
Structural and Computational Studies of the SARS-CoV-2 Spike Protein Binding Mechanisms with Nanobodies: From Structure and Dynamics to Avidity-Driven Nanobody Engineering


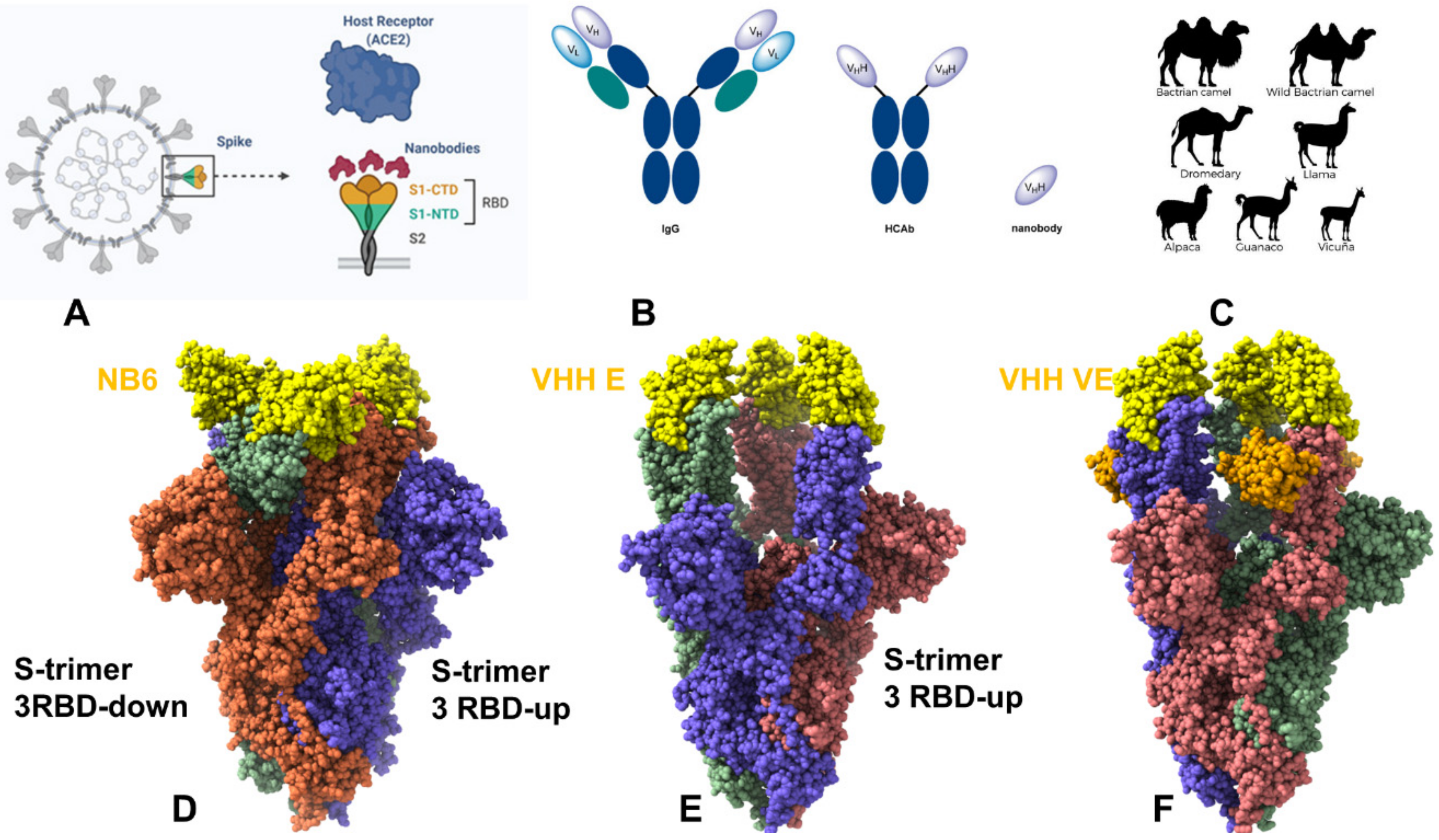{kind=link}
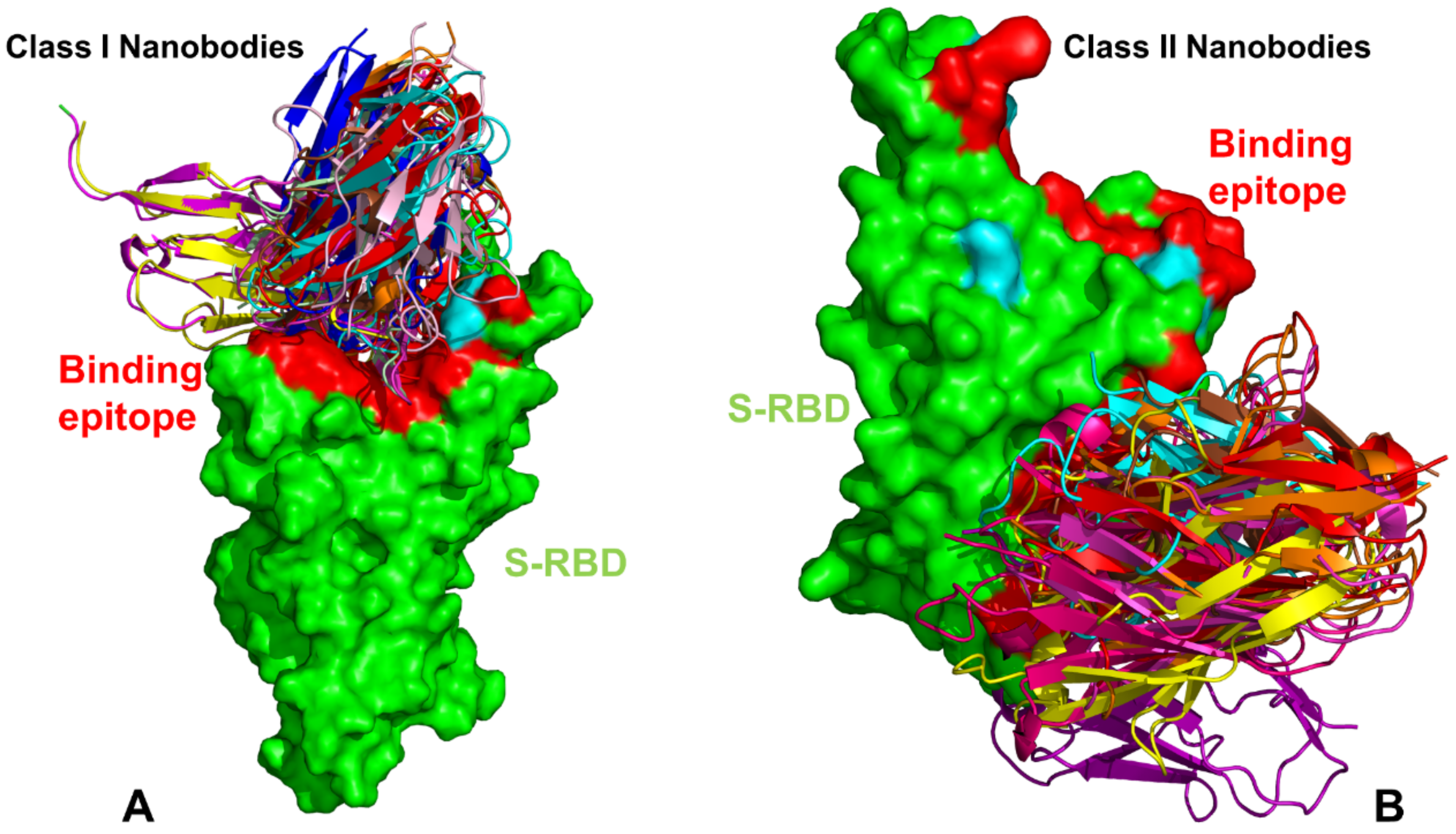{kind=link}
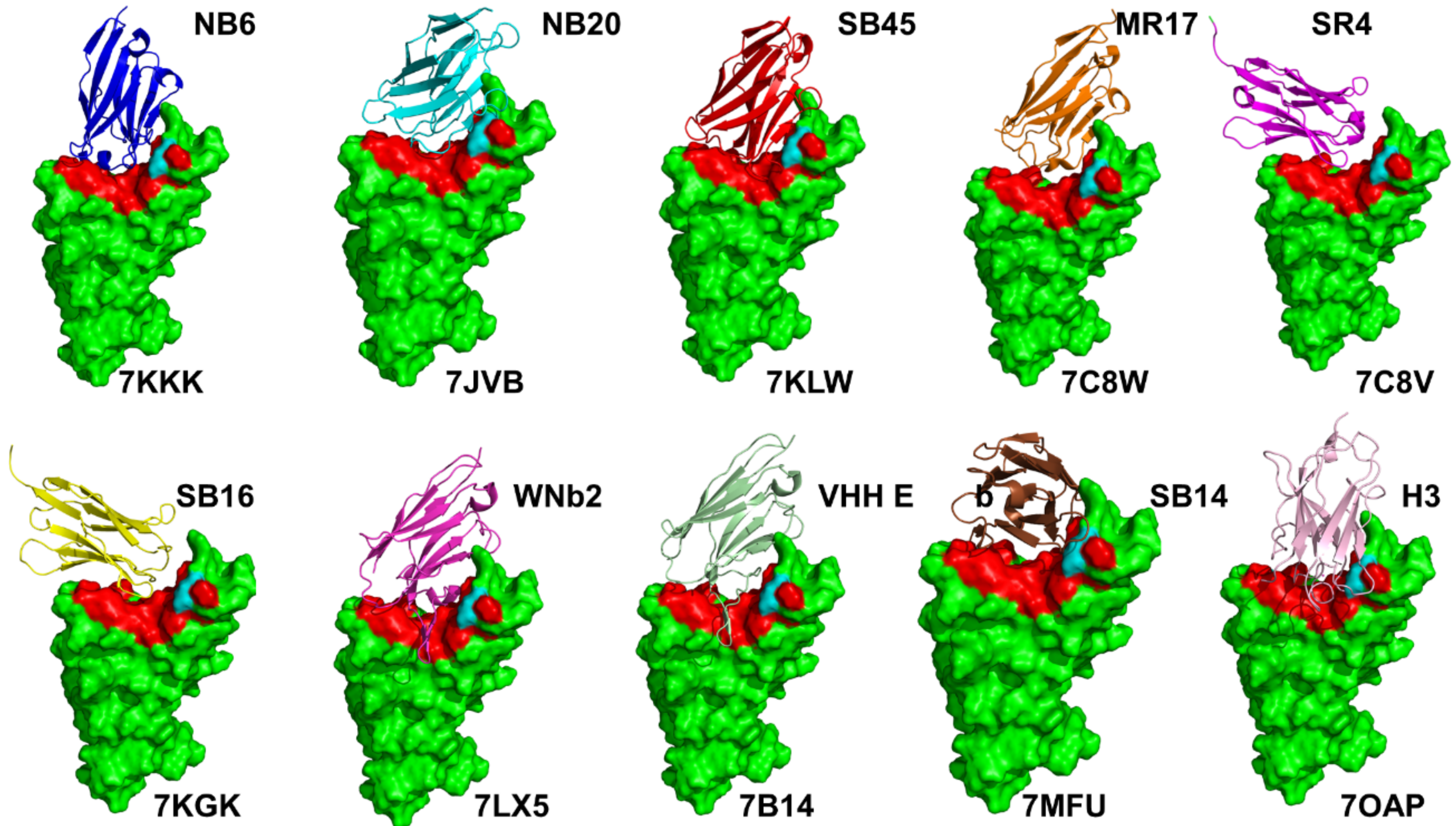{kind=link}
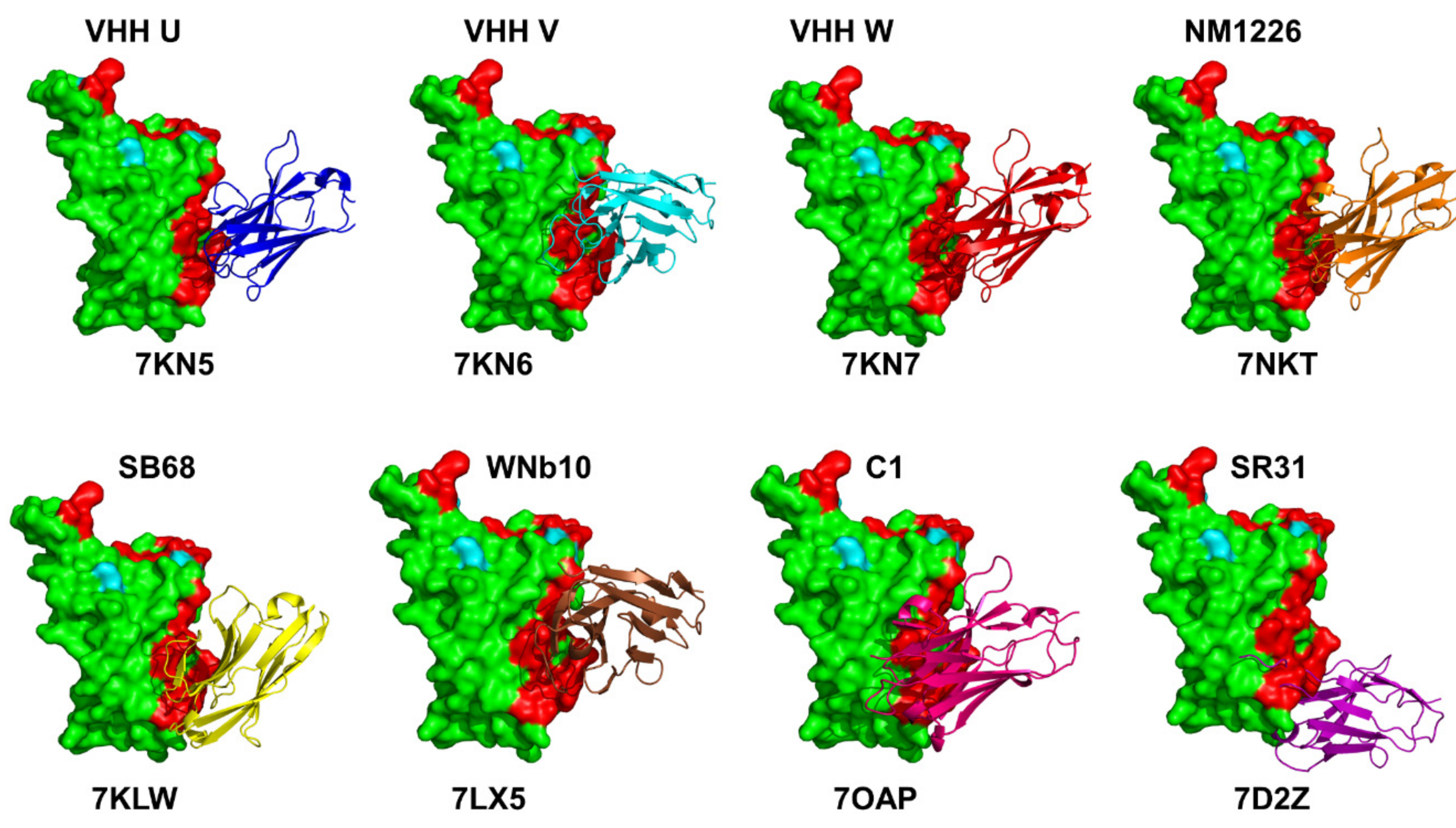{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
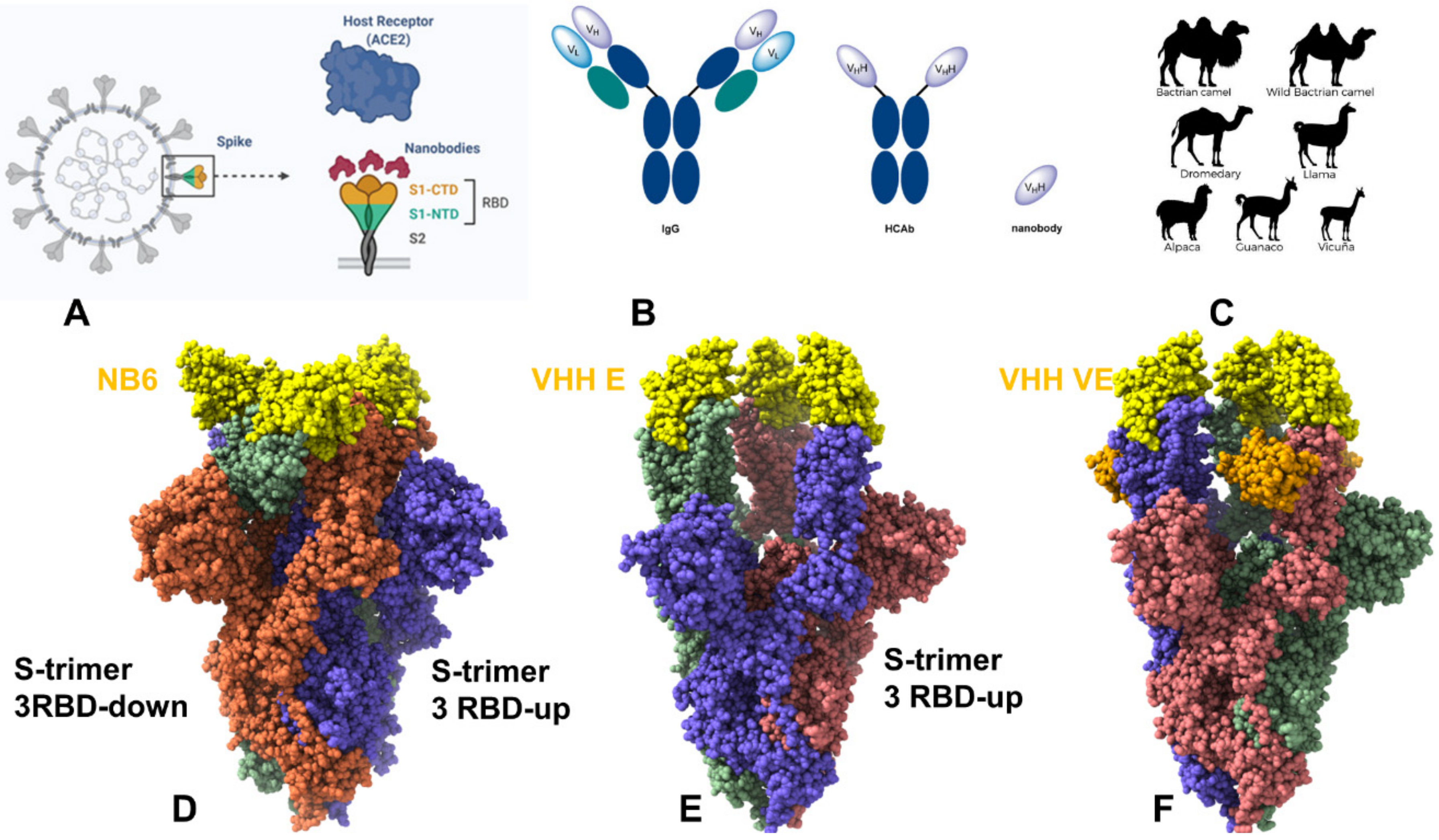
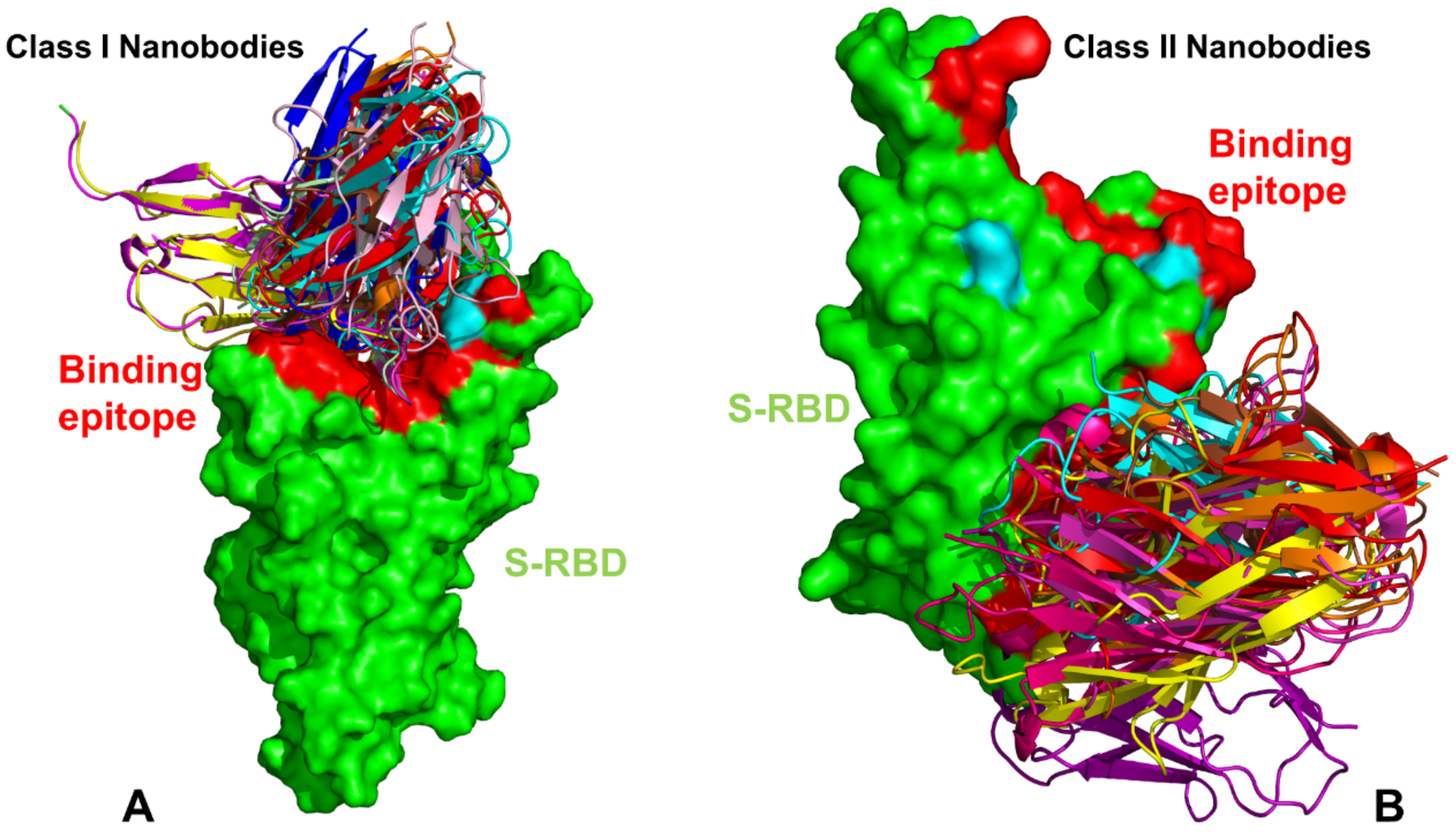
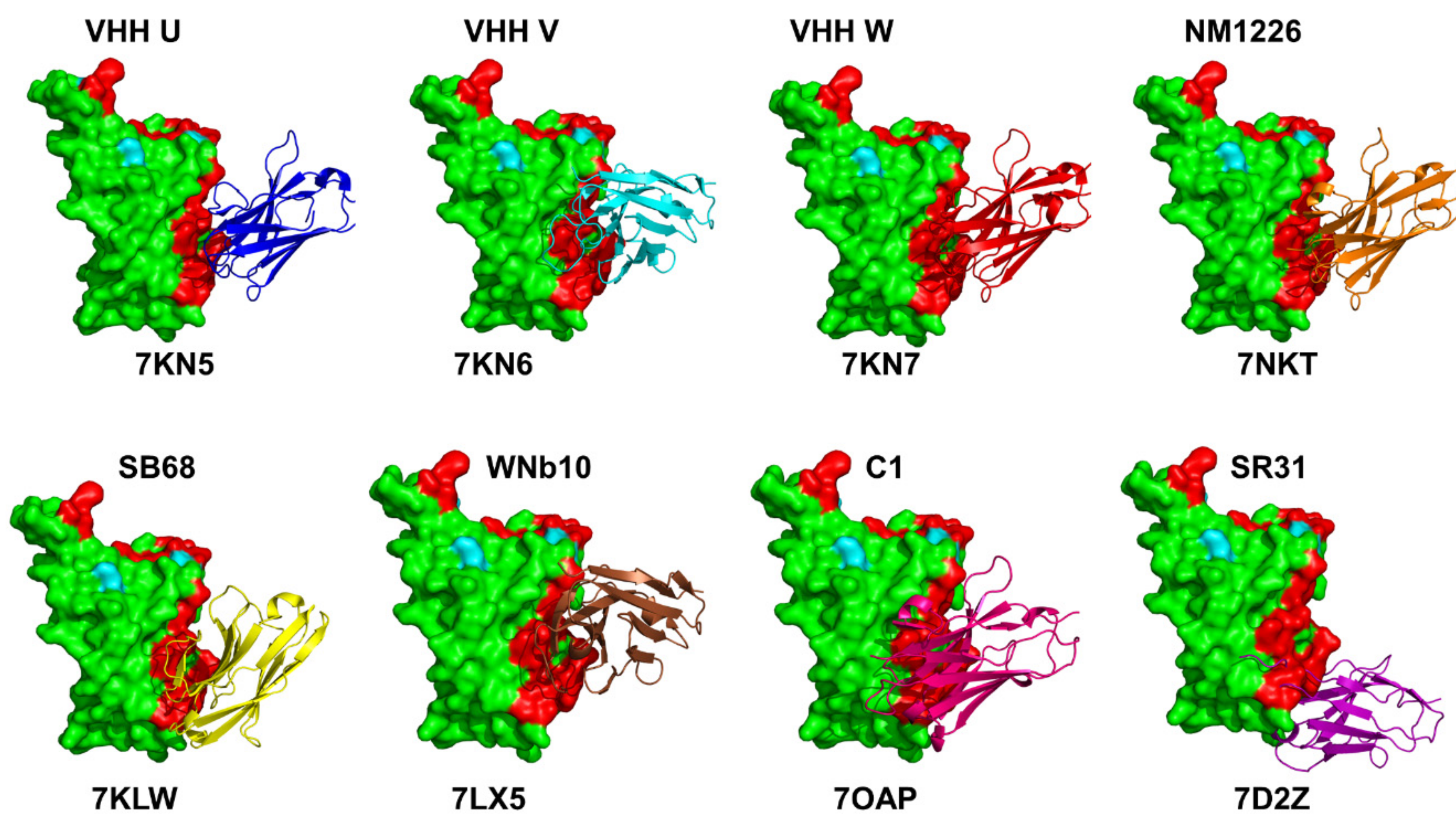
2. Structural Biology of the SARS-CoV-2 S Protein Complexes with Nanobodies: Classification, Binding Epitopes, and Molecular Analyses
3. Biophysical Characterization and Protein Engineering of SARS-CoV-2 Complexes with Nanobodies: From Monomeric to Multivalent Formats and Synergistic Combinations in the Quest for High-Affinity and Mutation-Resistant Nanobodies
4. Computational Studies of SARS-CoV-2 S Protein Binding Mechanisms: Structure, Dynamics, and Allostery
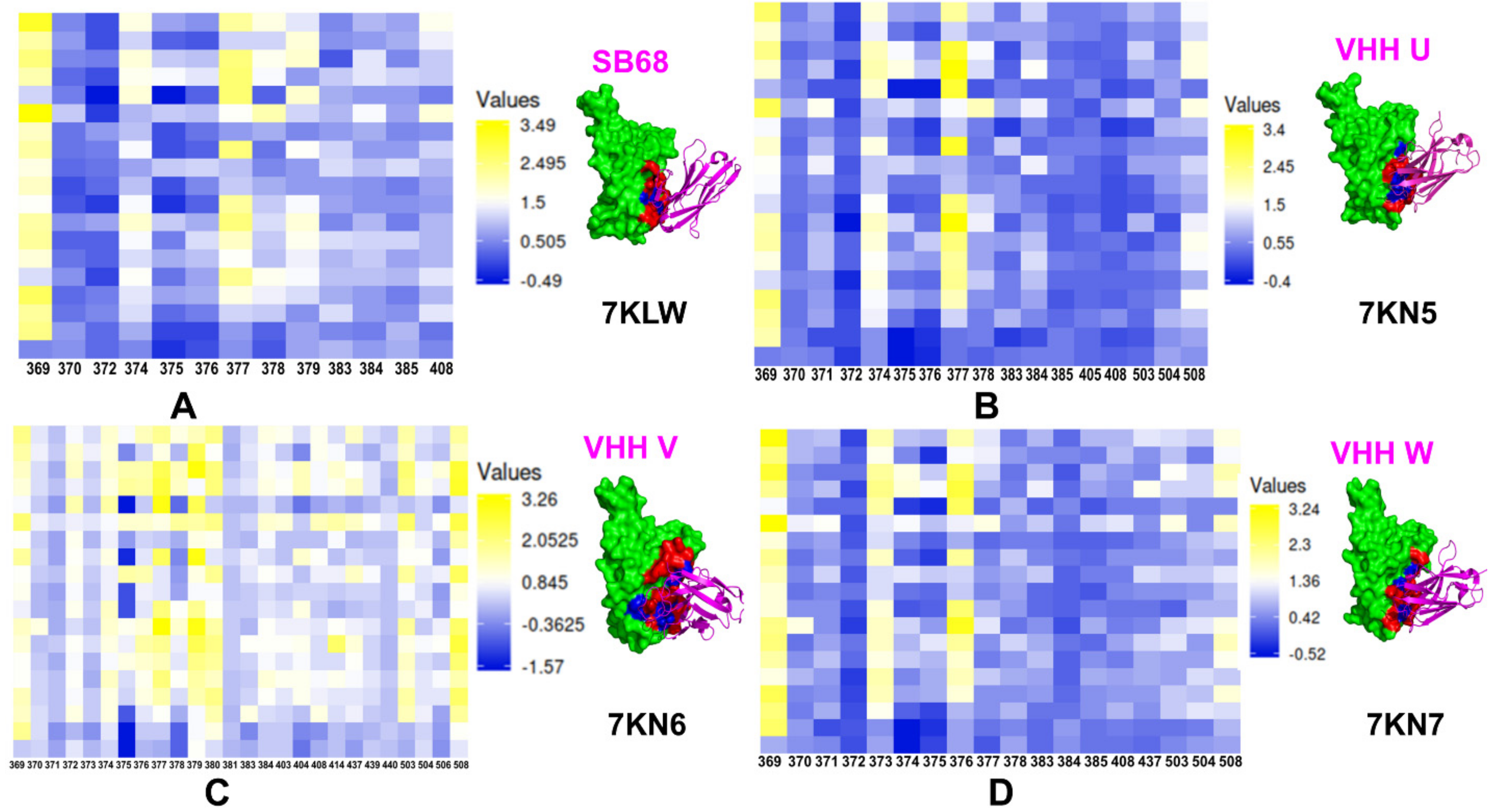
5. Exploring Computational Approaches for Mutational Profiling of SARS-CoV-2 S Protein Binding with Nanobodies and Characterization of the Binding Energy Hotspots
6. Integration of Structural and Computational Approaches for Nanobody Engineering and Avidity-Based Control of Binding Mechanisms: Outlook and Future Directions
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS | Severe Acute Respiratory Syndrome |
| RBD | Receptor Binding Domain |
| ACE2 | Angiotensin-Converting Enzyme 2 (ACE2) |
| NTD | N-terminal domain |
| RBD | receptor-binding domain |
| CTD1 | C-terminal domain 1 |
| CTD2 | C-terminal domain 2 |
| FP | fusion peptide |
| FPPR | fusion peptide proximal region |
| HR1 | heptad repeat 1 |
| CH | central helix region |
| CD | connector domain |
| HR2 | heptad repeat 2 |
| TM | transmembrane anchor |
| CT | cytoplasmic tail |
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.; Lagniton, P.N.; Ye, S.; Li, E.; Xu, R.-H. COVID-19: What has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 2020, 16, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.-C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; Von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Gavor, E.; Choong, Y.K.; Er, S.Y.; Sivaraman, H.; Sivaraman, J. Structural Basis of SARS-CoV-2 and SARS-CoV Antibody Interactions. Trends Immunol. 2020, 41, 1006–1022. [Google Scholar] [CrossRef]
- Finkelstein, M.; Mermelstein, A.; Miller, E.; Seth, P.; Stancofski, E.-S.; Fera, D. Structural Analysis of Neutralizing Epitopes of the SARS-CoV-2 Spike to Guide Therapy and Vaccine Design Strategies. Viruses 2021, 13, 134. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Barnes, C.O.; West, A.P., Jr.; Huey-Tubman, K.E.; Hoffmann, M.A.; Sharaf, N.G.; Hoffman, P.R.; Koranda, N.; Gristick, H.B.; Gaebler, C.; Muecksch, F.; et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182, 828–842.e16. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell 2020, 183, 1013–1023.e13. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef]
