
Stimulation of Osteoclast Formation by Oncostatin M and the Role of WNT16 as a Negative Feedback Regulator


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
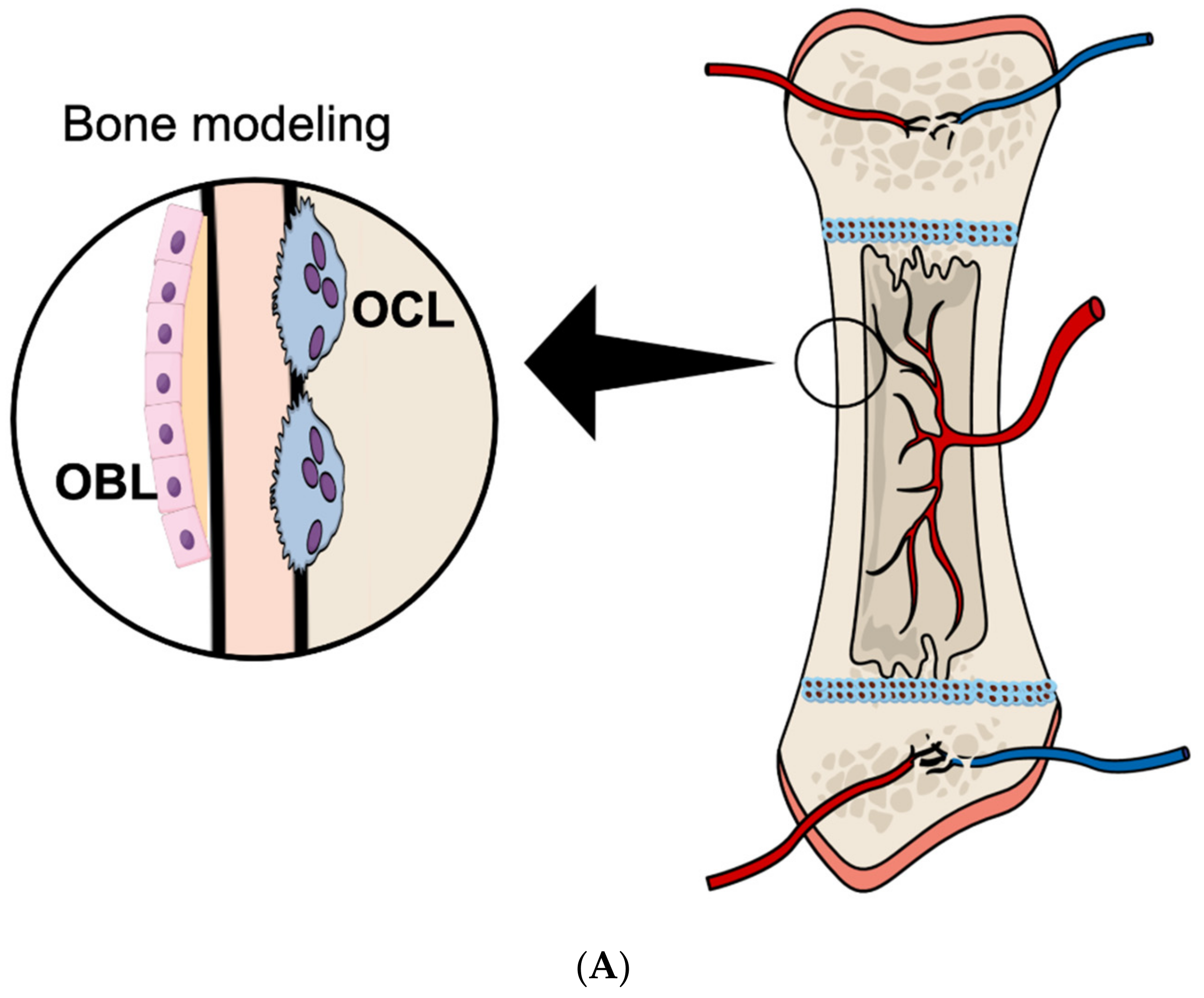
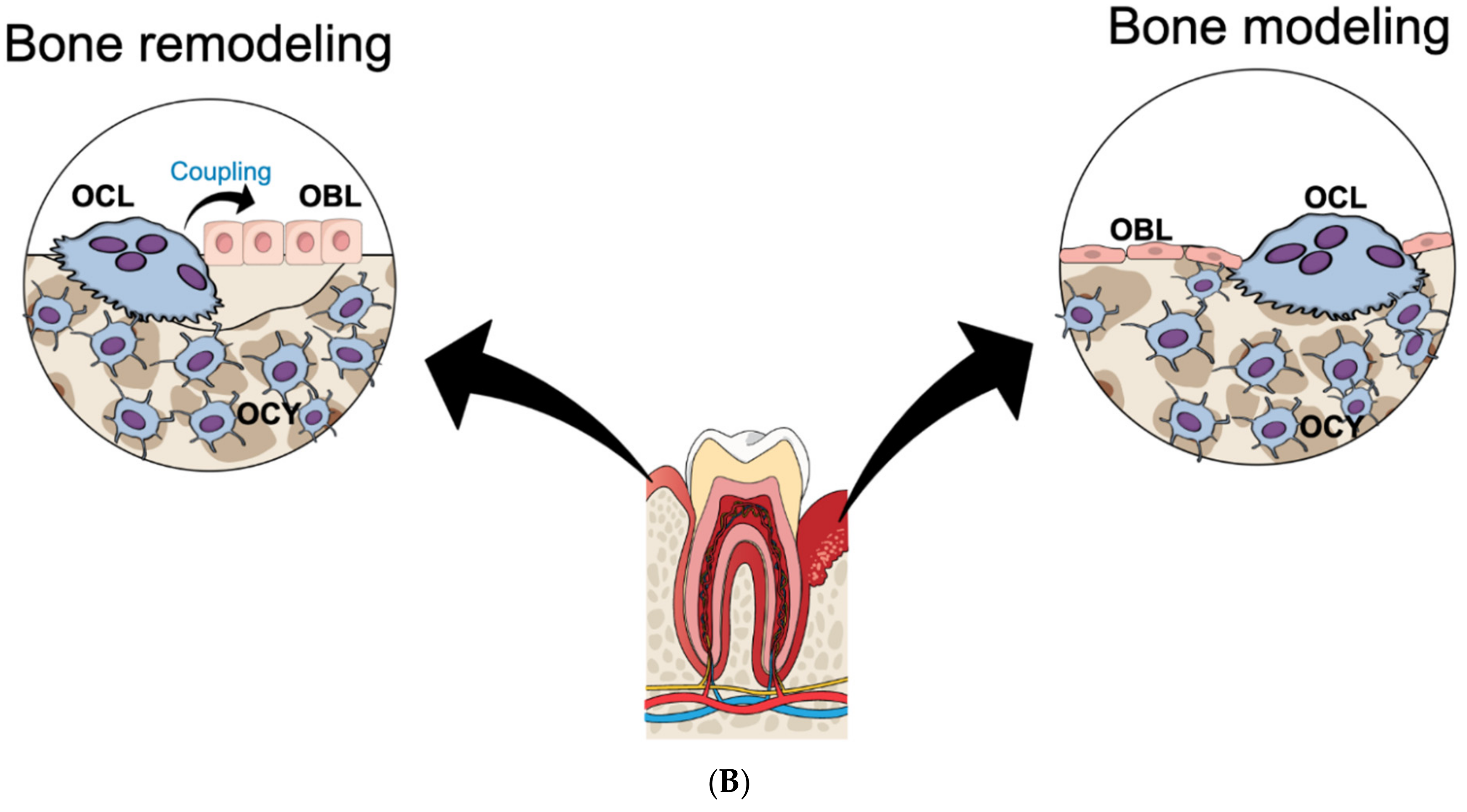
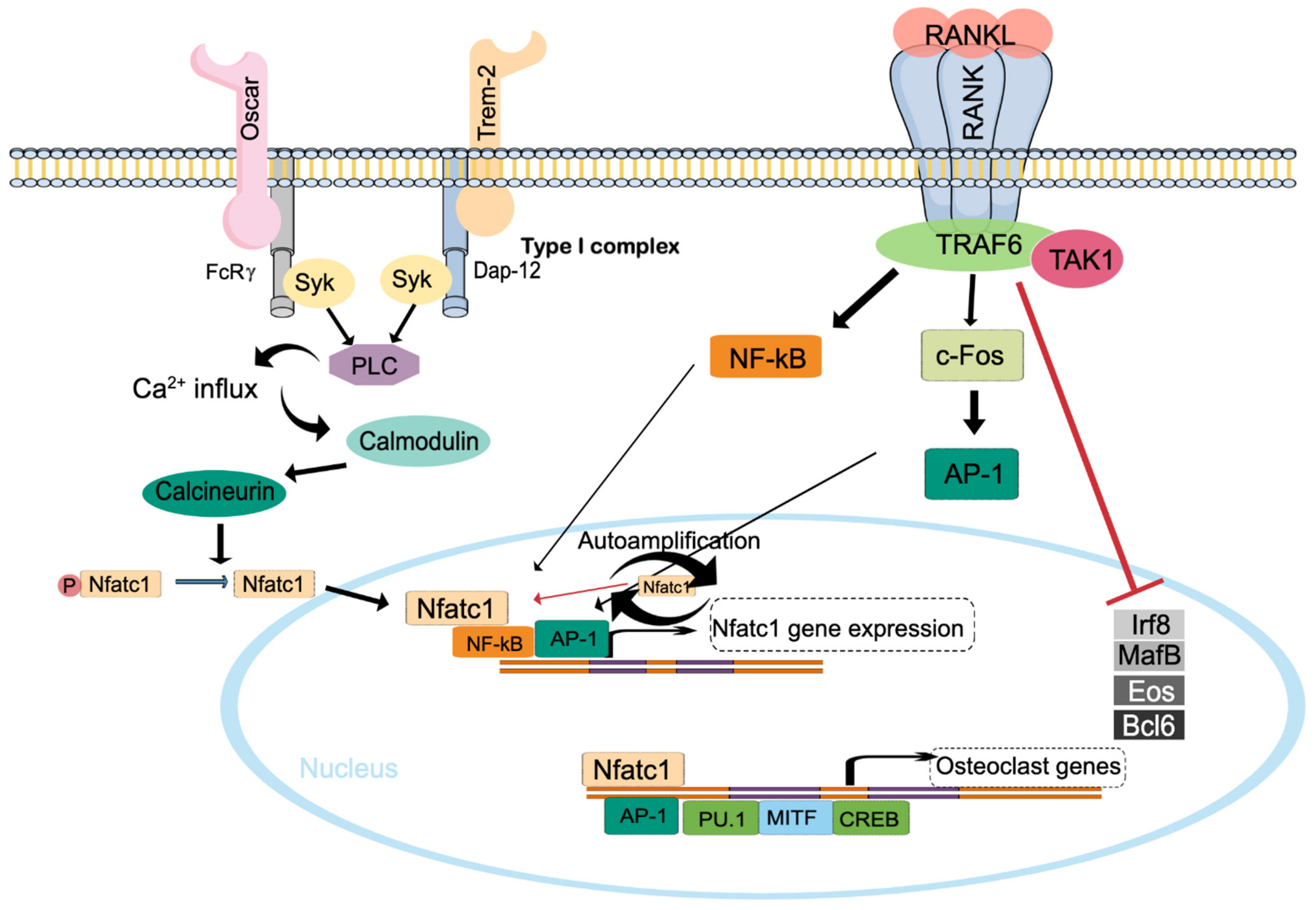
2. Osteoclast Formation
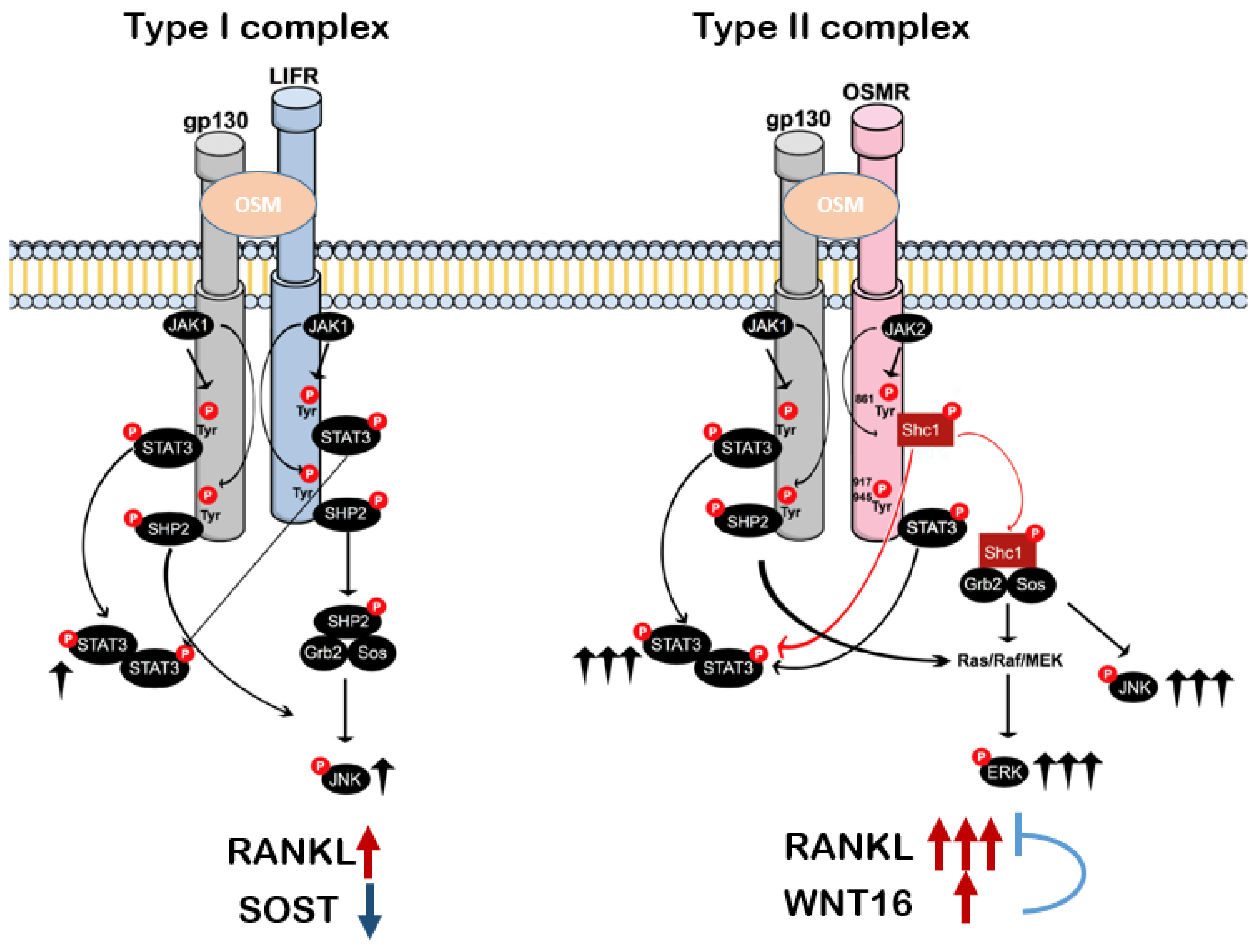
3. Bone Cell Expression and Binding Specificity of OSM, OSMR and LIFR
4. Stimulation of Osteoclastogenesis by OSM through RANKL
5. OSMR Signaling Inducing Osteoclast Formation
6. Effects of OSM on Bone In Vivo
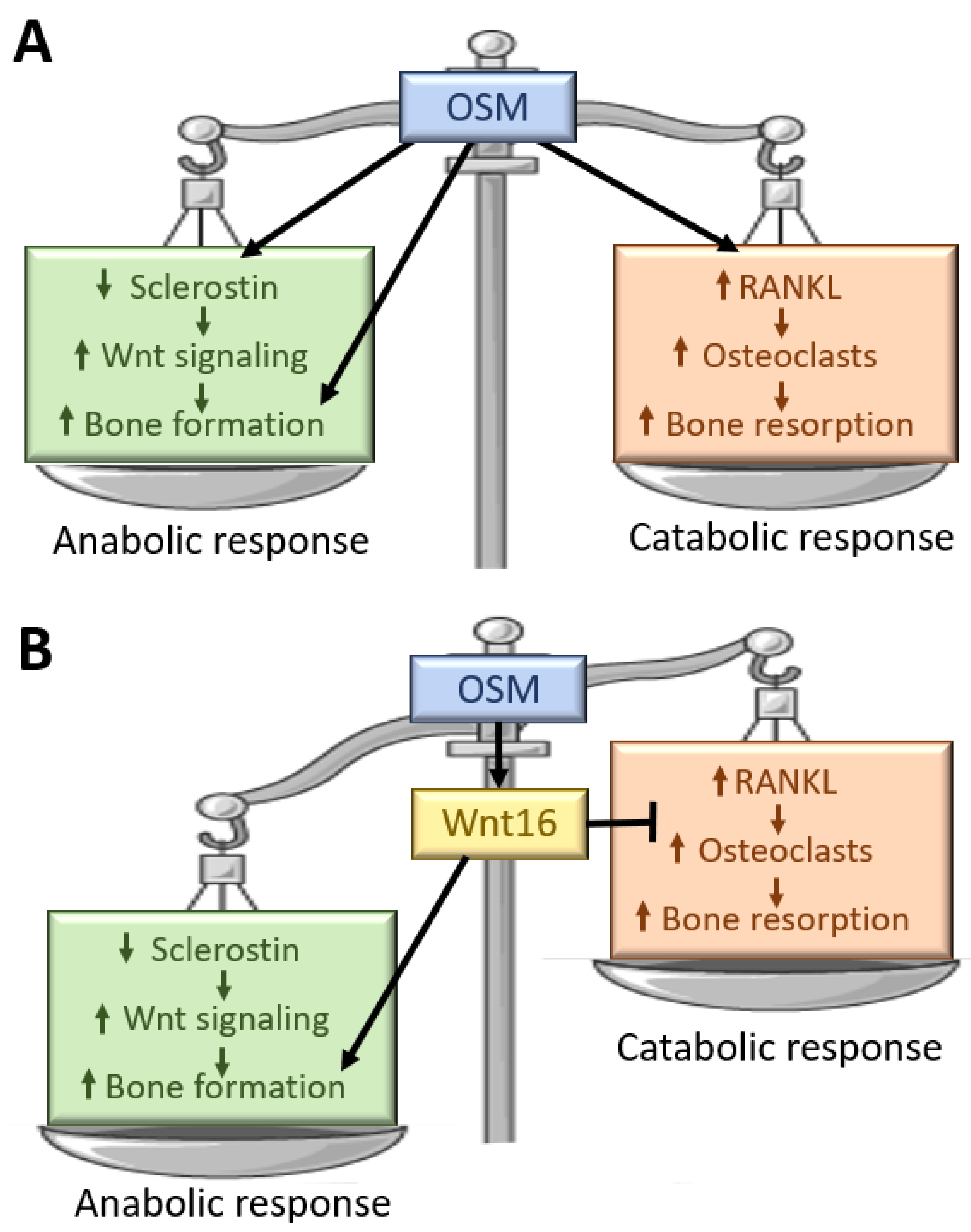
7. WNT16 as an Inhibitor of OSM-Induced Osteoclast Formation
8. Effects of Other Members of the IL-6 Family on Bone Resorption
9. Concluding Remarks
Funding
Conflicts of Interest
References
- Karsenty, G.; Kronenberg, H.M.; Settembre, C. Genetic Control of Bone Formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 629–648. [Google Scholar] [CrossRef] [PubMed]
- Buenzli, P.; Sims, N.A. Quantifying the osteocyte network in the human skeleton. Bone 2015, 75, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The Osteocyte: An Endocrine Cell … and More. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 2012, 23, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.P.; Lerner, U.H. The role of cytokines in inflammatory bone loss. Immunol. Investig. 2013, 42, 555–622. [Google Scholar] [CrossRef]
- McDonald, M.M.; Khoo, W.H.; Ng, P.Y.; Xiao, Y.; Zamerli, J.; Thatcher, P.; Kyaw, W.; Pathmanandavel, K.; Grootveld, A.K.; Moran, I.; et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 2021, 184, 1330–1347. [Google Scholar] [CrossRef] [PubMed]
- Søe, K.; Delaisse, J.-M.; Borggaard, X.G. Osteoclast formation at the bone marrow/bone surface interface: Importance of structural elements, matrix, and intercellular communication. Semin. Cell Dev. Biol. 2021, 112, 8–15. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef]
- Khosla, S.; Monroe, D.G. Regulation of Bone Metabolism by Sex Steroids. Cold Spring Harb. Perspect. Med. 2018, 8, a031211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, U.H. Role of Interleukins on Physiological and Pathological Bone Resorption and Bone Formation: Effects by Cytokines in The IL-6 and IL-10 Families. In Encyclopedia of Bone Biology; Zaidi, M., Ed.; Academic Press: Oxford, UK, 2020; pp. 67–87. [Google Scholar]
- Sims, N.A. Influences of the IL-6 cytokine family on bone structure and function. Cytokine 2021, 146, 155655. [Google Scholar] [CrossRef] [PubMed]
- Henning, P.; Movérare-Skrtic, S.; Westerlund, A.; de Souza, P.P.C.; Floriano-Marcelino, T.; Nilsson, K.H.; El Shahawy, M.; Ohlsson, C.; Lerner, U.H. WNT16 is Robustly Increased by Oncostatin M in Mouse Calvarial Osteoblasts and Acts as a Negative Feedback Regulator of Osteoclast Formation Induced by Oncostatin M. J. Inflamm. Res. 2021, 14, 4723–4741. [Google Scholar] [CrossRef]
- Yoshida, H.; Hayashi, S.-I.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S.-I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a Cytokine and Its Receptor by Functional Screening of the Extracellular Proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Buki, K.; Vääräniemi, J.; Gu, G.; Väänänen, H.K. The Critical Role of IL-34 in Osteoclastogenesis. PLoS ONE 2011, 6, e18689. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J.; Sims, N.A. RANKL/OPG: Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef]
- Simonet, W.; Lacey, D.; Dunstan, C.; Kelley, M.; Chang, M.-S.; Lüthy, R.; Nguyen, H.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-Dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; de Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Asano, T.; Muro, R.; Huynh, N.C.-N.; Komatsu, N.; Okamoto, K.; Nakano, K.; Okamura, T.; Nitta, T.; Takayanagi, H. OPG Production Matters Where It Happened. Cell Rep. 2020, 32, 108124. [Google Scholar] [CrossRef] [PubMed]
