
Dysregulation of mTOR Signaling after Brain Ischemia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
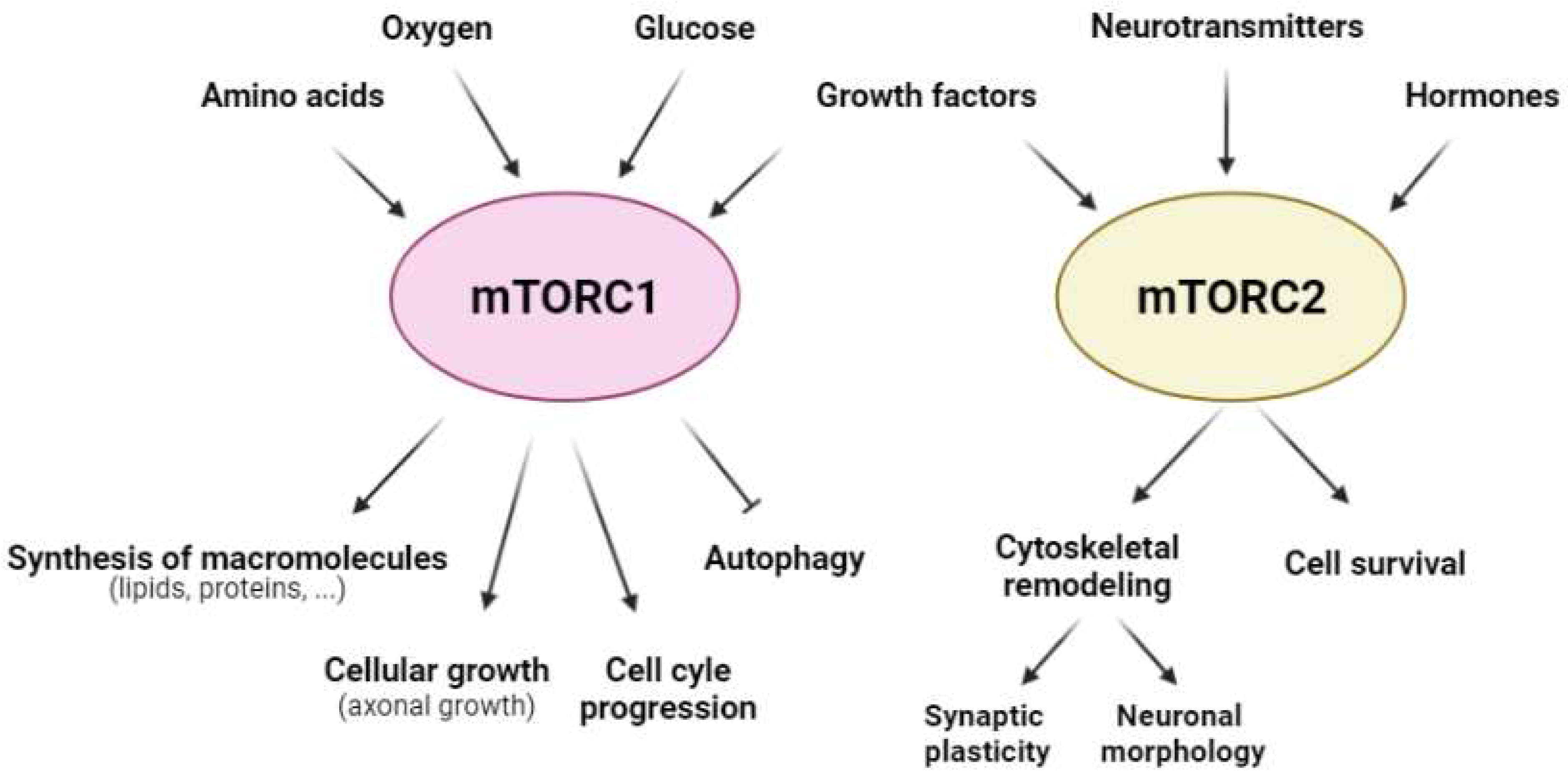
:1. mTOR in the Brain under Physiological Conditions
