uses a porphyrin compound as a targeting moiety in anticancer drug delivery.
6. Perspective on Future of Anti-Cancer Drug Development and Conclusions
The metabolism of cancer cells and their subsequent signaling transductions are altered in comparison with normal cells to support the elevated energy demand, as well as the acquisition and maintenance of their malignant properties. The metabolic alterations, such as the Warburg effect, ROS increase, and glutamine dependence, are the prominent features after the transformation of normal cells into cancer cells. Because researchers discovered that the Warburg effect and ROS increase first, there have been countless attempts to develop anti-cancer drugs that target them (Table 1). However, these attempts have fallen short of expectations.

Table 1.
Therapeutic strategies for targeting cancer metabolism.
Despite the fact that glutamine dependence is a hallmark of cancer cell metabolism, the significance of glutamine dependance in cancer metabolism has been realized relatively recently compared to the Warburg effect and ROS increase. As a result, the drug development of anti-cancer drugs targeting the glutamine dependence has recently started. CB-839, Sirpiglenastat (DRP-104) and DON showed excellent therapeutic efficacies in the clinical trials [285,291,294,295]. The clinical successes promoted a burst of development of anticancer drugs targeting glutamine dependence by inhibiting glutaminase or the glutamine transporter (Table 1). Unlike the anticancer drugs targeting the Warburg effect and ROS increase, these drugs do not affect the metabolism of normal cells because glutamine dependence is the uniquely limited to cancer cells. Because normal cells are not addicted to glutamine, the anticancer drugs targeting glutamine dependence showed a negligible side effect in clinical trial, which means that the drugs are ideal for combination therapies with other anticancer drugs. Because there is a plethora of cytotoxic and immunologic anticancer drugs, the development of combination therapies that use anticancer drugs that target glutamine inhibition with cytotoxic or immunologic anticancer drugs should be actively investigated as potential novel cancer treatment strategies.
Author Contributions
H.-S.C. and S.-T.H. wrote and edited the manuscript. H.-S.C. drew the figures and H.-S.C. made table. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that there are no conflict of interest.
References
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Bouchez, C.L.; Hammad, N.; Cuvellier, S.; Ransac, S.; Rigoulet, M.; Devin, A. The Warburg Effect in Yeast: Repression of Mitochondrial Metabolism Is Not a Prerequisite to Promote Cell Proliferation. Front. Oncol. 2020, 10, 1333. [Google Scholar] [CrossRef]
- Gentric, G.; Mieulet, V.; Mechta-Grigorou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox. Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 325–334. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Lui, V.W.Y.; Grandis, J.R. EGFR-mediated cell cycle regulation. Anticancer Res. 2002, 22, 1–11. [Google Scholar]
- Lo, H.-W.; Hung, M.-C. Nuclear EGFR signalling network in cancers: Linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 2007, 96, R16–R20. [Google Scholar] [CrossRef]
- Zhu, X.F.; Liu, Z.C.; Xie, B.F.; Li, Z.M.; Feng, G.K.; Yang, D.; Zeng, Y.X. EGFR tyrosine kinase inhibitor AG1478 inhibits cell proliferation and arrests cell cycle in nasopharyngeal carcinoma cells. Cancer Lett. 2001, 169, 27–32. [Google Scholar] [CrossRef]
- Chou, Y.-T.; Lin, H.-H.; Lein, Y.-C.; Wang, Y.-H.; Hong, C.-F.; Kao, Y.-R.; Lin, S.-C.; Chang, Y.-C.; Lin, S.-Y.; Chen, S.-J.; et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010, 70, 8822–8831. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal. Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Iwamoto, M.; Kawada, K.; Nakamoto, Y.; Itatani, Y.; Inamoto, S.; Toda, K.; Kimura, H.; Sasazuki, T.; Shirasawa, S.; Okuyama, H.; et al. Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J. Nucl. Med. 2014, 55, 2038–2044. [Google Scholar] [CrossRef]
- Wang, P.; Song, M.; Zeng, Z.-I.; Zhu, C.-F.; Lu, W.-H.; Yang, J.; Ma, M.-Z.; Huang, A.-M.; Hu, Y.; Huang, P. Identification of NDUFAF1 in mediating K-Ras induced mitochondrial dysfuction by a proteomic screening approach. Oncotarget 2015, 6, 3947–3962. [Google Scholar] [CrossRef]
- Neuzil, J.; Rohlena, J.; Dong, L.-F. K-Ras and mitochondria: Dangerous liaisons. Cell Res. 2012, 22, 285–287. [Google Scholar] [CrossRef][Green Version]
- Storz, P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases 2017, 8, 38–42. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancers Res. 1989, 49, 6449–6465. [Google Scholar]
- Semenza, G.L. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef]
- Soga, T. Cancer metabolism: Key players in metabolic reprogramming. Cancer Sci. 2013, 104, 275–281. [Google Scholar] [CrossRef]
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 31, 409. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Lopez-Gonzalez, J.S.; Báez-Viveros, J.L.; Aguilar-Cazares, D.; Prado-Garcia, H. Tumor cell metabolism: An integral view. Cancer Biol. Ther. 2011, 12, 939–948. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, X.; Chakravarty, H.; Yang, Z.; Tam, K.Y. Identification of Novel Pyruvate Dehydrogenase Kinase 1 (PDK1) Inhibitors by Kinase Activity-Based High-Throughput Screening for Anticancer Therapeutics. ACS Comb. Sci. 2018, 20, 660–671. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.-X.; Qian, D.Z.; Dai, M.-S. Molecular Crosstalk Between Myc and HIF in Cancer. Front. Cell Dev. Biol. 2020, 8, 590576. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Biodot, R.; Végran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN-and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signaling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jian, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef]
