
Oncostatin M Induces Lipolysis and Suppresses Insulin Response in 3T3-L1 Adipocytes



Abstract
1. Introduction
2. Results
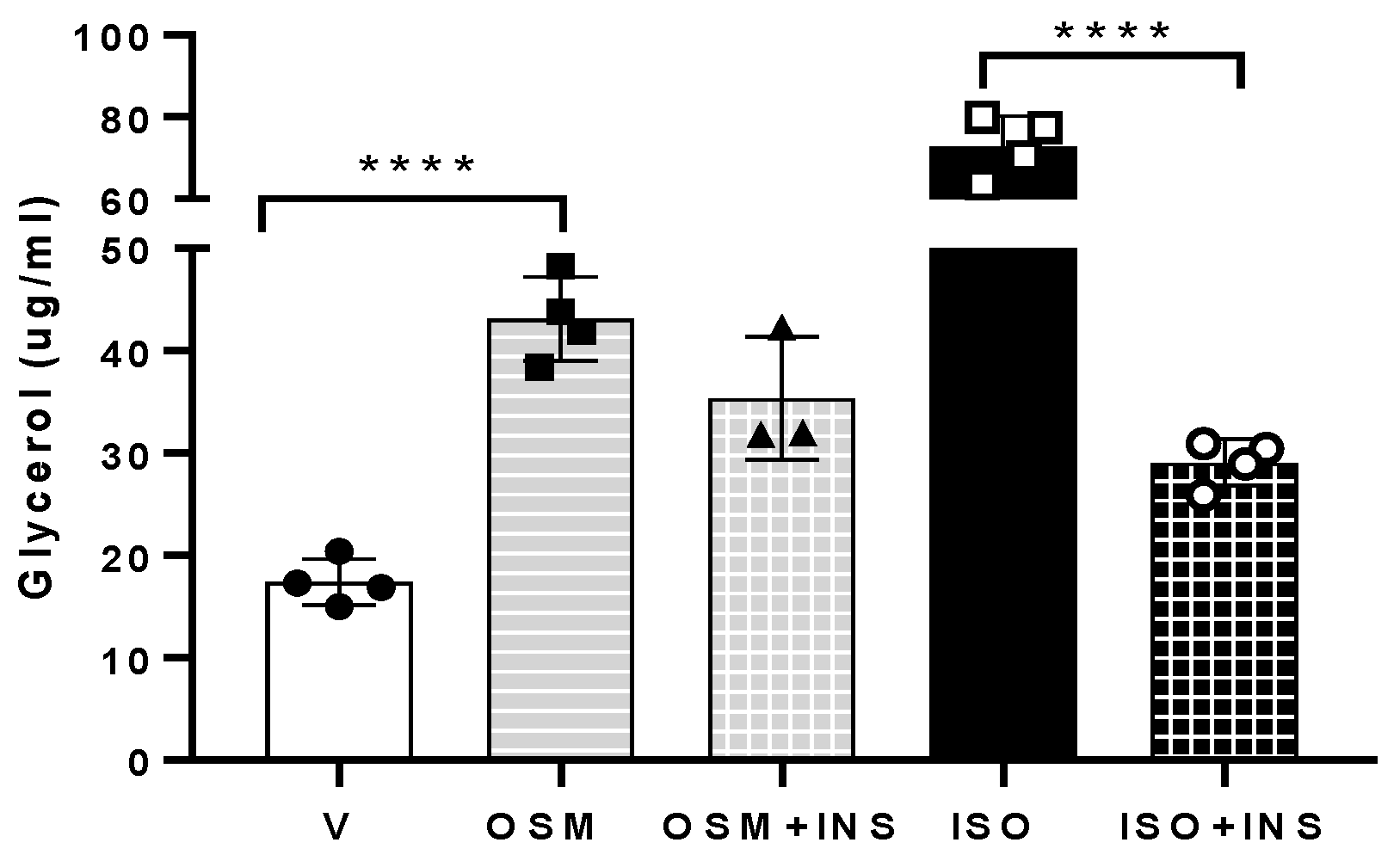
2.1. OSM Stimulates Adipocyte Lipolysis and Diminishes the Inhibitory Effects of Insulin on Lipolysis
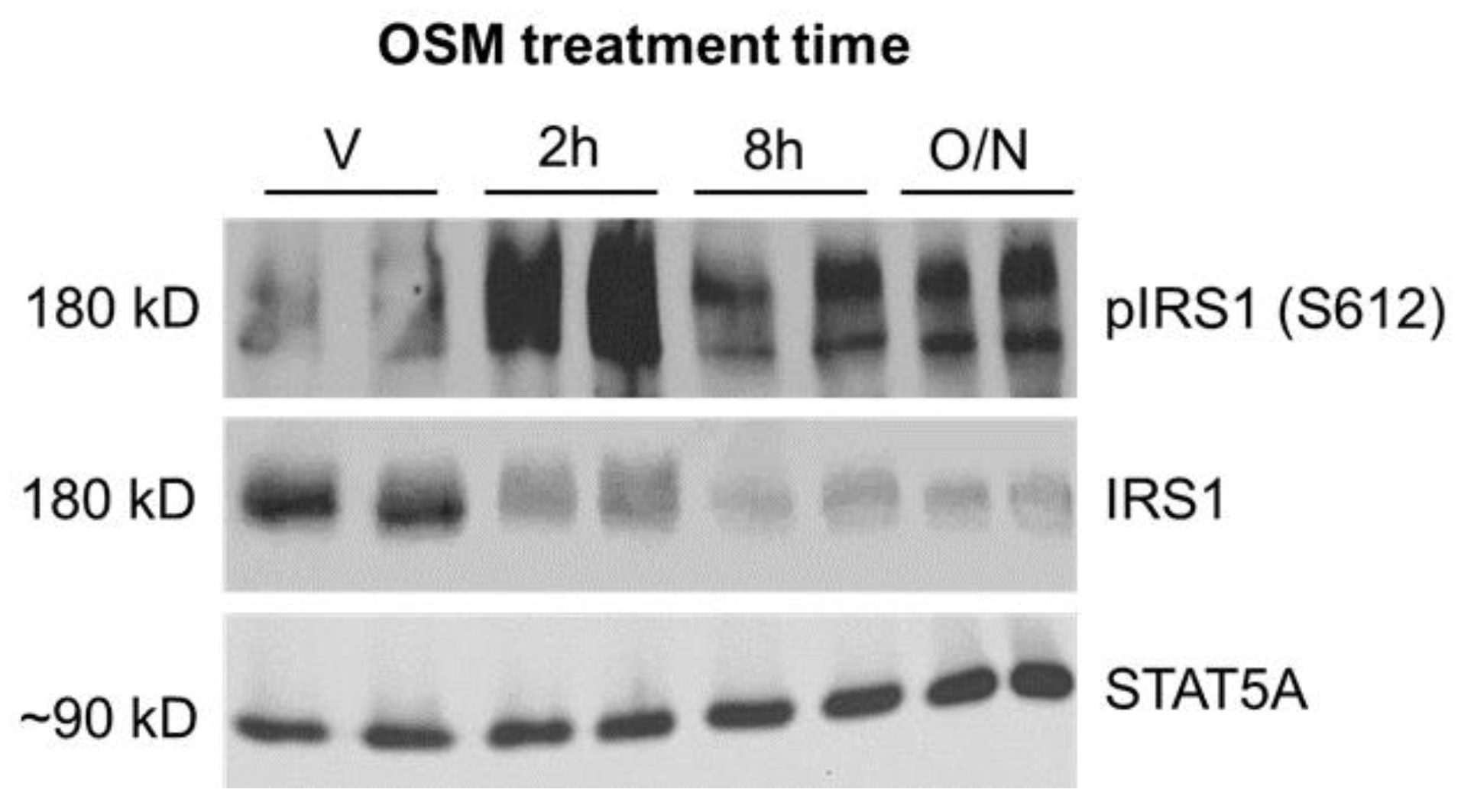
2.2. Inhibitory Phosphorylation of Insulin Receptor Substrate 1 (IRS1) Is Significantly Increased in OSM-Exposed Adipocytes
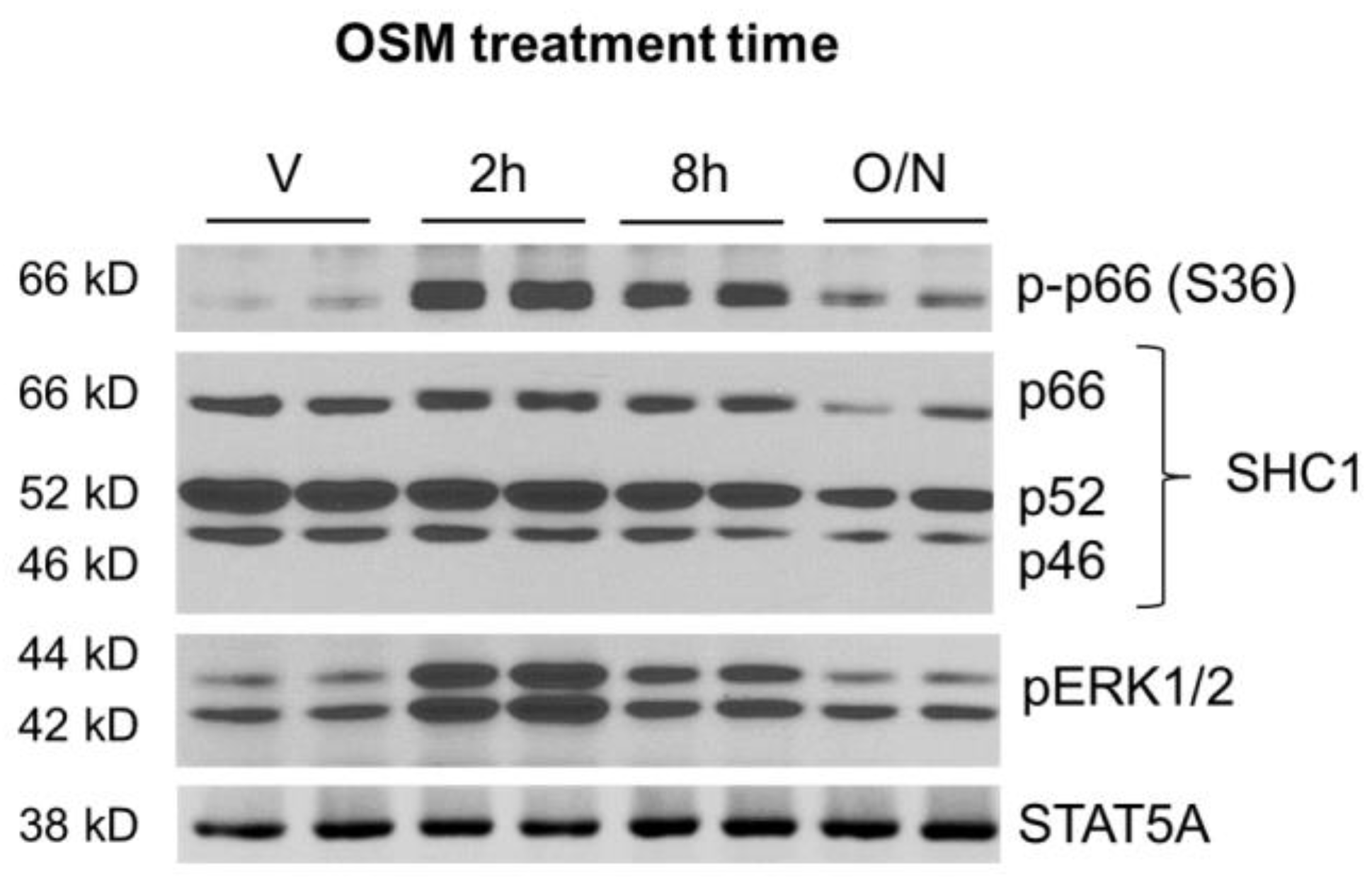
2.3. OSM Induces p66Shc Phosphorylation and ERK 1/2 Activation in Adipocytes
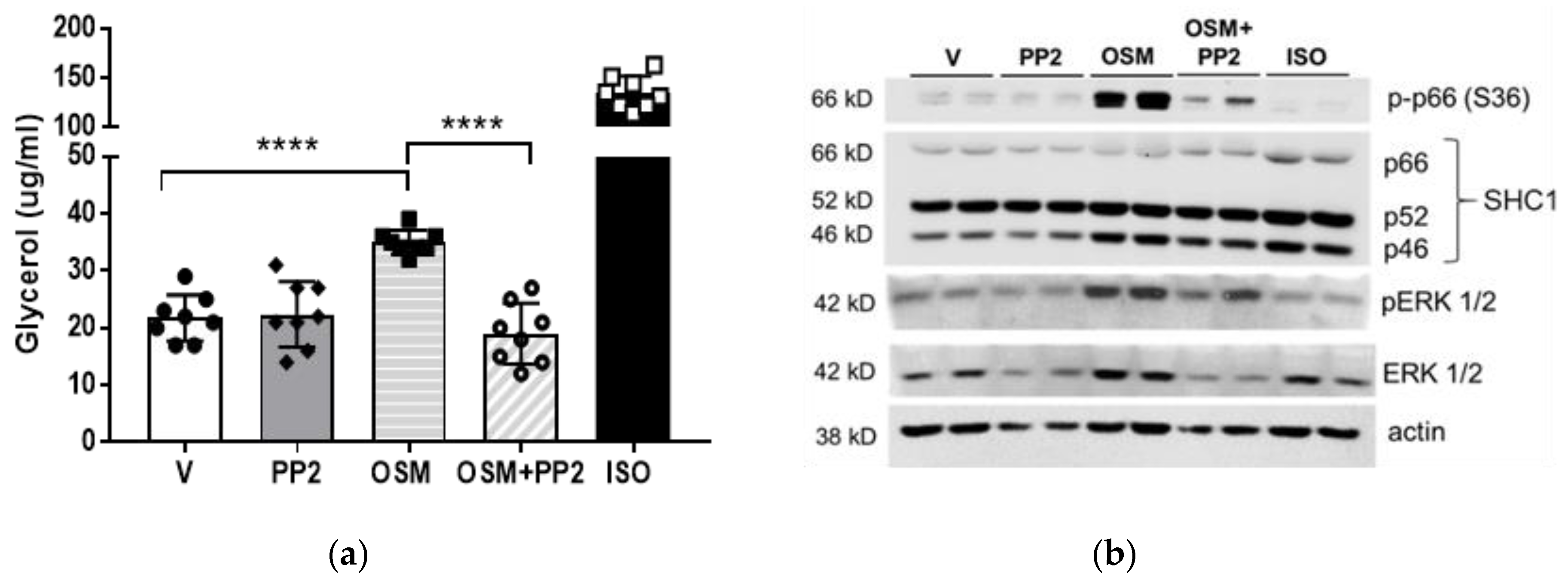
2.4. SHC1 Inhibition Abrogates OSM-Induced Adipocyte Lipolysis
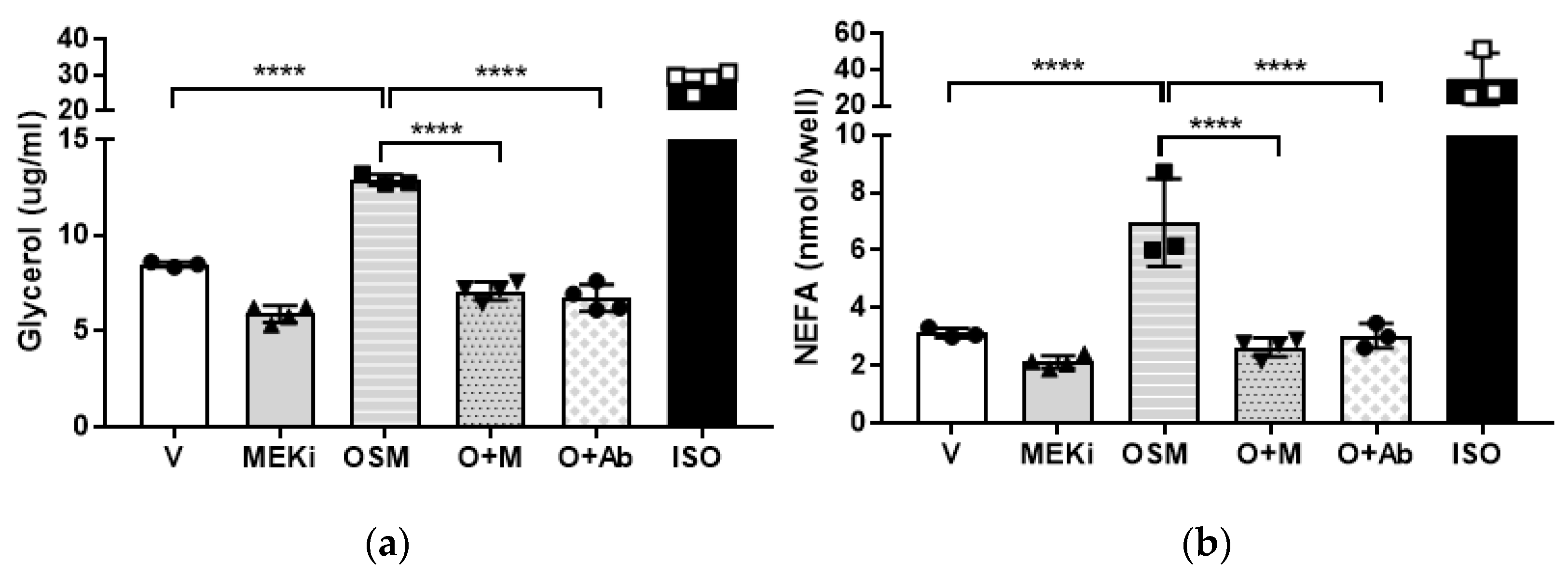
2.5. Mitogen-Activated Protein Kinase-Kinase (MEK) or OSMR Blockade Inhibits OSM-Induced Adipocyte Lipolysis
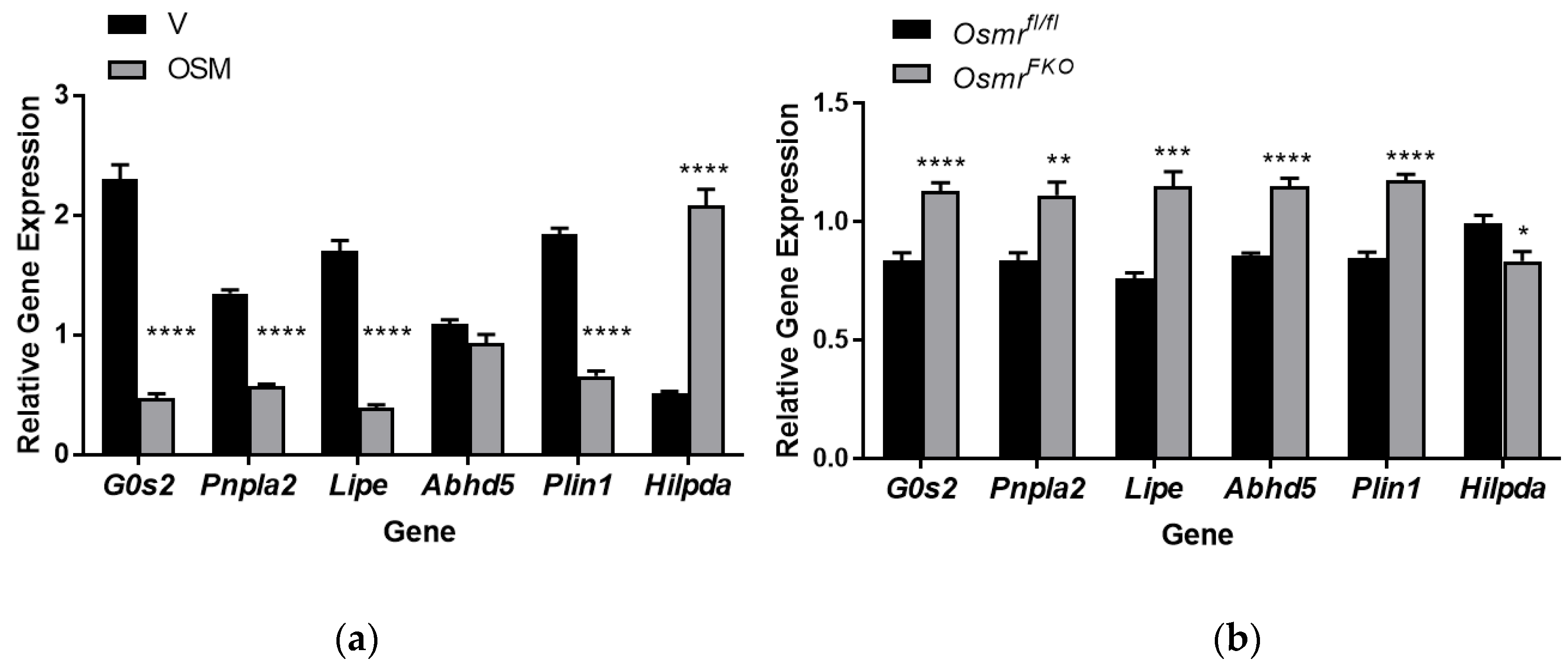
2.6. OSM Exposure and OSMR Deletion Have Opposing Effects on Lipolysis-Associated Genes in Adipocytes
3. Discussion
4. Materials and Methods
4.1. 3T3-L1 Adipocyte Culture and OSM Exposure Time Course
4.2. Glycerol and NEFA Assays
4.3. Immunoblotting
4.4. Primary Adipocyte Culture from Osmrfl/fl and OsmrFKO Mice
4.5. Gene Expression Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grant, R.W.; Stephens, J.M. Fat in flames: Influence of cytokines and pattern recognition receptors on adipocyte lipolysis. Am. J. Physiol.-Endocrinol. Metab. 2015, 309, E205–E213. [Google Scholar] [CrossRef] [PubMed]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Eichmann, T.O.; Taschler, U.; Zimmermann, R.; Zechner, R.; Lass, A. Measurement of lipolysis. Methods Enzymol. 2014, 538, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef]