- Cawley, K.M.; Bustamante-Gomez, N.C.; Guha, A.G.; MacLeod, R.S.; Xiong, J.; Gubrij, I.; Liu, Y.; Mulkey, R.; Palmieri, M.; Thostenson, J.D.; et al. Local Production of Osteoprotegerin by Osteoblasts Suppresses Bone Resorption. Cell Rep. 2020, 32, 108052. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone–immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L.J. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Iii, F.S.B.; Frankel, W.N.; et al. TRANCE Is a Novel Ligand of the Tumor Necrosis Factor Receptor Family That Activates c-Jun N-terminal Kinase in T Cells. J. Biol. Chem. 1997, 272, 25190–25194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.-I.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.; Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kato, S.; Kodama, T.; Takahashi, S.; Calame, K.; Takayanagi, H. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 3117–3122. [Google Scholar] [CrossRef] [Green Version]
- Danks, L.; Komatsu, N.; Guerrini, M.M.; Sawa, S.; Armaka, M.; Kollias, G.; Nakashima, T.; Takayanagi, H. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann. Rheum. Dis. 2016, 75, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Win, S.; Yan, M.; Huynh, N.C.; Sawa, S.; Tsukasaki, M.; Terashima, A.; Pluemsakunthai, W.; Kollias, G.; Nakashima, T.; et al. Plasma cells promote osteoclastogenesis and periarticular bone loss in autoimmune arthritis. J. Clin. Investig. 2021, 131, e143060. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zarling, J.M.; Shoyab, M.; Marquardt, H.; Hanson, M.B.; Lioubin, M.N.; Todaro, G.J. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9739–9743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskela, K.; Pelliniemi, T.-T.; Remes, K.; Rajamäki, A.; Pulkki, K. Serum oncostatin M in multiple myeloma: Association with prognostic factors. Br. J. Haematol. 1997, 96, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Gurluler, E.; Tumay, L.V.; Guner, O.S.; Kucukmetin, N.T.; Hizli, B.; Zorluoglu, A. Oncostatin-M as a novel biomarker in colon cancer patients and its association with clinicopathologic variables. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2042–2047. [Google Scholar] [PubMed]
- Torres, C.; Perales, S.; Alejandre, M.J.; Iglesias, J.; Palomino-Morales, R.; Martin, M.; Caba, O.; Prados, J.; Aránega, A.; Delgado, J.R.; et al. Serum Cytokine Profile in Patients with Pancreatic Cancer. Pancreas 2014, 43, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.P.M.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tsang, O.T.-Y.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Cross, A.; Edwards, S.W.; Bucknall, R.C.; Moots, R.J. Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis Care Res. 2004, 50, 1430–1436. [Google Scholar] [CrossRef]
- Pradeep, A.; Manojkumar, S.T.; Garima, G.; Raju, A. Serum levels of oncostatin M (a gp 130 cytokine): An inflammatory biomarker in periodontal disease. Biomarkers 2009, 15, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Grenier, A.; Combaux, D.; Chastre, J.; Gougerot-Pocidalo, M.A.; Gibert, C.; Dehoux, M.; Chollet-Martin, S. Oncostatin M Production by Blood and Alveolar Neutrophils during Acute Lung Injury. Lab. Investig. 2001, 81, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Chida, K.; Todate, A.; Ide, K.; Asada, K.; Nakamura, Y.; Suzuki, K.; Kuwata, H.; Nakamura, H. Oncostatin M Production by Human Dendritic Cells in Response to Bacterial Products. Cytokine 2002, 17, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.J.; Lioubin, M.N.; Marquardt, H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and oncostatin M for human melanoma cells. J. Immunol. 1987, 139, 2977–2983. [Google Scholar]
- Guihard, P.; Boutet, M.-A.; Royer, B.B.-L.; Gamblin, A.-L.; Amiaud, J.; Renaud, A.; Berreur, M.; Rédini, F.; Heymann, D.; Layrolle, P.; et al. Oncostatin M, an Inflammatory Cytokine Produced by Macrophages, Supports Intramembranous Bone Healing in a Mouse Model of Tibia Injury. Am. J. Pathol. 2015, 185, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.-G.; Nicola, N.A.; et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auguste, P.; Guillet, C.; Fourcin, M.; Olivier, C.; Veziers, J.; Pouplard-Barthelaix, A.; Gascan, H. Signaling of Type II Oncostatin M Receptor. J. Biol. Chem. 1997, 272, 15760–15764. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, T.M.; Lagrou, M.J.; Fransson, I.; Werelius, B.; Delattre, O.; Thomas, G.; de Jong, P.J.; Todaro, G.J.; Dumanski, J.P. The Genes for Oncostatin M (OSM) and Leukemia Inhibitory Factor (LIF) Are Tightly Linked on Human Chromosome 22. Genomics 1993, 17, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.; Hudson, K.R.; Ikemizu, S.; Bravo, J.; Jones, E.Y.; Heath, J.K. Crystal structure and functional dissection of the cytostatic cytokine oncostatin M. Structure 2000, 8, 863–874. [Google Scholar] [CrossRef]