- Flöter, J.; Kaymak, I.; Schulze, A. Regulation of Metabolic Activity by p53. Metabolites 2017, 7, 21. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Targeted knockdown of polo-like kinase 1 alters metabolic regulation in melanoma. Cancer Lett. 2017, 394, 13–21. [Google Scholar] [CrossRef]
- Ou, B.; Zhao, J.; Guan, S.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Zheng, M.; Feng, H.; et al. Plk2 promotes tumor growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in colorectal cancer. Cancer Lett. 2016, 380, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Yu, H.; Li, W.; Li, F.; Wang, S.; Yu, N.; Jiang, Q. Plk1 promotes the migration of human lung adenocarcinoma epithelial cells via STAT3 signaling. Onco. Lett. 2018, 16, 6801–6807. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Sun, H.; Zhao, J.; Xu, Z.; Liu, Y.; Feng, H.; Peng, Z. Polo-like kinase 3 inhibits glucose metabolism in colorectal cancer by targeting HSP90/STAT3/HK2 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 426. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Kletzien, R.F.; Harris, P.K.; Foellmi, L.A. Glucose-6-phosphate dehydrogenase: A “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J. 1994, 8, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kloska, S.; Palczyński, K.; Mariciniak, T.; Talaśka, T.; Miller, M.; Wysocki, B.J.; Davis, P.; Wysocki, T.A. Queueing theory model of pentose phosphate pathway. Sci. Rep. 2022, 12, 4601. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.; Krüger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2014, 90, 927–963. [Google Scholar] [CrossRef]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stem, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Kamieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Calvo Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; Vander Heiden, M.G.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, X.; Liang, T.; Bai, X. O-GlcNAcylation: An important post-translational modification and a potential therapeutic target for cancer therapy. Mol. Med. 2022, 28, 115. [Google Scholar] [CrossRef]
- Butkinaree, C.; Park, K.; Hart, G.W. O-Linked β-N-Acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta 2010, 1800, 96–106. [Google Scholar] [CrossRef]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef]
- Gu, Y.; Mi, W.; Ge, Y.; Liu, H.; Fan, Q.; Han, C.; Yang, J.; Han, F.; Lu, X.; Yu, W. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res. 2010, 70, 6344–6351. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.P.; Ferrer, C.M.; Jackson, S.R.; Shahriari, K.S.; Vosseller, K.; Reginato, M.J. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J. Biol. Chem. 2012, 287, 11070–11081. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard 3rd, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Gu, Y.; Han, C.; Liu, H.; Fan, Q.; Zhang, X.; Cong, Q.; Yu, W. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim. Biophys. Acta 2011, 1812, 514–519. [Google Scholar] [CrossRef]
- Shi, Y.; Tomic, J.; Wen, F.; Shaha, S.; Bahlo, A.; Harrison, R.; Dennis, J.W.; Williams, R.; Gross, B.J.; Waller, S.; et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 2010, 24, 1588–1598. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Minner, S.; Guldvik, I.J.; Sandmann, M.J.; Tsourlakis, M.C.; Berge, V.; Svindland, A.; Schlomm, T.; Mills, I.G. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013, 73, 5277–5287. [Google Scholar] [CrossRef]
- Sodi, V.L.; Khaku, S.; Krutilina, R.; Schwab, L.P.; Vodadlo, D.J.; Seagroves, T.N.; Reginato, M.J. mTOR/MYC Axis Regulates O-GlcNAc Transferase (OGT) Expression and O-GlcNAcylation in Breast Cancer. Mol. Cancer Res. 2015, 13, 923–933. [Google Scholar] [CrossRef]
- Li, M.; Wu, C.; Yang, Y.; Zheng, Y.; Zheng, M.; Yu, S.; Wang, J.; Chen, L.; Li, H. 3-Phosphoglycerate dehydrogenase: A potential target for cancer treatment. Cell. Oncology 2021, 44, 541–556. [Google Scholar] [CrossRef]
- Luu, H.N.; Paragomi, P.; Wang, R.; Huang, J.Y.; Adams-Haduch, J.; Midttun, Q.; Ulvik, A.; Nguyen, T.C.; Brand, R.E.; Gao, Y. The Association between Serum and Glycine and Related-Metabolites with Pancreatic Cancer in a Prospective Cohort Study. Cancers 2022, 14, 2199. [Google Scholar] [CrossRef]
- McNamee, M.J.; Michod, D.; Niklison-Chirou, M.V. Can small molecular inhibitors that stop de novo serine synthesis be used in cancer treatment? Cell Death Discov. 2021, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Sowers, M.; Herring, J.; Zhang, W.; Tang, H.; Ou, Y.; Gu, W.; Zhang, K. Analysis of glucose-derived amino acids involved in one-carbon and cancer metabolism by stable-isotope labeling and gas chromatography mass spectrometry. Anal. Biochem. 2019, 566, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Hobson, K.; Ji, X. Methionine Restriction and Cancer Biology. Nutrients 2020, 12, 684. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phophoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Forshell, T.Z.P.; Rimpi, S.; Kreutzer, C.; Pretsch, W.; Bornkamm, G.W.; Nilsson, J.A. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012, 8, e1002573. [Google Scholar] [CrossRef]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zeng, L.-W.; Gong, R.; Yuan, F.; Shu, H.-B.; Li, S. mTORC1 activity regulates post-translational modifications of glycine decarboxylase to modulate glycine metabolism and tumorigenesis. Nat. Commun. 2021, 12, 4227. [Google Scholar] [CrossRef] [PubMed]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Ramapriyan, R.; Caetano, M.S.; Barsoumian, H.B.; Mafra, A.C.P.; Zambalde, E.P.; Menon, H.; Tsouko, E.; Welsh, J.W.; Cortez, M.A. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol. Ther. 2019, 195, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabό, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naïve T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.-Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 2007, 117, 2570–2582. [Google Scholar] [CrossRef]