- Hong, S.; Song, W.; Zushin, P.-J.H.; Liu, B.; Jedrychowski, M.P.; Mina, A.I.; Deng, Z.; Cabarkapa, D.; Hall, J.A.; Palmer, C.J.; et al. Phosphorylation of Beta-3 adrenergic receptor at serine 247 by ERK MAP kinase drives lipolysis in obese adipocytes. Mol. Metab. 2018, 12, 25–38. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Sethi, J.K. TNF-alpha and adipocyte biology. FEBS Lett. 2008, 582, 117–131. [Google Scholar] [CrossRef]
- Sharma, V.M.; Puri, V. Mechanism of TNF-α-induced lipolysis in human adipocytes uncovered. Obesity 2016, 24, 990. [Google Scholar] [CrossRef][Green Version]
- Kawakami, M.; Murase, T.; Ogawa, H.; Ishibashi, S.; Mori, N.; Takaku, F.; Shibata, S. Human recombinant TNF suppresses lipoprotein lipase activity and stimulates lipolysis in 3T3-L1 cells. J. Biochem. 1987, 101, 331–338. [Google Scholar] [CrossRef]
- Engin, A. The Pathogenesis of Obesity-Associated Adipose Tissue Inflammation. Adv. Exp. Med. Biol. 2017, 960, 221–245. [Google Scholar] [CrossRef]
- Guo, T.; Gupta, A.; Yu, J.; Granados, J.Z.; Gandhi, A.Y.; Evers, B.M.; Iyengar, P.; Infante, R.E. LIFR-α-dependent adipocyte signaling in obesity limits adipose expansion contributing to fatty liver disease. iScience 2021, 24, 102227. [Google Scholar] [CrossRef]
- Issa, N.; Lachance, G.; Bellmann, K.; Laplante, M.; Stadler, K.; Marette, A. Cytokines promote lipolysis in 3T3-L1 adipocytes through induction of NADPH oxidase 3 expression and superoxide production. J. Lipid Res. 2018, 59, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.D. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Owens, B.M.J.; Hegazy, A.N. The oncostatin M-stromal cell axis in health and disease. Scand. J. Immunol. 2018, 88, e12694. [Google Scholar] [CrossRef] [PubMed]