- Ge, J.; Wang, R.; Ju, B.; Zhang, Q.; Sun, J.; Chen, P.; Zhang, S.; Tian, Y.; Shan, S.; Cheng, L.; et al. Antibody neutralization of SARS-CoV-2 through ACE2 receptor mimicry. Nat. Commun. 2021, 12, 250. [Google Scholar] [CrossRef]
- Ku, Z.; Xie, X.; Davidson, E.; Ye, X.; Su, H.; Menachery, V.D.; Li, Y.; Yuan, Z.; Zhang, X.; Muruato, A.E.; et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat. Commun. 2021, 12, 469. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, H.; Wu, N.C.; Lee, C.-C.D.; Zhu, X.; Zhao, F.; Huang, D.; Yu, W.; Hua, Y.; Tien, H.; et al. Structural basis of a shared antibody response to SARS-CoV-2. Science 2020, 369, 1119–1123. [Google Scholar] [CrossRef]
- Zhou, D.; Duyvesteyn, H.M.E.; Chen, C.-P.; Huang, C.-G.; Chen, T.-H.; Shih, S.-R.; Lin, Y.-C.; Cheng, C.-Y.; Cheng, S.-H.; Huang, Y.-C.; et al. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat. Struct. Mol. Biol. 2020, 27, 950–958. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Lv, Z.; Deng, Y.-Q.; Ye, Q.; Cao, L.; Sun, C.-Y.; Fan, C.; Huang, W.; Sun, S.; Sun, Y.; Zhu, L.; et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV 2 by a potent therapeutic antibody. Science 2020, 369, 1505–1509. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a hu-man monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- Asarnow, D.; Wang, B.; Lee, W.-H.; Hu, Y.; Huang, C.-W.; Faust, B.; Ng, P.M.L.; Ngoh, E.Z.X.; Bohn, M.; Bulkley, D.; et al. Structural insight into SARS-CoV-2 neutralizing antibodies and modulation of syncytia. Cell 2021, 184, 3192–3204.e16. [Google Scholar] [CrossRef]
- Hurlburt, N.K.; Seydoux, E.; Wan, Y.-H.; Edara, V.V.; Stuart, A.B.; Feng, J.; Suthar, M.S.; McGuire, A.T.; Stamatatos, L.; Pancera, M. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat. Commun. 2020, 11, 5413. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Starr, T.N.; Czudnochowski, N.; Liu, Z.; Zatta, F.; Park, Y.-J.; Addetia, A.; Pinto, D.; Beltramello, M.; Hernandez, P.; Greaney, A.J.; et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 2021, 597, 97–102. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional importance of the D614G mutation in the SARS-CoV-2 spike protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. [Google Scholar] [CrossRef]