- Gearing, D.P.; Comeau, M.R.; Friend, D.J.; Gimpel, S.D.; Thut, C.J.; McGourty, J.; Brasher, K.K.; King, J.A.; Gillis, S.; Mosley, B.; et al. The IL-6 signal transducer, gp130: An oncostatin M receptor and affinity converter for the LIF receptor. Science 1992, 255, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Mosley, B.; de Imus, C.; Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporeno, E.; Paonessa, G.; Salvati, A.L.; Graziani, R.; Delmastro, P.; Ciliberto, G.; Toniatti, C. Oncostatin M binds directly to gp130 and behaves as interleukin-6 antagonist on a cell line expressing gp130 but lacking functional oncostatin M receptors. J. Biol. Chem. 1994, 269, 10991–10995. [Google Scholar] [CrossRef]
- Underhill-Day, N.; Heath, J.K. Oncostatin M (OSM) cytostasis of breast tumor cells: Characterization of an OSM receptor beta-specific kernel. Cancer Res. 2006, 66, 10891–10901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellido, T.; Stahl, N.; Farruggella, T.J.; Borba, V.; Yancopoulos, G.D.; Manolagas, S.C. Detection of receptors for interleukin-6, interleukin-11, leukemia inhibitory factor, oncostatin M, and ciliary neurotrophic factor in bone marrow stromal/osteoblastic cells. J. Clin. Investig. 1996, 97, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Rodan, S.B.; Wesolowski, G.; Hilton, D.J.; Nicola, N.A.; Rodan, G.A. Leukemia Inhibitory Factor Binds with High Affinity to Preosteoblastic RCT-1 Cells and Potentiates the Retinoic Acid Induction of Alkaline Phosphatase. Endocrinology 1990, 127, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef] [Green Version]
- Allan, E.H.; Hilton, D.J.; Brown, M.A.; Evely, R.S.; Yumita, S.; Metcalf, D.; Gough, N.M.; Ng, K.W.; Nicola, N.A.; Martin, T.J. Osteoblasts display receptors for and responses to leukemia-inhibitory factor. J. Cell. Physiol. 1990, 145, 110–119. [Google Scholar] [CrossRef]
- Persson, E.; Souza, P.P.C.; Floriano-Marcelino, T.; Conaway, H.H.; Henning, P.; Lerner, U.H. Activation of Shc1 Allows Oncostatin M to Induce RANKL and Osteoclast Formation More Effectively Than Leukemia Inhibitory Factor. Front. Immunol. 2019, 10, 1164. [Google Scholar] [CrossRef] [Green Version]
- le Goff, B.; Singbrant, S.; Tonkin, B.A.; Martin, T.J.; Romas, E.; Sims, N.; Walsh, N.C. Oncostatin M acting via OSMR, augments the actions of IL-1 and TNF in synovial fibroblasts. Cytokine 2014, 68, 101–109. [Google Scholar] [CrossRef]
- Du, Q.; Qian, Y.; Xue, W. Cross-reactivity of two human IL-6 family cytokines OSM and LIF explored by protein-protein docking and molecular dynamics simulation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129907. [Google Scholar] [CrossRef] [PubMed]
- Huyton, T.; Zhang, J.-G.; Luo, C.S.; Lou, M.-Z.; Hilton, D.J.; Nicola, N.A.; Garrett, T.P.J. An unusual cytokine: Ig-domain interaction revealed in the crystal structure of leukemia inhibitory factor (LIF) in complex with the LIF receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 12737–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrian-Segarra, J.M.; Schindler, N.; Gajawada, P.; Lörchner, H.; Braun, T.; Pöling, J. The AB loop and D-helix in binding site III of human Oncostatin M (OSM) are required for OSM receptor activation. J. Biol. Chem. 2018, 293, 7017–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrian-Segarra, J.M.; Sreenivasan, K.; Gajawada, P.; Lörchner, H.; Braun, T.; Pöling, J. The AB loop of oncostatin M (OSM) determines species-specific signaling in humans and mice. J. Biol. Chem. 2018, 293, 20181–20199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, T.M.; Bruce, A.G. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc. Natl. Acad. Sci. USA 1991, 88, 8641–8645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R. The crystal structure and biological function of leukemia inhibitory factor: Implications for receptor binding. Cell 1994, 77, 1101–1116. [Google Scholar] [CrossRef]