- Elks, C.M.; Zhao, P.; Grant, R.W.; Hang, H.; Bailey, J.L.; Burk, D.H.; McNulty, M.A.; Mynatt, R.L.; Stephens, J.M. Loss of Oncostatin M Signaling in Adipocytes Induces Insulin Resistance and Adipose Tissue Inflammation in Vivo. J. Biol. Chem. 2016, 291, 17066–17076. [Google Scholar] [CrossRef] [PubMed]
- Hang, H.; Bailey, J.L.; Elks, C.M. Oncostatin M Mediates Adipocyte Expression and Secretion of Stromal-Derived Factor 1. Biology 2019, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Infantes, D.; White, U.A.; Elks, C.M.; Morrison, R.F.; Gimble, J.M.; Considine, R.V.; Ferrante, A.W.; Ravussin, E.; Stephens, J.M. Oncostatin M Is Produced in Adipose Tissue and Is Regulated in Conditions of Obesity and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E217–E225. [Google Scholar] [CrossRef]
- Mosley, B.; De Imus, C.; Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef]
- Wong, S.; Botelho, F.M.; Rodrigues, R.M.; Richards, C.D. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 Dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Investig. 2014, 94, 1003–1016. [Google Scholar] [CrossRef]
- Halfter, H.; Friedrich, M.; Postert, C.; Ringelstein, E.B.; Stögbauer, F. Activation of Jak-Stat and MAPK2 pathways by oncostatin M leads to growth inhibition of human glioma cells. Mol. Cell Biol. Res. Commun. 1999, 1, 109–116. [Google Scholar] [CrossRef]
- Levy, J.B.; Schindler, C.; Raz, R.; Levy, D.E.; Baron, R.; Horowitz, M.C. Activation of the JAK-STAT signal transduction pathway by oncostatin-M cultured human and mouse osteoblastic cells. Endocrinology 1996, 137, 1159–1165. [Google Scholar] [CrossRef]