- Fiorentini, S.; Messali, S.; Zani, A.; Caccuri, F.; Giovanetti, M.; Ciccozzi, M.; Caruso, A. First detection of SARS-CoV-2 spike protein N501 mutation in Italy in August, 2020. Lancet Infect. Dis. 2021, 21, e147. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Muik, A.; Wallisch, A.-K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; Türeci, Ö.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine–elicited human sera. Science 2021, 371, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Hogan, C.A.; Jassem, A.N.; Sbihi, H.; Joffres, Y.; Tyson, J.R.; Noftall, K.; Taylor, M.; Lee, T.; Fjell, C.; Wilmer, A.; et al. Rapid Increase in SARS-CoV-2 P.1 Lineage Leading to Codominance with B.1.1.7 Lineage, British Columbia, Canada, January–April 2021. Emerg. Infect. Dis. 2021, 27, 2802–2809. [Google Scholar] [CrossRef] [PubMed]
- Javanmardi, K.; Chou, C.-W.; Terrace, C.I.; Annapareddy, A.; Kaoud, T.S.; Guo, Q.; Lutgens, J.; Zorkic, H.; Horton, A.P.; Gardner, E.C.; et al. Rapid characterization of spike variants via mammalian cell surface display. Mol. Cell 2021, 81, 5099–5111.e8. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Czudnochowski, N.; Starr, T.N.; Marzi, R.; Walls, A.C.; Zatta, F.; Bowen, J.E.; Jaconi, S.; Di Iulio, J.; Wang, Z.; et al. Broad sarbecovirus neutralization by a human monoclonal antibody. Nature 2021, 597, 103–108. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Saunders, K.; Edwards, R.J.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation and antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2021, 602, 671–675. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e15. [Google Scholar] [CrossRef]
- Chen, F.; Liu, Z.; Jiang, F. Prospects of Neutralizing Nanobodies Against SARS-CoV-2. Front. Immunol. 2021, 12, 690742. [Google Scholar] [CrossRef]
- Aria, H.; Mahmoodi, F.; Ghaheh, H.S.; Mavandadnejad, F.; Zare, H.; Heiat, M.; Bakherad, H. Outlook of therapeutic and diagnostic competency of nanobodies against SARS-CoV-2: A systematic review. Anal. Biochem. 2022, 640, 114546. [Google Scholar] [CrossRef]
- Niu, L.; Wittrock, K.N.; Clabaugh, G.C.; Srivastava, V.; Cho, M.W. A Structural Landscape of Neutralizing Antibodies against SARS-CoV-2 Receptor Binding Domain. Front. Immunol. 2021, 12, 647934. [Google Scholar] [CrossRef]
- Sasisekharan, R. Preparing for the Future—Nanobodies for Covid-19? N. Engl. J. Med. 2021, 384, 1568–1571. [Google Scholar] [CrossRef]
- Labroussaa, F.; Jores, J. SARS-CoV-2 nanobodies 2.0. Signal Transduct. Target. Ther. 2021, 6, 202. [Google Scholar] [CrossRef]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; Van Schie, L.; Hoffmann, M.; Pöhlmann, S.; et al. Structural Basis for Potent Neutralization of Betacoronaviruses by Single-Domain Camelid Antibodies. Cell 2020, 181, 1436–1441. [Google Scholar] [CrossRef]