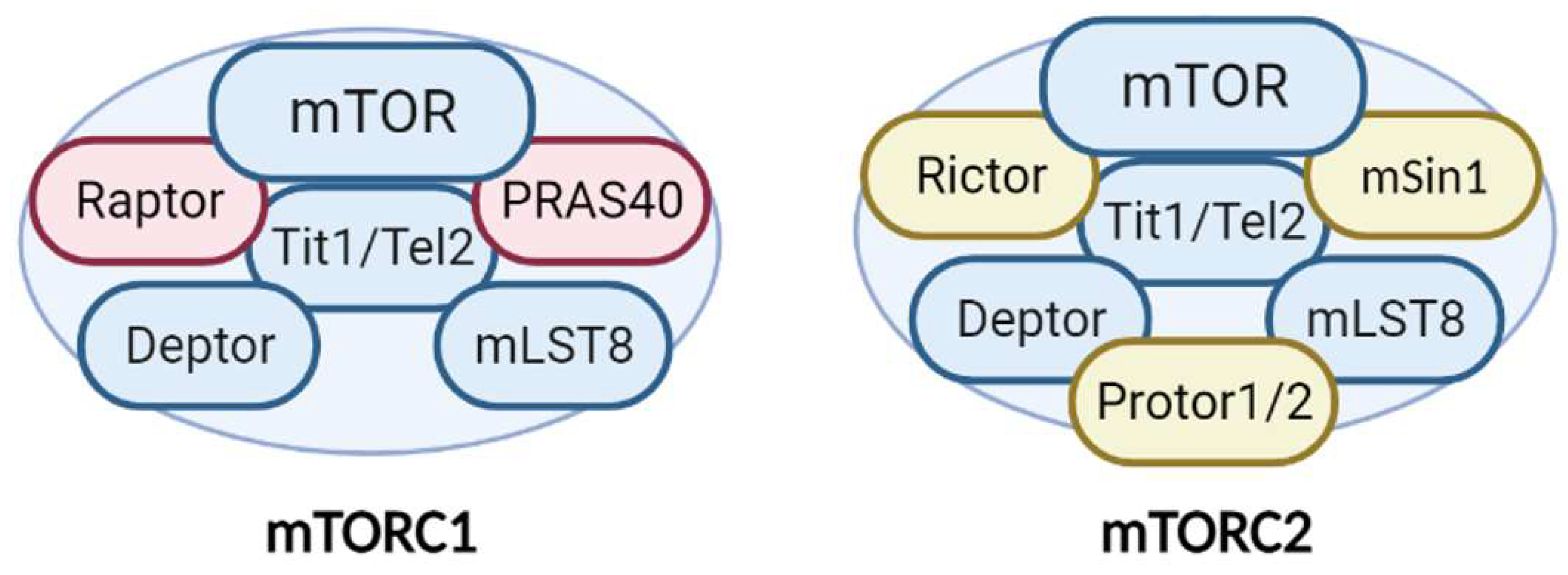
1.1. The Structure of mTOR and Its Complexes in the Brain
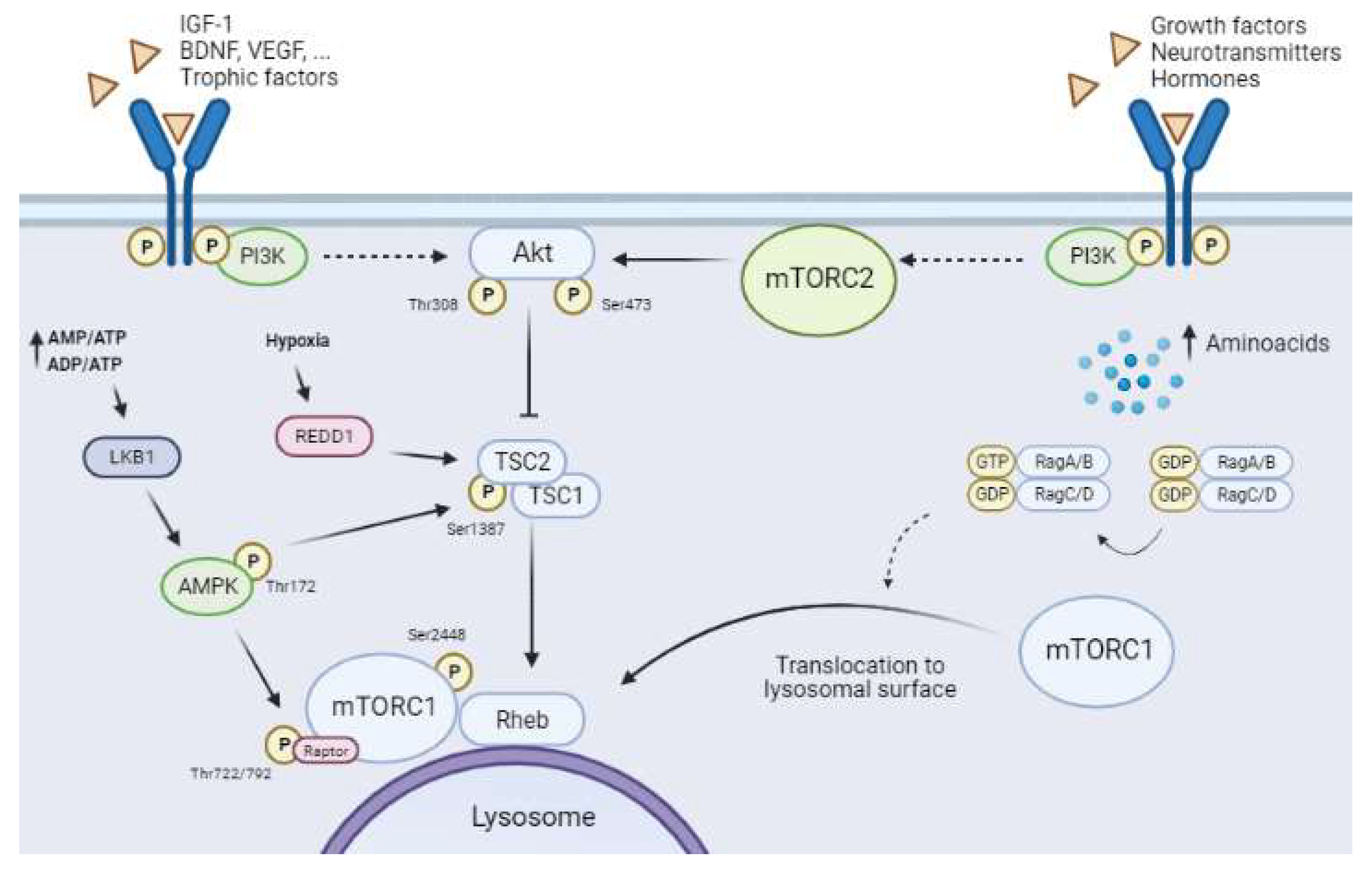
1.2. Upstream Regulatory Pathways of mTORCs