- Henning, P.; Movérare-Skrtic, S.; Westerlund, A.; Chaves de Souza, P.P.; Floriano-Marcelino, T.; Nilsson, K.H.; El Shahawy, M.; Ohlsson, C.; Lerner, U.H. WNT16 is Robustly Increased by Oncostatin M in Mouse Calvarial Osteoblasts and Acts as a Negative Feedback Regulator of Osteoclast Formation Induced by Oncostatin M. J. Inflamm. Res. 2021, 14, 4723–4741. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, H.M.; Radtke, S.; Schaper, F.; Heinrich, P.C.; Behrmann, I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J. Biol. Chem. 2000, 275, 40742–40748. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.; Souza, P.P.C.; Floriano-Marcelino, T.; Conaway, H.H.; Henning, P.; Lerner, U.H. Activation of Shc1 Allows Oncostatin M to Induce RANKL and Osteoclast Formation More Effectively Than Leukemia Inhibitory Factor. Front. Immunol. 2019, 10, 1164. [Google Scholar] [CrossRef] [PubMed]
- Stephens, J.M.; Bailey, J.L.; Hang, H.; Rittell, V.; Dietrich, M.A.; Mynatt, R.L.; Elks, C.M. Adipose Tissue Dysfunction Occurs Independently of Obesity in Adipocyte-Specific Oncostatin Receptor Knockout Mice. Obesity 2018, 26, 1439–1447. [Google Scholar] [CrossRef]
- Zhou, Y.; Abidi, P.; Kim, A.; Chen, W.; Huang, T.-T.; Kraemer, F.B.; Liu, J. Transcriptional Activation of Hepatic ACSL3 and ACSL5 by Oncostatin M Reduces Hypertriglyceridemia through Enhanced β-Oxidation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2198–2205. [Google Scholar] [CrossRef]
- Pollack, V.; Sarközi, R.; Banki, Z.; Feifel, E.; Wehn, S.; Gstraunthaler, G.; Stoiber, H.; Mayer, G.; Montesano, R.; Strutz, F.; et al. Oncostatin M-induced effects on EMT in human proximal tubular cells: Differential role of ERK signaling. Am. J. Physiol.-Ren. Physiol. 2007, 293, F1714–F1726. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Tanaka, M.; Naiki, T.; Miyajima, A. Oncostatin M Inhibits Adipogenesis through the RAS/ERK and STAT5 Signaling Pathways. J. Biol. Chem. 2006, 281, 37913–37920. [Google Scholar] [CrossRef]