- Dong, J.; Huang, B.; Jia, Z.; Wang, B.; Kankanamalage, S.G.; Titong, A.; Liu, Y. Development of multi-specific humanized llama antibodies blocking SARS-CoV-2/ACE2 interaction with high affinity and avidity. Emerg. Microbes Infect. 2020, 9, 1034–1036. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Jovcevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Owens, R.J.; Naismith, J.H. Structural Biology of Nanobodies against the Spike Protein of SARS-CoV-2. Viruses 2021, 13, 2214. [Google Scholar] [CrossRef] [PubMed]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbølle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Nambulli, S.; Xiao, Z.; Liu, H.; Sang, Z.; Duprex, W.P.; Schneidman-Duhovny, D.; Zhang, C.; Shi, Y. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science 2020, 370, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sang, Z.; Kim, Y.J.; Xiang, Y.; Cohen, T.; Belford, A.K.; Huet, A.; Conway, J.F.; Sun, J.; Taylor, D.J.; et al. Potent neutralizing nanobodies resist convergent circulating variants of SARS-CoV-2 by targeting diverse and conserved epitopes. Nat. Commun. 2021, 12, 4676. [Google Scholar] [CrossRef]
- Mast, F.D.; Fridy, P.C.; Ketaren, N.E.; Wang, J.; Jacobs, E.Y.; Olivier, J.P.; Sanyal, T.; Molloy, K.R.; Schmidt, F.; Rutkowska, M.; et al. Highly synergistic combinations of nanobodies that target SARS-CoV-2 and are resistant to escape. eLife 2021, 10, e73027. [Google Scholar] [CrossRef]
- Bracken, C.J.; Lim, S.A.; Solomon, P.; Rettko, N.J.; Nguyen, D.P.; Zha, B.S.; Schaefer, K.; Byrnes, J.R.; Zhou, J.; Lui, I.; et al. Bi-paratopic and multivalent VH domains block ACE2 binding and neutralize SARS-CoV-2. Nat. Chem. Biol. 2021, 17, 113–121. [Google Scholar] [CrossRef]
- Francino-Urdaniz, I.M.; Steiner, P.J.; Kirby, M.B.; Zhao, F.; Haas, C.M.; Barman, S.; Rhodes, E.R.; Leonard, A.C.; Peng, L.; Sprenger, K.G.; et al. One-shot identification of SARS-CoV-2 S RBD escape mutants using yeast screening. Cell Rep. 2021, 36, 109627. [Google Scholar] [CrossRef]
- Koenig, P.-A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.-M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef]
- Pymm, P.; Adair, A.; Chan, L.-J.; Cooney, J.P.; Mordant, F.L.; Allison, C.C.; Lopez, E.; Haycroft, E.R.; O’Neill, M.T.; Tan, L.L.; et al. Nanobody cocktails potently neutralize SARS-CoV-2 D614G N501Y variant and protect mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2101918118. [Google Scholar] [CrossRef]
- Güttler, T.; Aksu, M.; Dickmanns, A.; Stegmann, K.M.; Gregor, K.; Rees, R.; Taxer, W.; Rymarenko, O.; Schünemann, J.; Dienemann, C.; et al. Neutralization of SARS-CoV-2 by highly potent, hyperthermostable, and mutation-tolerant nanobodies. EMBO J. 2021, 40, e107985. [Google Scholar] [CrossRef]
- Xu, J.; Xu, K.; Jung, S.; Conte, A.; Lieberman, J.; Muecksch, F.; Lorenzi, J.C.C.; Park, S.; Schmidt, F.; Wang, Z.; et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature 2021, 595, 278–282. [Google Scholar] [CrossRef]
- Ma, H.; Zeng, W.; Meng, X.; Huang, X.; Yang, Y.; Zhao, D.; Zhou, P.; Wang, X.; Zhao, C.; Sun, Y.; et al. Potent Neutralization of SARS-CoV-2 by Hetero-Bivalent Alpaca Nanobodies Targeting the Spike Receptor-Binding Domain. J. Virol. 2021, 95, e02438-20. [Google Scholar] [CrossRef]