- Bergström, J.; Fürst, P.; Norée, L.O.; Vinnars, E. Intracellular free amino acid concentration in human muscle tissue. J. Appl. Physiol. 1974, 36, 693–697. [Google Scholar] [CrossRef]
- Deberadinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Invest. 2013, 123, 3678–3684. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2011, 1807, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Jiang, Y.; Sun, N.; Chen, Z.; Lv, Y.; Shao, K.; Li, N.; Qiu, B.; Gao, Y.; Tan, X.; et al. Identification of isocitrate dehydrogenase 1 as a potential diagnostic and prognostic biomarker for non-small cell lung cancer by proteomic analysis. Mol. Cell Proteomics 2012, 11, M111.008821. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is turned for proliferation and vulnerable to disruption. Nature. 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Corbet, C.; Pinto, A.; Martherus, R.; de Jesus, J.P.S.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 16, 523. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 773. [Google Scholar] [CrossRef]
- Bush, A.; Mateyak, M.; Dugan, K.; Obaya, A.; Adachi, S.; Sedivy, J.; Cole, M. c-myc null cells misregulate cad and gadd45 but not other proposed c-Myc targets. Genes Dev. 1998, 12, 3797–3802. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads do glutamine addition. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Goa, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and –independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NAPDH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Csibi, A.; Fendt, S.-M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTOR1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2021, 184, 2256. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Huang, C.; Freter, C. Lipid metabolism, apoptosis and cancer therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar] [CrossRef] [PubMed]
- Kuhajda, F.P.; Pízer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prof. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef]
- Shurbaji, M.S.; Kalbfleisch, J.H.; Thurmond, T.S. Immuohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum. Pathol. 1996, 27, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Gansler, T.S.; Hardman, W., 3rd; Hunt, D.A.; Schaffel, S.; Henninger, R.A. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum. Pathol. 1997, 28, 686–692. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Pizer, E.S.; Lax, S.F.; Kuhajda, F.P.; Pasternack, G.R.; Kurman, R.J. Fatty acid synthase expression in endometrial carcinoma: Correlation with cell proliferation and hormone receptors. Cancer 1998, 83, 528–537. [Google Scholar] [CrossRef]
- Rashid, A.; Pizer, E.S.; Moga, M.; Milgraum, L.Z.; Zahurak, M.; Pasternack, G.R.; Kuhajda, F.P.; Hamilton, S.R. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am. J. Pathol. 1997, 150, 201–208. [Google Scholar] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under contruction. Trends Endocrinol. Metab. 2008, 20, 72–77. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Calvo, D.; Gomez-Coronado, D.; Suarez, Y.; Lasuncion, M.A.; Vega, M.A. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J. Lipid Res. 1998, 39, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef]
- Neiman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Li, X.; Qian, X.; Lu, Z. Local histone acetylation by ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Autophagy 2017, 13, 1790–1791. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 functions as a Protein Kinase to Coordinate Glycolysis and TCA cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Luo, D.-X.; Tong, D.-J.; Rajput, S.; Wang, C.; Liao, D.-F.; Cao, D.; Maser, E. Targeting acetyl-CoA carboxylases: Small molecular inhibitors and their therapeutic potential. Recent Pat. Anticancer Drug Discov. 2012, 7, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ariel Igal, R. Stearoyl-CoA desaturase-1: A novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010, 31, 1509–1515. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Yabe, D.; Brown, M.S.; Goldstein, J.L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 12753–12758. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavare, J.M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.-Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Gammon, S.R.; Knippers, J.D.; Paulsen, S.R.; Rubink, D.S.; Winder, W.W. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 1985 2002, 92, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Flavin, R.; Zadra, G.; Loda, M. Metabolic alterations and targeted therapies in prostate cancer. J. Pathol. 2011, 223, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-A.; Han, W.-F.; Morin, P.J.; Chrest, F.J.; Pizer, E.S. Activation of Fatty Acid Synthesis during Neoplastic Transformation: Role of Mitogen-Activated Protein Kinase and Phosphatidylinositol 3-Kinase. Exp. Cell Res. 2002, 279, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.-J.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Liu, L.; Yan, H.; Ruan, M.; Yang, H.; Wang, L.; Lei, B.; Sun, X.; Chang, C.; Huang, G.; Xie, W. An AKT/PRMT5/SREBP1 axis in lung adenocarcinoma regulates de novo lipogenesis and tumor growth. Cancer Sci. 2021, 112, 3083–3098. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Oh, Y.-T.; Yue, P.; Khuri, F.R.; Sun, S.-Y. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 2016, 35, 642–650. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V., Jr. The life of lipid droplets. Biochim. Biophys. Acta 2009, 179, 459–466. [Google Scholar] [CrossRef]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth vis Autophagic Release of Stored Fatty Acids. IScience 2020, 23, 101569. [Google Scholar] [CrossRef]
- Harris, C.A.; Haas, J.T.; Streeper, R.S.; Stone, S.J.; Kumari, M.; Yang, K.; Han, X.; Brownell, N.; Gross, R.W.; Zechner, R.; et al. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J. Lipid Res. 2011, 52, 657–667. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Guan, L.; Chen, Y.; Chen, P.; Sun, J.; Gonzalez, F.J.; Huang, M.; Bi, H. Lipidomics reveals carnitine palmitoyltransferase 1C protects cancer cells from lipotoxicity and senescence. J. Pharm. Anal. 2021, 11, 340–350. [Google Scholar] [CrossRef]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes. Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef]
- Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activators and cancers: Complex stories. Nat. Rev. Cancer 2004, 4, 61–70. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef]
- Sanchez-Macedo, N.; Feng, J.; Faubert, B.; Chang, N.; Elia, A.; Rushing, E.J.; Tsuchihara, K.; Bungard, D.; Berger, S.L.; Jones, R.G.; et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013, 20, 659–668. [Google Scholar] [CrossRef]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mark, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef]
- Xing, F.; Geng, L.; Guan, H.; Liu, D.; Li, Y.; Zeng, L.; Chen, Y.; Tian, R.; Li, Z.; Cao, R.; et al. Astragalin mitigates inflammatory osteolysis by negatively modulating osteoclastogenesis via ROS and MAPK signaling pathway. Int. Immunopharmacol. 2022, 112, 109278. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Rojas, L.; Casillas, F.; López, A.; Betancourt, M.; Ommati, M.M.; Retana-Márquez, S. Physiological role of reactive oxygen species in testis and epididymal spermatozoa. Andrologia 2022, 54, e14367. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Murtaza, G.; Metwally, E.; Kalhoro, D.H.; Kalhoro, M.S.; Rahu, B.A.; Sahito, R.G.A.; Yin, Y.; Yang, H.; Chughtai, M.I.; et al. The Role of Oxidative Stress and Antioxidant Balance in Pregnancy. Mediat. Inflamm. 2021, 2021, 9962860. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Molecular and cellular mechanisms in vascular injury in hypertension: Role of angiotensin II-editorial review. Curr. Opin. Nephrol. Hypertens. 2005, 14, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.F.; Laude, K.; McNally, J.S.; Harrison, D.G. ATVB in focus: Redox mechanisms in blood vessels. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Abdulfattah, A.Y.; Warren, B.O.; Shu, J.; Said, N. Redox Homeostasis and Metabolism in Cancer: A Complex Mechanism and Potential Targeted Therapeutics. Int. J. Mol. Sci. 2020, 21, 3100. [Google Scholar] [CrossRef]
- Sarkar, D.; Fisher, P.B. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2006, 236, 13–23. [Google Scholar] [CrossRef]
- Wilcox, J.N.; Subramanian, R.R.; Sundell, C.L.; Tracey, W.R.; Pollock, J.S.; Harrison, D.G.; Marsden, P.A. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2479–2488. [Google Scholar] [CrossRef]
- Lagorce, J.F.; Thomes, J.C.; Catanzano, G.; Buxeraud, J.; Raby, M.; Raby, C. Formation of molecular iodine during oxidation of iodide by the peroxidase/H2O2 system. Implications for antithyroid therapy. Biochem. Pharmacol. 1991, 42, S89–S92. [Google Scholar] [CrossRef]
- Trigo, D.; Nadais, A.; Carvalho, A.; Morgado, B.; Santos, F.; Pereira, S.; da Cruz, E.; Silva, O.A.B. Mitochondria dysfunction and impaired response to oxidative stress promotes proteostasis disruption in aged human cells. Mitochondrion 2022, 20, S1567–S7249. [Google Scholar] [CrossRef]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. Computational Prediction of Inhibitors and Inducers of the Major Isoforms of Cytochrome P450. Molecules 2022, 27, 5875. [Google Scholar] [CrossRef] [PubMed]
- Akintade, D.D.; Chaudhuri, B. Sensing the Generation of Intracellular Free Electrons Using the Inactive Catalytic Subunit of Cytochrome P450s as a Sink. Sensors 2020, 20, 4050. [Google Scholar] [CrossRef] [PubMed]
- Oosterheert, W.; Reis, J.; Gros, P.; Mattevi, A. An Elegant Four-Helical Fold in NOX and STEAP Enzymes Facilitates Electron Transport across Biomembranes-Similar Vehicle, Different Destination. Acc. Chem. Res. 2020, 53, 1969–1980. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Zatti, M. Biochemical Aspects of Phagocytosis in Polymorphonuclear Leucocytes. NADH and NADPH Oxidation by the Granules of Resting and Phagocytizing Cells. Experientia 1964, 20, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.A.; Savelieff, M.G.; Eid, A.A.; Feldman, E.L. Nox, Nox, Are You There? The Role of NADPH Oxidases in the Peripheral Nervous System. Antioxid. Redox. Signal. 2022, 37, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, T.; Angeloni, C.; Prata, C.; Hrelia, S. NADPH Oxidases: Redox Regulators of Stem Cell Fate and Function. Antioxidants 2021, 10, 973. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Wang, Z.; Wu, P.; Su, X.; Wu, Z.; Gan, J.; Zhang, D. NOX4 has the potential to be a biomarker associated with colon cancer ferroptosis and immune infiltration based on bioinformatics analysis. Front. Oncol. 2022, 12, 968043. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef]
- Arsova-Sarafinovska, Z.; Matevska, N.; Petrovski, D.; Banev, S.; Dzikova, S.; Georgiev, V.; Sikole, A.; Sayal, A.; Aydin, A.; Suturkova, L.; et al. Manganese superoxide dismutase (MnSOD) genetic polymorphism is associated with risk of early-onset prostate cancer. Cell Biochem. Funct. 2008, 26, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Bakker, A.C.; Vanhaesebroeck, B.; Beyaert, R.; Jacob, W.A.; Fiers, W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 1992, 267, 5317–5323. [Google Scholar] [CrossRef] [PubMed]
- Graff, S.; Barach, A.; Bickerman, H.A.; Beck, G.J.; Eisenberg, M.; Freedman, A.D. Hypoxia in cancer. Acta Unio. Int. Contra. Cancrum. 1960, 16, 651–655. [Google Scholar] [PubMed]
- Muraoka, K.; Shimizu, K.; Sun, X.; Zhang, Y.K.; Tani, T.; Hashimoto, T.; Yagi, M.; Miyazaki, I.; Yamamoto, K. Hypoxia, but not reoxygenation, induces interleukin 6 gene expression through NF-kappa B activation. Transplantation 1997, 63, 466–470. [Google Scholar] [CrossRef]
- Klassen, D.K.; Sagone, A.L., Jr. Evidence for both oxygen and non-oxygen dependent mechanisms of antibody sensitized target cell lysis by human monocytes. Blood 1980, 56, 985–992. [Google Scholar] [CrossRef]