- Burfoot, M.S.; Rogers, N.C.; Watling, D.; Smith, J.M.; Pons, S.; Paonessaw, G.; Pellegrini, S.; White, M.F.; Kerr, I.M. Janus Kinase-dependent Activation of Insulin Receptor Substrate 1 in Response to Interleukin-4, Oncostatin M, and the Interferons. J. Biol. Chem. 1997, 272, 24183–24190. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Eblen, S.T. Chapter Four—Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 138, pp. 99–142. [Google Scholar]
- Daniele, G.; Eldor, R.; Merovci, A.; Clarke, G.D.; Xiong, J.; Tripathy, D.; Taranova, A.; Abdul-Ghani, M.; DeFronzo, R.A. Chronic reduction of plasma free fatty acid improves mitochondrial function and whole-body insulin sensitivity in obese and type 2 diabetic individuals. Diabetes 2014, 63, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Jelenik, T.; Nowotny, P.; Szendroedi, J.; Roden, M. Reduction of non-esterified fatty acids improves insulin sensitivity and lowers oxidative stress, but fails to restore oxidative capacity in type 2 diabetes: A randomised clinical trial. Diabetologia 2014, 57, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Makimura, H.; Stanley, T.L.; Suresh, C.; De Sousa-Coelho, A.L.; Frontera, W.R.; Syu, S.; Braun, L.R.; Looby, S.E.; Feldpausch, M.N.; Torriani, M.; et al. Metabolic Effects of Long-Term Reduction in Free Fatty Acids with Acipimox in Obesity: A Randomized Trial. J. Clin. Endocrinol. Metab. 2016, 101, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Romauch, M.; Schreiber, R.; Grabner, G.F.; Hütter, S.; Kotzbeck, P.; Benedikt, P.; Eichmann, T.O.; Yamada, S.; Knittelfelder, O.; et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat. Commun. 2017, 8, 14859. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Hung, C.W.; Ahmadian, M.; Zhao, P.; Keinan, O.; Gomez, A.V.; DeLuca, J.H.; Dadpey, B.; Lu, D.; Zaid, J.; et al. Catecholamines suppress fatty acid re-esterification and increase oxidation in white adipocytes via STAT3. Nat. Metab. 2020, 2, 620–634. [Google Scholar] [CrossRef]
- Richard, A.J.; Hang, H.; Allerton, T.D.; Zhao, P.; Mendoza, T.; Ghosh, S.; Elks, C.M.; Stephens, J.M. Loss of Adipocyte STAT5 Confers Increased Depot-Specific Adiposity in Male and Female Mice That Is Not Associated With Altered Adipose Tissue Lipolysis. Front. Endocrinol. 2022, 13, 438. [Google Scholar] [CrossRef]
- Hjelholt, A.J.; Lee, K.Y.; Arlien-Søborg, M.C.; Pedersen, S.B.; Kopchick, J.J.; Puri, V.; Jessen, N.; Jørgensen, J.O.L. Temporal patterns of lipolytic regulators in adipose tissue after acute growth hormone exposure in human subjects: A randomized controlled crossover trial. Mol. Metab. 2019, 29, 65–75. [Google Scholar] [CrossRef]
- Kopchick, J.J.; Berryman, D.E.; Puri, V.; Lee, K.Y.; Jorgensen, J.O.L. The effects of growth hormone on adipose tissue: Old observations, new mechanisms. Nat. Rev. Endocrinol. 2020, 16, 135–146. [Google Scholar] [CrossRef]
- Grove, R.I.; Mazzucco, C.E.; Radka, S.F.; Shoyab, M.; Kiener, P.A. Oncostatin M up-regulates low density lipoprotein receptors in HepG2 cells by a novel mechanism. J. Biol. Chem. 1991, 266, 18194–18199. [Google Scholar] [CrossRef]
- Li, C.; Kraemer, F.B.; Ahlborn, T.E.; Liu, J. Induction of Low Density Lipoprotein Receptor (LDLR) Transcription by Oncostatin M Is Mediated by the Extracellular Signal-regulated Kinase Signaling Pathway and the Repeat 3 Element of the LDLR Promoter. J. Biol. Chem. 1999, 274, 6747–6753. [Google Scholar] [CrossRef] [PubMed]