- Tamura, T.; Udagawa, N.; Takahashi, N.; Miyaura, C.; Tanaka, S.; Yamada, Y.; Koishihara, Y.; Ohsugi, Y.; Kumaki, K.; Taga, T. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 11924–11928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Gubrij, I.; Lin, S.C.; Saylors, R.L.; Manolagas, S.C. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-kappaB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J. Biol. Chem. 1999, 274, 19301–19308. [Google Scholar]
- Richards, C.; Langdona, C.; Deschampsab, P.; Pennicac, D.; Shaughnessy, S.G. Stimulation of Osteoclast Differentiation In Vitro by Mouse Oncostatin M, Leukaemia Inhibitory Factor, Cardiotrophin-1 and Interleukin 6: Synergy with Dexamethasone. Cytokine 2000, 12, 613–621. [Google Scholar] [CrossRef]
- Blackwell, K.A.; Raisz, L.G.; Pilbeam, C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010, 21, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.C.; Johnson, R.W.; Hu, Y.; Brennan, H.J.; Poulton, I.J.; Zhang, J.-G.; Jenkins, B.; Smyth, G.K.; Nicola, N.A.; Sims, N.A. Murine Oncostatin M Acts via Leukemia Inhibitory Factor Receptor to Phosphorylate Signal Transducer and Activator of Transcription 3 (STAT3) but Not STAT1, an Effect That Protects Bone Mass. J. Biol. Chem. 2016, 291, 21703–21716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabacco, G.; Bilezikian, J.P. Osteoanabolic and dual action drugs. Br. J. Clin. Pharmacol. 2019, 85, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.C.; Poulton, I.J.; McGregor, N.; Ho, P.W.M.; Allan, E.H.; Quach, J.M.; Martin, T.J.; Sims, N.A. Sustained RANKL response to parathyroid hormone in oncostatin M receptor-deficient osteoblasts converts anabolic treatment to a catabolic effect in vivo. J. Bone Miner. Res. 2012, 27, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Jay, P.R.; Centrella, M.; Lorenzo, J.; Bruce, A.G.; Horowitz, M.C. Oncostatin-M: A new bone active cytokine that activates osteoblasts and inhibits bone resorption. Endocrinology 1996, 137, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, N.A.; Jenkins, B.J.; Quinn, J.M.; Nakamura, A.; Glatt, M.; Gillespie, M.T.; Ernst, M.; Martin, T.J. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J. Clin. Investig. 2004, 113, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Udagawa, N.; Takahashi, N.; Yoshitake, F.; Narita, H.; Ebisu, S.; Ishihara, K. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone 2006, 39, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, R.A.; Juan, T.S.-C.; Welcher, A.A.; Sun, Y.; Cupples, R.; Guthrie, B.; Fletcher, F.A. Cloning and Characterization of a Specific Receptor for Mouse Oncostatin M. Mol. Cell. Biol. 1998, 18, 3357–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanns, H.M. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015, 26, 545–558. [Google Scholar] [CrossRef]
- Hermanns, H.M.; Radtke, S.; Haan, C.; de Leur, H.S.-V.; Tavernier, J.; Heinrich, P.C.; Behrmann, I. Contributions of leukemia inhibitory factor receptor and oncostatin M receptor to signal transduction in heterodimeric complexes with glycoprotein 130. J. Immunol. 1999, 163, 6651–6658. [Google Scholar]
- Lütticken, C.; Wegenka, U.M.; Yuan, J.; Buschmann, J.; Schindler, C.; Ziemiecki, A.; Harpur, A.G.; Wilks, A.F.; Yasukawa, K.; Taga, T.; et al. Association of Transcription Factor APRF and Protein Kinase Jak1 with the Interleukin-6 Signal Transducer gp130. Science 1994, 263, 89–92. [Google Scholar] [CrossRef]