- Littauer, A.; de Groot, H. Release of reactive oxygen by hepatocytes on reoxygenation: Three phases and role of mitochondria. Am. J. Physiol. 1992, 262, G1015–G1020. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef]
- Maranchie, J.K.; Zhan, Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von Hippel-Lindau-deficient renal cell carcinoma. Cancer Res. 2005, 65, 9190–9193. [Google Scholar] [CrossRef]
- Diebold, I.; Flugel, D.; Becht, S.; Belaiba, R.S.; Bonello, S.; Hess, J.; Kietzmann, T.; Gorlach, A. The hypoxia-inducible factor-2alpha is stabilized by oxidative stress involving NOX4. Antioxid. Redox. Signal. 2010, 13, 425–436. [Google Scholar] [CrossRef]
- Tang, P.; Dang, H.; Huang, J.; Xu, T.; Yuan, P.; Hu, J.; Sheng, J.F. NADPH oxidase NOX4 is a glycolytic regulator through mROS-HIF1alpha axis in thyroid carcinomas. Sci. Rep. 2018, 8, 15897. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Yin, Q.; Fujio, Y.; Walsh, K.; Griendling, K.K. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 22699–22704. [Google Scholar] [CrossRef] [PubMed]
- Kosmidou, I.; Xagorari, A.; Roussos, C.; Papapetropoulos, A. Reactive oxygen species stimulate VEGF production from C(2)C(12) skeletal myotubes through a PI3K/Akt pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L585–L592. [Google Scholar] [CrossRef] [PubMed]
- Endoh, H.; Sasaki, H.; Maruyama, K.; Takeyama, K.; Waga, I.; Shimizu, T.; Kato, S.; Kawashima, H. Rapid activation of MAP kinase by estrogen in the bone cell line. Biochem. Biophys. Res. Commun. 1997, 235, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Vallyathan, V.; Ding, M.; Shi, X.; Castranova, V. Molecular Activation of Activator Protein-1 In Silica and Asbestos-Induced Carcinogenesis. Inhal. Toxicol. 2000, 12, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Young, M.R.; Colburn, N.H. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: The JB6 model is predictive. Mol. Cell Biochem. 2002, 234, 185–193. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nakajima, M.; Morimoto, K. Establishment of a human system that generates O2- and induces 8-hydroxydeoxyguanosine, typical of oxidative DNA damage, by a tumor promotor. Cancer Res. 1994, 54, 5837–5840. [Google Scholar]
- Capitanio, G.; Papa, F.; Papa, S. The allosteric protein interactions in the proton-motive function of mammalian redox enzymes of the respiratory chain. Biochimie 2021, 189, 1–12. [Google Scholar] [CrossRef]
- Chung, H.-J.; Lee, H.-K.; Kwon, K.B.; Kim, H.-J.; Hong, S.-T. Transferrin as a thermosensitizer in radiofrequency hyperthermia for cancer treatment. Sci. Rep. 2018, 8, 13505. [Google Scholar] [CrossRef]
- Chung, H.-J.; Kim, H.-J.; Hong, S.-T. Tumor-specific delivery of a paclitaxel-loading HSA-haemin nanoparticle for cancer treatment. Nanomedicine 2020, 23, 102089. [Google Scholar] [CrossRef]
- Chung, H.-J.; Kim, H.-J.; Hong, S.-T. Iron-dextran as a thermosensitizer in radiofrequency hyperthermia for cancer treatment. Appl. Biol. Chem. 2019, 62, 1–9. [Google Scholar] [CrossRef]
- Yoshioka, E.; Chelakkot, V.S.; Licursi, M.; Rutihinda, S.G.; Som, J.; Derwish, L.; King, J.J.; Pongnopparat, T.; Mearow, K.; Larijani, M.; et al. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics 2018, 8, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.C.; Du, G.; Fukuda, Y.; Sun, D.; Sampath, J.; Mercer, K.E.; Wang, J.; Sosa-Pineda, B.; Murti, K.G.; Schuetz, J.D. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006, 443, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, M.; Matsuo, H.; Toyoda, Y.; Yonekura, Y.; Ishikawa, T.; Nakayama, A.; Higashino, T.; Kawamura, Y.; Fujimoto, N.; Shinomiya, N.; et al. Porphyrin accumulation in humans with common dysfunctional variants of ABCG2, a porphyrin transporter: Potential association with acquired photosensitivity. Hum. Cell 2021, 34, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Tsiftsoglou, A.S.; Tsamadou, A.T.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006, 111, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J.; et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef]
- Wu, K.-H.; Ho, C.-T.; Chen, Z.-F.; Chen, L.-C.; Whang-Peng, J.; Lin, T.-N.; Ho, Y.-S. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J. Food Drug. Anal. 2018, 26, 221–231. [Google Scholar] [CrossRef]
- Yang, K.-C.; Tsai, C.-Y.; Wang, Y.-J.; Wei, P.-L.; Lee, C.-H.; Chen, J.-H.; Wu, C.-H.; Ho, Y.-S. Apple polyphenol phloretin potentiates the anticancer actions of paclitaxel through induction of apoptosis in human hep G2 cells. Mol. Carcinog. 2009, 48, 420–431. [Google Scholar] [CrossRef]
- Granchi, C.; Fortunato, S.; Minutolo, F. Anticancer agents interacting with membrane glucose transporters. MedChemComm 2016, 7, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: Implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.A.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.R.E.; Zhang, Y.; Tao, J.; Shen, R.; Zhao, X.; Cleary, M.P.; Wang, T.; Yang, D.-Q. ATM inhibitor KU-55933 induces apoptosis and inhibits motility by blocking GLUT1-mediated glucose uptake in aggressive cancer cells with sustained activation of Akt. FASEB J. 2021, 35, e21264. [Google Scholar] [CrossRef]
- Gwak, H.; Haegeman, G.; Tsang, B.K.; Song, Y.S. Cancer-specific interruption of glucose metabolism by resveratrol is mediated through inhibition of Akt/GLUT1 axis in ovarian cancer cells. Mol. Carcinog. 2015, 54, 1529–1540. [Google Scholar] [CrossRef]
- Zhang, X.D.; Deslandes, E.; Villedieu, M.; Poulain, L.; Duval, M.; Gauduchon, P.; Schwartz, L.; Icard, P. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res. 2006, 26, 3561–3566. [Google Scholar]
- Chen, X.-S.; Li, L.-Y.; Guan, Y.-D.; Yang, J.-M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Zhang, P.; Chao, Z.; Xia, F.; Jiang, C.; Zhang, X.; Jiang, Z.; Liu, H. Hexokinase II inhibitor, 3-BrPA induced autophagy by stimulating ROS formation in human breast cancer cells. Genes. Cancer 2014, 5, 100–112. [Google Scholar] [CrossRef]