- Grove, R.I.; Eberhardt, C.; Abid, S.; Mazzucco, C.; Liu, J.; Kiener, P.; Todaro, G.; Shoyab, M. Oncostatin M is a mitogen for rabbit vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 1993, 90, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Tanaka, M.; Senba, E.; Miyajima, A.; Morikawa, Y. Deficiency of Oncostatin M Receptor B (OSMRB) Exacerbates High-fat Diet-induced Obesity and Related Metabolic Disorders in Mice. J. Biol. Chem. 2014, 289, 13821–13837. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Wang, P.-X.; Li, Z.-Z.; Zhang, X.-J.; Jiang, X.; Gong, J.; Qin, J.-j.; Guo, J.; Zhu, X.; Yang, S.; et al. Hepatic Oncostatin M Receptor β Regulates Obesity-Induced Steatosis and Insulin Resistance. Am. J. Pathol. 2016, 186, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Fukuhara, A.; Onodera, T.; Kita, S.; Yokoyama, C.; Otsuki, M.; Shimomura, I. SDF-1 Is an Autocrine Insulin-Desensitizing Factor in Adipocytes. Diabetes 2018, 67, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2020, 10, 2993. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Dunphy, G.; Heras-Murillo, I.; Mastrangelo, A.; Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell. Mol. Immunol. 2022, 19, 384–408. [Google Scholar] [CrossRef]
- Yu, G.; Wu, X.; Kilroy, G.; Halvorsen, Y.D.; Gimble, J.M.; Floyd, Z.E. Isolation of murine adipose-derived stem cells. Methods Mol. Biol. 2011, 702, 29–36. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer | Catalog Number | Dilution (Diluent) |
|---|---|---|---|
| anti-IRS1 | Cell Signaling 1 | 3194 | 1:1000 (5% NFDM) |
| anti-pIRS1 (S612) | Cell Signaling | 2386 | 1:1000 (5% BSA-TBST) 2 |
| anti-STAT5A | Abcam 3 | ab32043 | 1:1000 (1% BSA- TBST) |
| anti-p-p66 (S36) | Millipore Sigma | 566807 | 1:500 (1% BSA-TBST) |