- Ahmad, J.; Jiang, J.; Boyd, L.F.; Zeher, A.; Huang, R.; Xia, D.; Natarajan, K.; Margulies, D.H. Structures of synthetic nanobody–SARS-CoV-2 receptor-binding domain complexes reveal distinct sites of interaction. J. Biol. Chem. 2021, 297, 101202. [Google Scholar] [CrossRef]
- Hanke, L.; Das, H.; Sheward, D.J.; Vidakovics, L.P.; Urgard, E.; Moliner-Morro, A.; Kim, C.; Karl, V.; Pankow, A.; Smith, N.L.; et al. A bispecific monomeric nanobody induces spike trimer dimers and neutralizes SARS-CoV-2 in vivo. Nat. Commun. 2022, 13, 155. [Google Scholar] [CrossRef]
- Huo, J.; Mikolajek, H.; Le Bas, A.; Clark, J.J.; Sharma, P.; Kipar, A.; Dormon, J.; Norman, C.; Weckener, M.; Clare, D.K.; et al. A potent SARS-CoV-2 neutralising nanobody shows therapeutic efficacy in the Syrian golden hamster model of COVID-19. Nat. Commun. 2021, 12, 5469. [Google Scholar] [CrossRef]
- Ye, G.; Gallant, J.; Zheng, J.; Massey, C.; Shi, K.; Tai, W.; Odle, A.; Vickers, M.; Shang, J.; Wan, Y.; et al. The development of Nanosota-1 as anti-SARS-CoV-2 nanobody drug candidates. eLife 2021, 10, e64815. [Google Scholar] [CrossRef]
- Li, T.; Cai, H.; Yao, H.; Zhou, B.; Zhang, N.; van Vlissingen, M.F.; Kuiken, T.; Han, W.; GeurtsvanKessel, C.H.; Gong, Y.; et al. A synthetic nanobody targeting RBD protects hamsters from SARS-CoV-2 infection. Nat. Commun. 2021, 12, 4635. [Google Scholar] [CrossRef]
- Obeng, E.M.; Dzuvor, C.K.; Danquah, M.K. Anti-SARS-CoV-1 and −2 nanobody engineering towards avidity-inspired therapeutics. Nano Today 2022, 42, 101350. [Google Scholar] [CrossRef]
- Walter, J.D.; Hutter, C.A.J.; Garaeva, A.A.; Scherer, M.; Zimmermann, I.; Wyss, M.; Rheinberger, J.; Ruedin, Y.; Earp, J.C.; Egloff, P.; et al. Biparatopic sybody constructs neutralize SARS-CoV-2 variants of concern and mitigate emergence of drug resistance. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wagner, T.R.; Ostertag, E.; Kaiser, P.D.; Gramlich, M.; Ruetalo, N.; Junker, D.; Haering, J.; Traenkle, B.; Becker, M.; Dulovic, A.; et al. NeutrobodyPlex—monitoring SARS-CoV-2 neutralizing immune responses using nanobodies. EMBO Rep. 2021, 22, e52325. [Google Scholar] [CrossRef]
- Rossotti, M.A.; van Faassen, H.; Tran, A.; Sheff, J.; Sandhu, J.K.; Duque, D.; Hewitt, M.; Wen, S.; Bavananthasivam, R.; Beitari, S.; et al. Arsenal of Nanobodies for Broad-Spectrum Countermeasures against Current and Future SARS-CoV-2 Variants of Concerns. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dong, J.; Huang, B.; Wang, B.; Titong, A.; Kankanamalage, S.G.; Jia, Z.; Wright, M.; Parthasarathy, P.; Liu, Y. Development of humanized tri-specific nanobodies with potent neutralization for SARS-CoV-2. Sci. Rep. 2020, 10, 17806. [Google Scholar] [CrossRef]