- Murakami, M.; Narazaki, M.; Hibi, M.; Yawata, H.; Yasukawa, K.; Hamaguchi, M.; Taga, T.; Kishimoto, T. Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in the cytokine receptor family. Proc. Natl. Acad. Sci. USA 1991, 88, 11349–11353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, L.C.; Hawley, T.S.; Lust, J.A.; Goldman, S.J.; Hawley, R.G. Tyrosine phosphorylation of JAK-TYK kinases in malignant plasma cell lines growth-stimulated by interleukins 6 and 11. Biochem. Biophys. Res. Commun. 1994, 202, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yamanaka, Y.; Hirano, T. Interleukin-6-Induced Tyrosine Phosphorylation of Multiple Proteins in Murine Hematopoietic Lineage Cells. Biochem. Biophys. Res. Commun. 1994, 200, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Hintzen, C.; Evers, C.; Lippok, B.E.; Volkmer, R.; Heinrich, P.C.; Radtke, S.; Hermanns, H.M. Box 2 Region of the Oncostatin M Receptor Determines Specificity for Recruitment of Janus Kinases and STAT5 Activation. J. Biol. Chem. 2008, 283, 19465–19477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guschin, D.; Rogers, N.; Briscoe, J.; Witthuhn, B.; Watling, D.; Horn, F.; Pellegrini, S.; Yasukawa, K.; Heinrich, P.; Stark, G.R. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 1995, 14, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Haan, S.; Keller, J.F.; Behrmann, I.; Heinrich, P.C.; Haan, C. Multiple reasons for an inefficient STAT1 response upon IL-6-type cytokine stimulation. Cell. Signal. 2005, 17, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Meraz, M.; White, J.M.; Lampe, P.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Takahashi, M.; Carpino, N.; Jou, S.-T.; Chao, J.-R.; Tanaka, S.; Shigeyoshi, Y.; Parganas, E.; Ihle, J.N. Leukemia Inhibitory Factor Regulates Trophoblast Giant Cell Differentiation via Janus Kinase 1-Signal Transducer and Activator of Transcription 3-Suppressor of Cytokine Signaling 3 Pathway. Mol. Endocrinol. 2008, 22, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Gerhartz, C.; Heesel, B.; Sasse, J.; Hemmann, U.; Landgraf, C.; Schneidermergener, J.; Horn, F.; Heinrich, P.C.; Graeve, L. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J. Biol. Chem. 1996, 271, 12991–12998. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Dahmen, H.; Grimm, C.; Gendo, C.; Müller-Newen, G.; Heinrich, P.C.; Schaper, F. The cytoplasmic tyrosine motifs in full-length glycoprotein 130 have different roles in IL-6 signal transduction. J. Immunol. 2000, 164, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, M.H.; Kerr, L.M.; Stark, G.R.; Darnell, J.E. Contribution of STAT SH2 Groups to Specific Interferon Signaling by the Jak-STAT pathway. Science 1995, 267, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Hemmann, U.; Gerhartz, C.; Heesel, B.; Sasse, J.; Kurapkat, G.; Grötzinger, J.; Wollmer, A.; Zhong, Z.; Darnell, J.E.; Graeve, L.; et al. Differential activation of acute phase response factor/Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. II. Src homology SH2 domains define the specificity of stat factor activation. J. Biol. Chem. 1996, 271, 12999–13007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukada, T.; Hibi, M.; Yamanaka, Y.; Takahashi-Tezuka, M.; Fujitani, Y.; Yamaguchi, T.; Nakajima, K.; Hirano, T. Two Signals Are Necessary for Cell Proliferation Induced by a Cytokine Receptor gp130: Involvement of STAT3 in Anti-Apoptosis. Immunity 1996, 5, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Fahmi, A.; Smart, N.; Punn, A.; Jabr, R.; Marber, M.; Heads, R. p42/p44-MAPK and PI3K are sufficient for IL-6 family cytokines/gp130 to signal to hypertrophy and survival in cardiomyocytes in the absence of JAK/STAT activation. Cell. Signal. 2013, 25, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Fujio, Y.; Kunisada, K.; Hirota, H.; Matsui, H.; Kishimoto, T.; Yamauchi-Takihara, K. Activation of Phosphatidylinositol 3-Kinase through Glycoprotein 130 Induces Protein Kinase B and p70 S6 Kinase Phosphorylation in Cardiac Myocytes. J. Biol. Chem. 1998, 273, 9703–9710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanns, H.M.; Radtke, S.; Schaper, F.; Heinrich, P.C.; Behrmann, I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J. Biol. Chem. 2000, 275, 40742–40748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Yamazaki, M.; Shevde, N.K.; Pike, J.W. Transcriptional control of receptor activator of nuclear factor-kappaB ligand by the protein kinase A activator forskolin and the transmembrane glycoprotein 130-activating cytokine, oncostatin M, is exerted through multiple distal enhancers. Mol. Endocrinol. 2007, 21, 197–214. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Robledo, O.; Kinzie, E.; Blanchard, F.; Richards, C.; Miyajima, A.; Baumann, H. Receptor Subunit-specific Action of Oncostatin M in Hepatic Cells and Its Modulation by Leukemia Inhibitory Factor. J. Biol. Chem. 2000, 275, 25273–25285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.B.M.; Prigent, S.A. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Ventura, A.; Luzi, L.; Pacini, S.; Baldari, C.; Pelicci, P.G. The p66Shc Longevity Gene Is Silenced through Epigenetic Modifications of an Alternative Promoter. J. Biol. Chem. 2002, 277, 22370–22376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, A.D.; Hui, W.; Cawston, T.E.; Richards, C.D. Adenoviral Gene Transfer of Interleukin-1 in Combination with Oncostatin M Induces Significant Joint Damage in a Murine Model. Am. J. Pathol. 2003, 162, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Hui, W.; Cawston, T.; Richards, C.D.; Rowan, A.D. A model of inflammatory arthritis highlights a role for oncostatin M in pro-inflammatory cytokine-induced bone destruction via RANK/RANKL. Arthritis Res. Ther. 2005, 7, R57–R64. [Google Scholar] [CrossRef] [Green Version]
- Malik, N.; Haugen, H.S.; Modrell, B.; Shoyab, M.; Clegg, C.H. Developmental abnormalities in mice transgenic for bovine oncostatin M. Mol. Cell. Biol. 1995, 15, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- de Hooge, A.S.; van de Loo, F.A.; Bennink, M.B.; de Jong, D.S.; Arntz, O.J.; Lubberts, E.; Richards, C.D.; van den Berg, W.B. Adenoviral transfer of murine oncostatin M elicits periosteal bone apposition in knee joints of mice, despite synovial inflammation and up-regulated expression of interleukin-6 and receptor activator of nuclear factor-kappa B ligand. Am. J. Pathol. 2002, 160, 1733–1743. [Google Scholar] [CrossRef]
- Nicolaidou, V.; Wong, M.M.; Redpath, A.N.; Ersek, A.; Baban, D.F.; Williams, L.M.; Cope, A.P.; Horwood, N.J. Monocytes Induce STAT3 Activation in Human Mesenchymal Stem Cells to Promote Osteoblast Formation. PLoS ONE 2012, 7, e39871. [Google Scholar] [CrossRef] [Green Version]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.-H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.E.; Amin, N.; Kemp, J.P.; et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-F.; Tobias, J.H.; Duncan, E.; Evans, D.M.; Eriksson, J.; Paternoster, L.; Yerges-Armstrong, L.M.; Lehtimäki, T.; Bergström, U.; Kähönen, M.; et al. WNT16 Influences Bone Mineral Density, Cortical Bone Thickness, Bone Strength, and Osteoporotic Fracture Risk. PLoS Genet. 2012, 8, e1002745. [Google Scholar] [CrossRef] [Green Version]
- Moverare-Skrtic, S.; Henning, P.; Liu, X.; Nagano, K.; Saito, H.; Börjesson, A.E.; Sjögren, K.; Windahl, S.H.; Farman, H.; Kindlund, B.; et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 2014, 20, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wergedal, J.E.; Kesavan, C.; Brommage, R.; Das, S.; Mohan, S. Role of WNT16 in the Regulation of Periosteal Bone Formation in Female Mice. Endocrinology 2015, 156, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Alam, I.; Alkhouli, M.; Gerard-O’Riley, R.L.; Wright, W.B.; Acton, D.; Gray, A.K.; Patel, B.; Reilly, A.M.; Lim, K.-E.; Robling, A.G.; et al. Osteoblast-Specific Overexpression of Human WNT16 Increases Both Cortical and Trabecular Bone Mass and Structure in Mice. Endocrinology 2016, 157, 722–736. [Google Scholar] [CrossRef]
- Ohlsson, C.; Henning, P.; Nilsson, K.H.; Wu, J.; Gustafsson, K.L.; Sjögren, K.; Törnqvist, A.; Koskela, A.; Zhang, F.-P.; Lagerquist, M.; et al. Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice. J. Endocrinol. 2018, 237, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löwik, C.; van der Pluijm, G.; Bloys, H.; Hoekman, K.; Bijvoet, O.; Aarden, L.; Papapoulos, S. Parathyroid hormone (PTH) and PTH-like protein (PLP) stimulate interleukin-6 production by osteogenic cells: A possible role of interleukin-6 in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1989, 162, 1546–1552. [Google Scholar] [CrossRef]
- Ishimi, Y.; Miyaura, C.; Jin, C.H.; Akatsu, T.; Abe, E.; Nakamura, Y.; Yamaguchi, A.; Yoshiki, S.; Matsuda, T.; Hirano, T. IL-6 is produced by osteoblasts and induces bone resorption. J. Immunol. 1990, 145, 3297–3303. [Google Scholar] [PubMed]