| anti-Shc1 | R&D Systems | MAB7129 | 1:500 (1% BSA-TBST) |
| anti-pERK1/2 | Promega 4 | V8031 | 1:5000 (1% BSA-TBST) |
| anti-ERK 1/2 | Cell Signaling | 4695 | 1:1000 (1% BSA-TBST) |
| anti-β actin | Santa Cruz | sc-47778 | 1:1000 (1% BSA-TBST) |
| Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| G0s2 | CCT GCA CAC TTT CCA TCT GA | CAA AGC CAG TCT GAC GCA A |
| Pnpla2 | CTC ATA AAG TGG CAA GTT GTC TG | GAG CTC ATC CAG GCC AAT |
| Lipe | CTG CAA GAG TAT GTC ACG CTA | CTC GTT GCG TTT GTA GTG C |
| Abhd5 | CCC ACA TCT ACA TCA CAC CTT | GAG AGA ACA TCA GCG TCC ATA |
| Plin1 | CGT GGA GAG TAA GGA TGT CAA TG | GTG CTG TTG TAG GTC TTC TGG |
| Hilpda | TTC CAC GTT GCG ACT CAG | GAA GTA GAG CTG CTT TCT GCT |
| Ppia | CCA CTG TCG CTT TTC GCC GC | TGC AAA CAG CTC GAA GGA GAC GC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, J.L.; Hang, H.; Boudreau, A.; Elks, C.M. Oncostatin M Induces Lipolysis and Suppresses Insulin Response in 3T3-L1 Adipocytes. Int. J. Mol. Sci. 2022, 23, 4689. https://doi.org/10.3390/ijms23094689
Bailey JL, Hang H, Boudreau A, Elks CM. Oncostatin M Induces Lipolysis and Suppresses Insulin Response in 3T3-L1 Adipocytes. International Journal of Molecular Sciences. 2022; 23(9):4689. https://doi.org/10.3390/ijms23094689
Chicago/Turabian StyleBailey, Jennifer L., Hardy Hang, Anik Boudreau, and Carrie M. Elks. 2022. "Oncostatin M Induces Lipolysis and Suppresses Insulin Response in 3T3-L1 Adipocytes" International Journal of Molecular Sciences 23, no. 9: 4689. https://doi.org/10.3390/ijms23094689
APA StyleBailey, J. L., Hang, H., Boudreau, A., & Elks, C. M. (2022). Oncostatin M Induces Lipolysis and Suppresses Insulin Response in 3T3-L1 Adipocytes. International Journal of Molecular Sciences, 23(9), 4689. https://doi.org/10.3390/ijms23094689