- Nambulli, S.; Xiang, Y.; Tilston-Lunel, N.L.; Rennick, L.J.; Sang, Z.; Klimstra, W.B.; Reed, D.S.; Crossland, N.A.; Shi, Y.; Duprex, W.P. Inhalable Nanobody (PiN-21) prevents and treats SARS-CoV-2 infections in Syrian hamsters at ultra-low doses. Sci. Adv. 2021, 7, eabh0319. [Google Scholar] [CrossRef]
- Yao, H.; Cai, H.; Li, T.; Zhou, B.; Qin, W.; Lavillette, D.; Li, D. A high-affinity RBD-targeting nanobody improves fusion partner’s potency against SARS-CoV-2. PLoS Pathog. 2021, 17, e1009328. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, Z.; Li, H.; Zhong, K.; Zhao, Q.; Wang, Z.; Wu, Z.; Yang, D.; Sun, S.; Yang, N.; et al. Development of multivalent nanobodies blocking SARS-CoV-2 infection by targeting RBD of spike protein. J. Nanobiotechnol. 2021, 19, 33. [Google Scholar] [CrossRef]
- Zupancic, J.M.; Schardt, J.S.; Desai, A.A.; Makowski, E.K.; Smith, M.D.; Pornnoppadol, G.; Barbosa, M.G.D.M.; Cascalho, M.; Lanigan, T.M.; Tessier, P.M. Engineered Multivalent Nanobodies Potently and Broadly Neutralize SARS-CoV-2 Variants. Adv. Ther. 2021, 4, 2100099. [Google Scholar] [CrossRef]
- Yin, V.; Lai, S.-H.; Caniels, T.G.; Brouwer, P.J.M.; Brinkkemper, M.; Aldon, Y.; Liu, H.; Yuan, M.; Wilson, I.A.; Sanders, R.W.; et al. Probing Affinity, Avidity, Anticooperativity, and Competition in Antibody and Receptor Binding to the SARS-CoV-2 Spike by Single Particle Mass Analyses. ACS Central Sci. 2021, 7, 1863–1873. [Google Scholar] [CrossRef]
- Erlendsson, S.; Teilum, K. Binding Revisited—Avidity in Cellular Function and Signaling. Front. Mol. Biosci. 2021, 7, 615565. [Google Scholar] [CrossRef]
- Divine, R.; Dang, H.V.; Ueda, G.; Fallas, J.A.; Vulovic, I.; Sheffler, W.; Saini, S.; Zhao, Y.T.; Raj, I.X.; Morawski, P.A.; et al. Designed proteins assemble antibodies into modular nanocages. Science 2021, 372, eabd9994. [Google Scholar] [CrossRef]
- Gur, M.; Taka, E.; Yilmaz, S.Z.; Kilinc, C.; Aktas, U.; Golcuk, M. Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. J. Chem. Phys. 2020, 153, 075101. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.; Park, S.-J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Central Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef]
- Sikora, M.; von Bülow, S.; Blanc, F.E.C.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef]
- Brielle, E.; Schneidman-Duhovny, D.; Linial, M. The SARS-CoV-2 Exerts a Distinctive Strategy for Interacting with the ACE2 Human Receptor. Viruses 2020, 12, 497. [Google Scholar] [CrossRef]
- Ghorbani, M.; Brooks, B.R.; Klauda, J.B. Critical Sequence Hotspots for Binding of Novel Coronavirus to Angiotensin Converter Enzyme as Evaluated by Molecular Simulations. J. Phys. Chem. B 2020, 124, 10034–10047. [Google Scholar] [CrossRef]
- Taka, E.; Yilmaz, S.Z.; Golcuk, M.; Kilinc, C.; Aktas, U.; Yildiz, A.; Gur, M. Critical Interactions Between the SARS-CoV-2 Spike Glycoprotein and the Human ACE2 Receptor. J. Phys. Chem. B 2021, 125, 5537–5548. [Google Scholar] [CrossRef]