- Sims, N.A. Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int. J. Biochem. Cell Biol. 2016, 79, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Rousseau, F.; Guilhot, F.; Malinge, P.; Magistrelli, G.; Herren, S.; Jones, S.A.; Jones, G.; Scheller, J.; Lissilaa, R.; et al. Novel Insights into Interleukin 6 (IL-6) Cis- and Trans-signaling Pathways by Differentially Manipulating the Assembly of the IL-6 Signaling Complex. J. Biol. Chem. 2015, 290, 26943–26953. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Kawata, H.; Takahashi, F.; Higuchi, Y.; Furuichi, T.; Ohkawa, H. Bone marrow neutrophilia and suppressed bone turnover in human interleukin-6 transgenic mice. A cellular relationship among hematopoietic cells, osteoblasts, and osteoclasts mediated by stromal cells in bone marrow. Am. J. Pathol. 1995, 147, 1682–1692. [Google Scholar]
- De Benedetti, F.; Rucci, N.; del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6–transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Care Res. 2006, 54, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Balena, R.; Fattori, E.; Markatos, A.; Yamamoto, M.; Tanaka, H.; Ciliberto, G.; Rodan, G.; Costantini, F. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994, 13, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; Hangoc, G.; Girasole, G.; Passeri, G.; Williams, D.C.; Abrams, J.S.; Boyce, B.; Broxmeyer, H.; Manolagas, S.C. Increased Osteoclast Development After Estrogen Loss: Mediation by Interleukin-6. Science 1992, 257, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, L.; Tonkin, B.A.; Poulton, I.J.; McGregor, N.E.; Ferlin, W.; Sims, N.A. IL-6 trans -signalling mediates trabecular, but not cortical, bone loss after ovariectomy. Bone 2018, 112, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Girasole, G.; Passeri, G.; Jilka, R.L.; Manolagas, S.C. Interleukin-11: A new cytokine critical for osteoclast development. J. Clin. Investig. 1994, 93, 1516–1524. [Google Scholar] [CrossRef] [Green Version]
- Ahlen, J.; Andersson, S.; Mukohyama, H.; Roth, C.; Bäckman, A.; Conaway, H.; Lerner, U. Characterization of the bone-resorptive effect of interleukin-11 in cultured mouse calvarial bones. Bone 2002, 31, 242–251. [Google Scholar] [CrossRef]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology 1998, 139, 4743–4746. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Jenkins, B.J.; Nakamura, A.; Quinn, J.M.; Li, R.; Gillespie, M.T.; Ernst, M.; Robb, L.; Martin, T.J. Interleukin-11 Receptor Signaling Is Required for Normal Bone Remodeling. J. Bone Miner. Res. 2005, 20, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Watanabe, S.; Ishii, G.; Takeda, S.; Nakayama, K.; Fukumoto, S.; Kaneta, Y.; Inoue, D.; Matsumoto, T.; Harigaya, K.; et al. Interleukin-11 as a Stimulatory Factor for Bone Formation Prevents Bone Loss with Advancing Age in Mice. J. Biol. Chem. 2002, 277, 49011–49018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaughnessy, S.G.; Walton, K.J.; Deschamps, P.; Butcher, M.; Beaudin, S.M. Neutralization of interleukin-11 activity decreases osteoclast formation and increases cancellous bone volume in ovariectomized mice. Cytokine 2002, 20, 78–85. [Google Scholar] [CrossRef]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Pompolo, S.; Allan, E.H.; Quinn, J.M.W.; Gillespie, M.T.; Martin, T.J.; Sims, N.A. Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J. Bone Miner. Res. 2008, 23, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- McGregor, N.E.; Poulton, I.J.; Walker, E.C.; Pompolo, S.; Quinn, J.M.W.; Martin, T.J.; Sims, N.A. Ciliary Neurotrophic Factor Inhibits Bone Formation and Plays a Sex-Specific Role in Bone Growth and Remodeling. Calcif. Tissue Res. 2010, 86, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Mansoori, M.N.; Kakaji, M.; Shukla, M.; Gupta, S.K.; Singh, D. Interleukin 27 (IL-27) Alleviates Bone Loss in Estrogen-deficient Conditions by Induction of Early Growth Response-2 Gene. J. Biol. Chem. 2017, 292, 4686–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, S.; Nakamura, C.; Fukawa, T.; Ono, K.; Ohwaki, T.; Yoshimoto, T.; Wada, S. Effects of IL-23 and IL-27 on osteoblasts and osteoclasts: Inhibitory effects on osteoclast differentiation. J. Bone Miner. Metab. 