- Jae, H.J.; Chung, J.W.; Park, H.S.; Lee, M.J.; Lee, K.C.; Kim, H.-C.; Yoon, J.H.; Chung, H.; Park, J.H. The antitumor effect and hepatotoxicity of a hexokinase II inhibitor 3-bromopyruvate: In vivo investigation of intraarterial administration in a rabbit VX2 hepatoma model. Korean J. Radiol. 2009, 10, 596–603. [Google Scholar] [CrossRef]
- Amadori, D.; Frassineti, G.L.; De Matteis, A.; Mustacchi, G.; Santoro, A.; Cariello, S.; Ferrari, M.; Nascimben, O.; Nanni, O.; Lombardi, A.; et al. Modulating effect of lonidamine on response to doxorubicin in metastatic breast cancer patients: Results from a multicenter prospective randomized trial. Breast Cancer Res. Treat. 1998, 49, 209–217. [Google Scholar] [CrossRef]
- Nath, K.; Nelson, D.S.; Heitjan, D.F.; Leeper, D.B.; Zhou, R.; Glickson, J.D. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015, 28, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wei, L.; Liu, Y.; Ding, Y.; Liu, X.; Zhang, X.; Wang, X.; Yao, Y.; Lu, J.; Wang, Q.; et al. Gen-27, a newly synthesized flavonoid, inhibits glycolysis and induces cell apoptosis via suppression of hexokinase II in human breast cancer cells. Biochem. Pharmacol. 2017, 125, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, Y.; Liu, C.; Wang, H.; Fan, H.; Li, Y.; Wang, J.; Zhang, X.; Lu, J.; Ji, H.; et al. Chemopreventive Activity of GEN-27, a genistein derivative, in colitis-associated cancer is mediated by p65-CDX2-β-catenin axis. Oncotarget 2016, 7, 17870–17884. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zheng, M.; Wu, S.; Gao, S.; Yang, M.; Li, Z.; Min, Q.; Sun, W.; Chen, L.; Xiang, G.; et al. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 2017, 36, 58. [Google Scholar] [CrossRef]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef]
- Li, W.; Hao, J.; Zhang, L.; Cheng, Z.; Deng, X.; Shu, G. Astragalin Reduces Hexokinase 2 through Increasing miR-125b to Inhibit the Proliferation of Hepatocellular Carcinoma Cells in Vitro and in Vivo. J. Agric. Food Chem. 2017, 65, 5961–5972. [Google Scholar] [CrossRef]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flus and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A., 2nd; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Rhier, N.J.; Molinier, N.; Long, C.; Deshmukh, S.K.; Kate, A.S.; Ranadive, P.; Verekar, S.A.; Jiotode, M.; Lavhale, R.R.; Tokdar, R.; et al. Anticancer activity of koningic acid and semisynthetic derivatives. Bioorg. Med. Chem. 2015, 23, 3712–3721. [Google Scholar] [CrossRef]
- Li, T.; Tan, X.; Yang, R.; Miao, Y.; Zhang, M.; Xi, Y.; Guo, R.; Zheng, M.; Li, B. Discovery of novel glyceraldehyde-3-phosphate dehydrogenase inhibitor via docking-based virtual screening. Bioorg. Chem. 2020, 96, 103620. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Howell, S.K.; Sanford, R.J.; Beisswenger, P.J. Methylglyoxal can modify GAPDH activity and structure. Ann. N. Y. Acad. Sci. 2005, 1043, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Steták, A.; Veress, R.; Ovádi, J.; Csermely, P.; Kéri, G.; Ullrich, A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007, 67, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Vitamin K(3) and K(5) are inhibitors of tumor pyruvate kinase M2. Cancer Lett. 2012, 16, 204–210. [Google Scholar] [CrossRef]
- Liu, J.; Wu, N.; Ma, L.; Liu, M.; Liu, G.; Zhang, Y.; Lin, X. Oleanolic acid suppresses aerobic glycolysis in cancer cells by switching pyruvate kinase type M isoforms. PLoS ONE 2014, 9, e91606. [Google Scholar] [CrossRef]
- Alistar, A.T.; Desnoyers, R.; D’Agostino, R.J.; Pasche, B. CPI-613 enhances FOLFIRINOX response rate in stage IV pancreatic cancer. Ann. Oncol. 2016, 27, VI228. [Google Scholar] [CrossRef]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef]
- Whitehouse, S.; Cooper, R.H.; Randle, P.J. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 1974, 141, 761–774. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef]
- Li, C.; Meng, G.; Su, L.; Chen, A.; Xia, M.; Xu, C.; Yu, D.; Jiang, A.; Wei, J. Dichloroacetate blocks aerobic glycolytic adaptation to attenuated measles virus and promotes viral replication leading to enhanced oncolysis in glioblastoma. Oncotarget 2015, 30, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. USA 2009, 106, 22199–22204. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Kulak, N.; Pridgen, E.M.; Farokhzad, O.C.; Langer, R.; Lippard, S.J. Nanoparticle encapsulation of mitaplatin and the effect thereof on in vivo properties. ACS Nano. 2013, 7, 5675–5683. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Ghosh, M.; Dutta, S.K. A potent tumoricidal co-drug ‘Bet-CA’-an ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Sci. Rep. 2015, 5, 7762. [Google Scholar] [CrossRef]
- PAPACONSTANTINOU, J.; COLOWICK, S.P. The role of glycolysis in the growth of tumor cells. II. The effect of oxamic acid on the growth of HeLa cells in tissue culture. J. Biol. Chem. 1961, 236, 285–288. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. Recent update on Human Lactate Dehydrogenase Enzyme 5 (hLDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Coyle, T.; Levante, S.; Shetler, M.; Winfield, J. In vitro and in vivo cytotoxicity of gossypol against central nervous system tumor cell lines. J. Neurooncol. 1994, 19, 25–35. [Google Scholar] [CrossRef]
- Van Poznak, C.; Seidman, A.D.; Reidenberg, M.M.; Moasser, M.M.; Sklarin, N.; Van Zee, K.; Borgen, P.; Gollub, M.; Bacotti, D.; Yao, T.J.; et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: A phase I/II clinical trial. Breast Cancer Res. Treat. 2001, 66, 239–248. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stenfano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxylate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Di Moanco, M.; Pizzini, A.; Gatto, V.; Leonardi, L.; Gallo, M.; Brignardello, E.; Boccuzzi, G. Role of glucose-6-phosphate dehydrogenase inhibition in the antiproliferative effects of dehydroepiandrosterone on human breast cancer cells. Br. J. Cancer 1997, 75, 589–592. [Google Scholar] [CrossRef]