- Fernández, A. Structural Impact of Mutation D614G in SARS-CoV-2 Spike Protein: Enhanced Infectivity and Therapeutic Opportunity. ACS Med. Chem. Lett. 2020, 11, 1667–1670. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Arantes, P.R.; Saha, A.; Palermo, G. Fighting COVID-19 Using Molecular Dynamics Simulations. ACS Central Sci. 2020, 6, 1654–1656. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Fatihi, S.; Rathore, S.; Pathak, A.K.; Gahlot, D.; Mukerji, M.; Jatana, N.; Thukral, L. A rigorous framework for detecting SARS-CoV-2 spike protein mutational ensemble from genomic and structural features. Curr. Res. Struct. Biol. 2021, 3, 290–300. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef]
- Fallon, L.; Belfon, K.A.A.; Raguette, L.; Wang, Y.; Stepanenko, D.; Cuomo, A.; Guerra, J.; Budhan, S.; Varghese, S.; Corbo, C.P.; et al. Free Energy Landscapes from SARS-CoV-2 Spike Glycoprotein Simulations Suggest that RBD Opening Can Be Modulated via Interactions in an Allosteric Pocket. J. Am. Chem. Soc. 2021, 143, 11349–11360. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Teruel, N.; Mailhot, O.; Najmanovich, R.J. Modelling conformational state dynamics and its role on infection for SARS-CoV-2 Spike protein variants. PLoS Comput. Biol. 2021, 17, e1009286. [Google Scholar] [CrossRef]
- Yazhini, A.; Sidhanta, D.; Srinivasan, N. D614G substitution at the hinge region enhances the stability of trimeric SARS-CoV-2 spike protein. Bioinformation 2021, 17, 439–445. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Aulic, S.; Fermeglia, M.; Pricl, S. Computational Alanine Scanning and Structural Analysis of the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Complex. ACS Nano 2020, 14, 11821–11830. [Google Scholar] [CrossRef]
- Verkhivker, G. Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2020, 21, 8268. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins. J. Proteome Res. 2020, 19, 4587–4608. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J. Phys. Chem. B 2021, 125, 850–873. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.; Agajanian, S.; Oztas, D.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Allosteric Control of Structural Mimicry and Mutational Escape in the SARS-CoV-2 Spike Protein Complexes with the ACE2 Decoys and Miniprotein Inhibitors: A Network-Based Approach for Mutational Profiling of Binding and Signaling. J. Chem. Inf. Model. 2021, 61, 5172–5191. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.; Gupta, G. Computational analysis of protein stability and allosteric interaction networks in distinct conformational forms of the SARS-CoV-2 spike D614G mutant: Reconciling functional mechanisms through allosteric model of spike regulation. J. Biomol. Struct. Dyn. 2021, 98, 1–18. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Landscape-Based Mutational Sensitivity Cartography and Network Community Analysis of the SARS-CoV-2 Spike Protein Structures: Quantifying Functional Effects of the Circulating D614G Variant. ACS Omega 2021, 6, 16216–16233. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Atomistic Simulations and In Silico Mutational Profiling of Protein Stability and Binding in the SARS-CoV-2 Spike Protein Complexes with Nanobodies: Molecular Determinants of Mutational Escape Mechanisms. ACS Omega 2021, 6, 26354–26371. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Yang, F.; Zhang, H.; Wu, H.; Zhu, F.; Xue, W. Computational design and modeling of nanobodies toward SARS-CoV-2 receptor binding domain. Chem. Biol. Drug Des. 2021, 98, 1–18. [Google Scholar] [CrossRef]
- Golcuk, M.; Hacisuleyman, A.; Erman, B.; Yildiz, A.; Gur, M. Binding Mechanism of Neutralizing Nanobodies Targeting SARS-CoV-2 Spike Glycoprotein. J. Chem. Inf. Model. 2021, 61, 5152–5160. [Google Scholar] [CrossRef]
- Vauquelin, G.; Charlton, S.J. Exploring avidity: Understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. J. Cereb. Blood Flow Metab. 2013, 168, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Vauquelin, G.; Bricca, G.; Van Liefde, I. Avidity and positive allosteric modulation/cooperativity act hand in hand to increase the residence time of bivalent receptor ligands. Fundam. Clin. Pharmacol. 2014, 28, 530–543. [Google Scholar] [CrossRef]
- Einav, T.; Yazdi, S.; Coey, A.; Bjorkman, P.J.; Phillips, R. Harnessing Avidity: Quantifying the Entropic and Energetic Effects of Linker Length and Rigidity for Multivalent Binding of Antibodies to HIV-1. Cell Syst. 2019, 9, 466–474.e7. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkhivker, G. Structural and Computational Studies of the SARS-CoV-2 Spike Protein Binding Mechanisms with Nanobodies: From Structure and Dynamics to Avidity-Driven Nanobody Engineering. Int. J. Mol. Sci. 2022, 23, 2928. https://doi.org/10.3390/ijms23062928
Verkhivker G. Structural and Computational Studies of the SARS-CoV-2 Spike Protein Binding Mechanisms with Nanobodies: From Structure and Dynamics to Avidity-Driven Nanobody Engineering. International Journal of Molecular Sciences. 2022; 23(6):2928. https://doi.org/10.3390/ijms23062928
Chicago/Turabian StyleVerkhivker, Gennady. 2022. "Structural and Computational Studies of the SARS-CoV-2 Spike Protein Binding Mechanisms with Nanobodies: From Structure and Dynamics to Avidity-Driven Nanobody Engineering" International Journal of Molecular Sciences 23, no. 6: 2928. https://doi.org/10.3390/ijms23062928
APA StyleVerkhivker, G. (2022). Structural and Computational Studies of the SARS-CoV-2 Spike Protein Binding Mechanisms with Nanobodies: From Structure and Dynamics to Avidity-Driven Nanobody Engineering. International Journal of Molecular Sciences, 23(6), 2928. https://doi.org/10.3390/ijms23062928