2007, 25, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Takaishi, H.; Takito, J.; Yoda, M.; Sakai, S.; Hikata, T.; Hakozaki, A.; Uchikawa, S.; Matsumoto, M.; Chiba, K.; et al. IL-27 abrogates receptor activator of NF-kappa B ligand-mediated osteoclastogenesis of human granulocyte-macrophage colony-forming unit cells through STAT1-dependent inhibition of c-Fos. J. Immunol. 2009, 183, 2397–2406. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Zhao, B.; Triantafyllopoulou, A.; Park-Min, K.H.; Ivashkiv, L.B. Interleukin-27 inhibits human osteoclastogenesis by abrogating RANKL-mediated induction of nuclear factor of activated T cells c1 and suppressing proximal RANK signaling. Arthritis Rheum. 2010, 62, 402–413. [Google Scholar] [PubMed] [Green Version]
- Park, J.S.; Jung, Y.O.; Oh, H.J.; Park, S.J.; Heo, Y.J.; Kang, C.M.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Cho, M.L.; et al. Interleukin-27 suppresses osteoclastogenesis via induction of interferon-gamma. Immunology 2012, 137, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Luo, W.; Hu, J.; Chen, Y.; Yu, T.; Yang, J.; Dong, S.; Tian, X.; Sun, L. Interleukin-27 prevents LPS-induced inflammatory osteolysis by inhibiting osteoclast formation and function. Am. J. Transl. Res. 2019, 11, 1154–1169. [Google Scholar]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Kobashigawa, T.; Yamanaka, H.; Kotake, S. IL-35 inhibits human osteoclastogenesis from monocytes induced by receptor-activator of NF-kappaB ligand. Cent. Eur. J. Immunol. 2018, 43, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Wang, Y.; Qiang, L.; Xu, Y.; Li, C.; Li, T.; Zhou, X.; Xiao, M.; Wang, J. Interleukin-35 Inhibits TNF-alpha-Induced Osteoclastogenesis and Promotes Apoptosis via Shifting the Activation from TNF Receptor-Associated Death Domain (TRADD)-TRAF2 to TRADD-Fas-Associated Death Domain by JAK1/STAT1. Front. Immunol. 2018, 9, 1417. [Google Scholar] [CrossRef] [Green Version]
- Lisignoli, G.; Piacentini, A.; Toneguzzi, S.; Grassi, F.; Cocchini, B.; Ferruzzi, A.; Gualtieri, G.; Facchini, A. Osteoblasts and stromal cells isolated from femora in rheumatoid arthritis (RA) and osteoarthritis (OA) patients express IL-11, leukaemia inhibitory factor and oncostatin M. Clin. Exp. Immunol. 2000, 119, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Torossian, F.; Guerton, B.; Anginot, A.; Alexander, K.; Desterke, C.; Soave, S.; Tseng, H.-W.; Arouche, N.; Boutin, L.; Kulina, I.; et al. Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight 2017, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolin, C.; Tawara, K.; Sutherland, C.; Redshaw, J.; Aranda, P.; Moselhy, J.; Anderson, R.; Jorcyk, C.L. Oncostatin M Promotes Mammary Tumor Metastasis to Bone and Osteolytic Bone Degradation. Genes Cancer 2012, 3, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omokehinde, T.; Jotte, A.; Johnson, R.W. gp130 Cytokines Activate Novel Signaling Pathways and Alter Bone Dissemination in ER+ Breast Cancer Cells. J. Bone Miner. Res. 2022, 7, 185–201. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, P.P.C.; Henning, P.; Lerner, U.H. Stimulation of Osteoclast Formation by Oncostatin M and the Role of WNT16 as a Negative Feedback Regulator. Int. J. Mol. Sci. 2022, 23, 3287. https://doi.org/10.3390/ijms23063287
de Souza PPC, Henning P, Lerner UH. Stimulation of Osteoclast Formation by Oncostatin M and the Role of WNT16 as a Negative Feedback Regulator. International Journal of Molecular Sciences. 2022; 23(6):3287. https://doi.org/10.3390/ijms23063287
Chicago/Turabian Stylede Souza, Pedro P. C., Petra Henning, and Ulf H. Lerner. 2022. "Stimulation of Osteoclast Formation by Oncostatin M and the Role of WNT16 as a Negative Feedback Regulator" International Journal of Molecular Sciences 23, no. 6: 3287. https://doi.org/10.3390/ijms23063287
APA Stylede Souza, P. P. C., Henning, P., & Lerner, U. H. (2022). Stimulation of Osteoclast Formation by Oncostatin M and the Role of WNT16 as a Negative Feedback Regulator. International Journal of Molecular Sciences, 23(6), 3287. https://doi.org/10.3390/ijms23063287