- Ho, H.-Y.; Cheng, M.-L.; Chiu, H.-Y.; Weng, S.-F.; Chiu, D.T.-Y. Dehydroepiandrosterone induces growth arrest of hepatoma cells via alteration of mitochondrial gene expression and function. Int. J. Oncol. 2008, 33, 969–977. [Google Scholar]
- Kaushik, N.; Kaushik, N.K.; Choi, E.H.; Kim, J.H. Blockade of Cellular Energy Metabolism through 6-Aminonicotinamide Reduces Proliferation of Non-Small Lung Cancer Cells by Inducing Endoplasmic Reticulum Stress. Biology 2021, 10, 1088. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, S.; Zhang, Y.; Wu, J.; Peng, H.; Fan, J.; Liao, J. Reactive oxygen species-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to polydatin-induced apoptosis in human nasopharyngeal carcinoma CNE cells. J. Cell Biochem. 2011, 112, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Luo, Y.; Lu, J.; Wang, Y.; Sheng, G. Polydatin Induces Apoptosis and Inhibits Growth of Acute Monocytic Leukemia Cells. J. Biochem. Mol. Toxicol. 2016, 30, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Mullarky, E.; Lucki, N.C.; Zavareh, R.B.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.Y.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.-K.; Lei, H.-M.; Liang, Q.; Tang, Y.-B.; Zhou, Y.; Wang, Y.; Zhang, S.; Li, W.-B.; Tong, Y.; Zhuang, G.; et al. Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics 2018, 8, 1808–1823. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Invest. 2021, 131, e140100. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLCa5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-J.; Meng, L.-Y.; Shen, Y.; Zhu, Y.-Z.; Liu, H.-R. S-benzyl-cysteine-mediated cell cycle arrest and apoptosis involving activation of mitochondrial-dependent caspase cascade through the p53 pathway in human gastric cancer SGC-7901 cells. Asian Pac. J. Cancer Prev. 2013, 14, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Van Geldermalsen, M.; Quek, L.-E.; Turner, N.; Freidman, N.; Pang, A.; Guan, Y.F.; Krycer, J.R.; Ryan, R.; Wang, Q.; Holst, J. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer 2018, 18, 689. [Google Scholar] [CrossRef]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K.; et al. Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 2010, 30, 4819–4828. [Google Scholar]
- Mueller, C.; Al-Batran, S.; Jaeger, E.; Schmidt, B.; Bausch, M.; Unger, C.; Sethuraman, N. A Phase IIa study of PEGylated glutaminase (PEG-PGA) plus 6-diazo-5-oxo-L-norleucine (DON) in patients with advanced refractory solid tumors. J. Clin. Oncol. 2008, 26, 2533. [Google Scholar] [CrossRef]
- Hanaford, A.R.; Alt, J.; Rais, R.; Wang, S.Z.; Kaur, H.; Thorek, D.L.J.; Eberhart, C.G.; Slusher, B.S.; Martin, A.M.; Raabe, E.H. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl. Oncol. 2019, 12, 1314–1322. [Google Scholar] [CrossRef]
- Yamashita, A.S.; da Costa Rosa, M.; Stumpo, V.; Rais, R.; Slusher, B.S.; Riggins, G.J. The glutamine anatagonist producing JHU-083 slows malignant glioma growth and disrupts mTOR signaling. Neurooncol. Adv. 2020, 3, vdaa149. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Estok, T.M.; Wild, R. Sirpiglenastat (DRP-104) Induces Antitumor Efficacry through Direct, Broad Antagonism of Glutamine Metabolism and Stimulation of the Innate and Adaptive Immune Systems. Mol. Cancer Ther. 2022, 21, 1561–1572. [Google Scholar] [CrossRef]
- Nedelcovych, M.T.; Tenora, L.; Kim, B.-H.; Kelschenbach, J.; Chao, W.; Hadas, E.; Jančařík, A.; Prchalová, E.; Zimmermann, S.C.; Dash, R.P.; et al. N-(Pivaloyloxy)alkoxy-carbonyl Prodrugs of the Glutamine Antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a Potential Treatment for HIV Associated Neurocognitive Disorders. J. Med. Chem. 2017, 60, 7186–7198. [Google Scholar] [CrossRef]
- Pham, K.; Maxwell, M.J.; Sweeney, H.; Alt, J.; Rais, R.; Eberhart, C.G.; Slusher, B.S.; Raabe, E.H. Novel Glutamine Antagonist JHU395 Suppresses MYC-Driven Medulloblastoma Growth and Induces Apoptosis. J. Neuropathol. Exp. Neurol. 2021, 80, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Rais, R.; Jančařík, A.; Tenora, L.; Nedelcovych, M.; Alt, J.; Englert, J.; Rojas, C.; Le, A.; Elgogary, A.; Tan, J.; et al. Discovery of 6-Diazo-5-oxo-I-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J. Med. Chem. 2016, 59, 8621–8633. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed]
- Masamha, C.P.; LaFontaine, P. Molecular targeting of glutaminase sensitizes ovarian cancer cells to chemotherapy. J. Cell Biochem. 2018, 119, 6136–6145. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Reis, L.M.D.; Adamoski, D.; Souza, R.O.O.; Ascencão, C.F.R.; de Oliveira, K.R.S.; Corrêa-da-Silva, F.; de Sá Patroni, F.M.; Dias, M.M.; Consonni, S.R.; de Moraes-Vieira, P.M.M.; et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 2019, 294, 9342–9357. [Google Scholar] [CrossRef]
- Simpson, N.E.; Tryndyak, V.P.; Pogribna, M.; Beland, F.A.; Pogribny, I.P. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics 2012, 7, 1413–1420. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar]
- Lee, Y.-Z.; Yang, C.-W.; Chang, H.-Y.; Hsu, H.-Y.; Chen, I.-S.; Chang, H.-S.; Lee, C.-H.; Lee, J.-C.; Kumar, C.R.; Qiu, Y.-Q.; et al. Discovery of selective inhibitors of Glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget 2014, 5, 6087–6101. [Google Scholar] [CrossRef]
- Yeh, T.-K.; Kuo, C.-C.; Lee, Y.-Z.; Ke, Y.-Y.; Chu, K.-F.; Hsu, H.-Y.; Chang, H.-Y.; Liu, Y.-W.; Song, J.-S.; Yang, C.-W.; et al. Design, Synthesis, and Evaluation of Thiazolidine-2,4,-dione Derivatives as a Novel Class of Glutaminase Inhibitors. J. Med. Chem. 2017, 60, 5599–5612. [Google Scholar] [CrossRef]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chun, J.; Pan, C.; Kumar, A.; Zhang, G.; Ha, Y.; Li, D.; Alesi, G.N.; Kang, Y.; Zhou, L.; et al. The PLAG1-GDH1 Axia Promotes Anoikis Resistance and Tumor Metastasis through CamKK2-AMPK Signaling in LKB1-Deficient Lung Cancer. Mol. Cell 2018, 69, 87–99.e7. [Google Scholar] [CrossRef] [PubMed]
- Vacanti, N.M.; Divakaruni, A.S.; Green, C.R.; Parker, S.J.; Henry, R.R.; Ciaraldi, T.P.; Murphy, A.N.; Metallo, C.M. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell 2014, 56, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijόn, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Xiao, L.; Tang, M.; Bai, F.; Li, J.; Li, L.; Shi, F.; Li, N.; Li, Y.; Du, Q.; et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics 2018, 8, 2329–2347. [Google Scholar] [CrossRef]
- Jariwala, N.; Mehta, G.A.; Bhatt, V.; Hussein, S.; Parker, K.A.; Yunus, N.; Parker, J.S.; Guo, J.Y.; Gatza, M.L. CPT1A and fatty acid β-oxidation are essential for tumor cell growth and survival in hormone receptor-positive breast cancer. NAR Cancer 2021, 3, zcab035. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, Q.; Zhao, C.; Li, J.; Yu, Z.; Zhu, Q. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce heptatocellular carcinoma cell apoptosis via p-elF2α/ATF4/CHOP axis. J. Cell Mol. Med. 2021, 25, 1468–1479. [Google Scholar] [CrossRef]
- Wei, X.; Shi, J.; Lin, Q.; Ma, X.; Pang, Y.; Mao, H.; Li, R.; Lu, W.; Wang, Y.; Liu, P. Targeting ACLY Attenuates Tumor Growth and Acquired Cisplatin Resistance in Ovarian Cancer by Inhibiting the PI3K-AKT Pathway and Activating the AMPK-ROS Pathway. Front. Oncol. 2021, 11, 642229. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Cuyàs, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, Á.G.; Menendez, J. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef]
- Shiragami, R.; Murata, S.; Kosugi, C.; Tezuka, T.; Yamazaki, M.; Hirano, A.; Yoshimura, Y.; Suzuki, M.; Shuto, K.; Koda, K. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int. J. Oncol. 2013, 43, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wu, P.; Senthikumar, R.; Tian, X.; Liu, H.; Shen, X.; Tao, Z.; Huang, P. Loss of fatty acid synthase suppresses the malignant phenotype of colorectal cancer cells by down-regulating energy metabolism and mTOR signaling pathway. J. Cancer Res. Clin. Oncol. 2016, 142, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.; Infante, J.; Arkenau, H.-T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef] [PubMed]
- Grube, S.; Dϋnisch, P.; Fretag, D.; Kausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J. Neurooncol. 2014, 118, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Kumar, A.; Singh, S.M. Fatty acid synthase inhibitor orlistat induces apoptosis in T cell lymphoma: Role of cell survival regulatory molecules. Biochim. Biophys. Acta 2012, 1820, 1754–1773. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005. [Google Scholar] [CrossRef]
- Mohammadzadeh, F.; Mosayebi, G.; Montazeri, V.; Darabi, M.; Fayezi, S.; Shaaker, M.; Rahmati, M.; Baradaran, B.; Mehdizadeh, A.; Darabi, M. Fatty Acid Composition of Tissue Cultured Breast Carcinoma and the Effect of Stearoyl-CoA Desaturase 1 Inhibition. J. Breast Cancer 2014, 17, 136–142. [Google Scholar] [CrossRef]
- Von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef]
- Piao, C.; Cui, X.; Zhan, B.; Li, J.; Li, Z.; Li, Z.; Liu, X.; Bi, J.; Zhang, Z.; Kong, C. Inhibition of stearoyl CoA desaturase-1 activity suppresses tumour progression and improves prognosis in human bladder cancer. J. Cell Mol. Med. 2019, 23, 2064–2076. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, T.; Yan, H.; Guo, K.; Liu, Z.; Wei, L.; Lu, W.; Qiu, C.; Jiang, J. Fatostatin reverses progesterone resistance by inhibiting the SREBP1-NF-ĸB pathway in endometrial carcinoma. Cell Death Dis. 2021, 12, 544. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, N.; Zhang, H.; Wang, L.; Duan, Y.; Wang, X.; Chen, T.; Liang, Y.; Li, Y.; Song, X.; et al. Fatostatin in Combination with Tamoxifen Induces Synergistic Inhibition in ER-Positive Breast Cancer. Drug Des. Devel. Ther. 2020, 14, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-H.; Mun, J.-G.; Jeon, H.-D.; Kee, J.-Y.; Hong, S.-H. Betulin inhibits Lung Metastasis by Inducing Cell Cycle Arrest, Autophagy, and Apoptosis of Metastatic Colorectal Cancer Cells. Nutrients 2020, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Siqingaowa; Sekar, S.; Gopalakrishnan, V.; Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 2017, 488, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-B.; Liu, X.-Y.; Jia, H.; Zhang, Y.; Jiang, Q.; Sun, H.; Li, X.; Sun, F.; Chai, Y.; Feng, F.; et al. A Novel Small-Molecule Inhibitor of SREBP-1 Based on Natural Product Monomers Upregulates the Sensitivity of Lung Squamous Cell Carcinoma Cells to Antitumor Drugs. Front. Pharmacol. 2022, 13, 895744. [Google Scholar] [CrossRef]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicenter, randomized, double-blind, placebo-controlled, phase 3 study. Lancet. Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Dogra, R.; Bhatia, R.; Shankar, R.; Bansal, P.; Rawal, R.K. Enasidenib: First Mutant IDH2 Inhibitor for the Treatment of Refractory and Relapsed Acute Myeloid Leukemia. Anticancer Agents Med. Chem. 2018, 18, 1936–1951. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.H.; de Botton, S.; Zeidan, A.M.; Fathi, A.T.; Kantarjian, H.M.; Bennett, J.M.; et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): A single-arm, phase 1b and randomized, phase 2 trial. Lancet Oncol. 2021, 22, 1597–1608. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.C.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A First-in Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Penas-Prado, M.; Peters, K.B.; Burris 3rd, H.A.; Maher, E.A.; Janku, F.; Cote, G.M.; de la Fuente, M.I.; Clarke, J.L.; Ellingson, B.M.; et al. Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-Human Phase 1 trial. Clin. Cancer Res. 2021, 27, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.-M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.I.; Colman, H.; Rosenthal, M.; Van Tine, B.A.; Levacic, D.; Walbert, T.; Gan, H.K.; Vieito, M.; Milhem, M.M.; Lipford, K.; et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: A multicenter, open-label, phase 1b/2 trial. Neuro. Oncol. 2022, 25, noac139. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).