1.2.1. The Canonical Pathway: PI3K/Akt/mTORC1 and Growth Factors
1.2.2. AMPK–mTORC1 Pathway: The Glucose Sensor
1.2.3. REDD1 and mTORC1
1.2.4. Regulation of mTORC1 by Amino Acid Levels
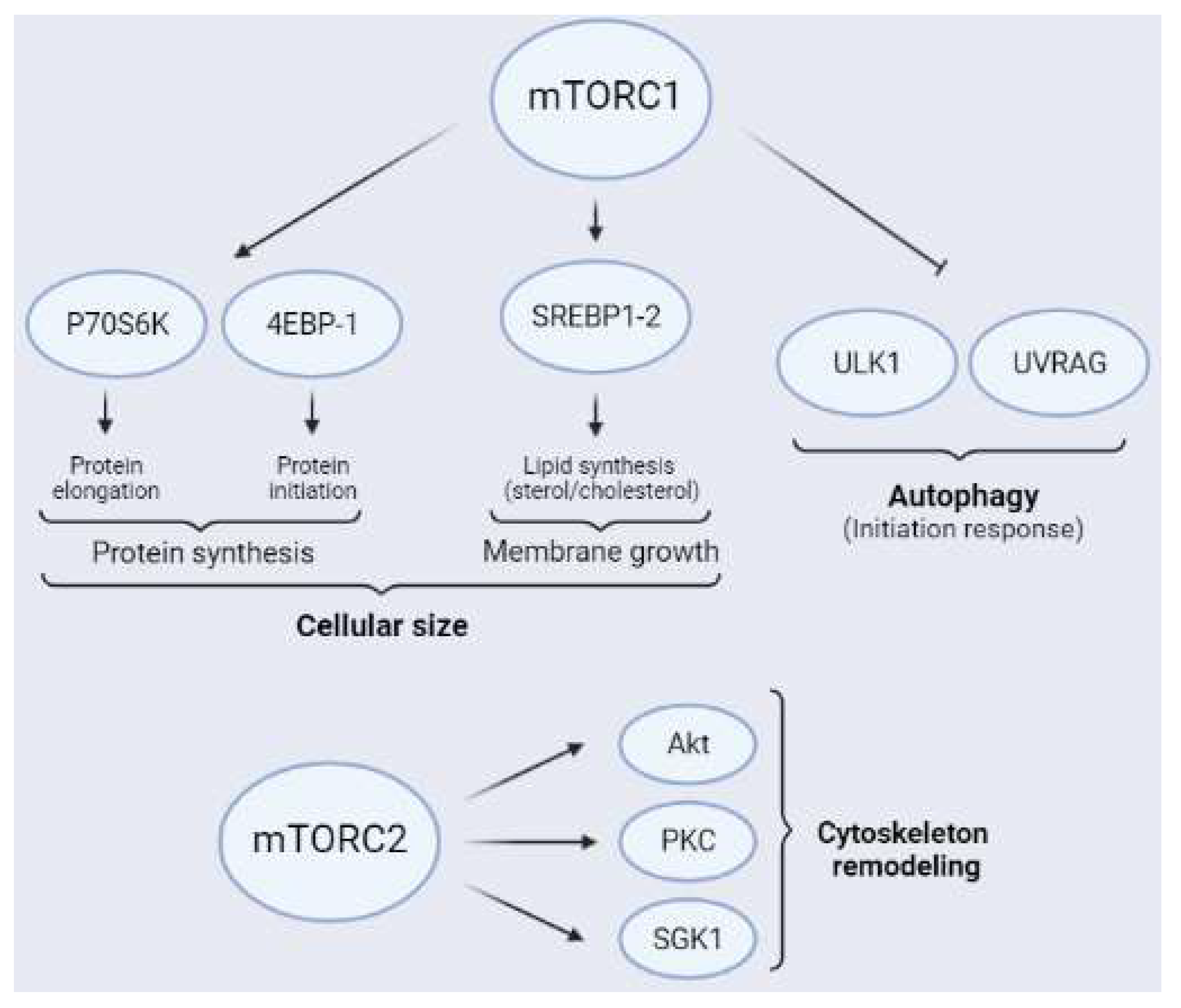
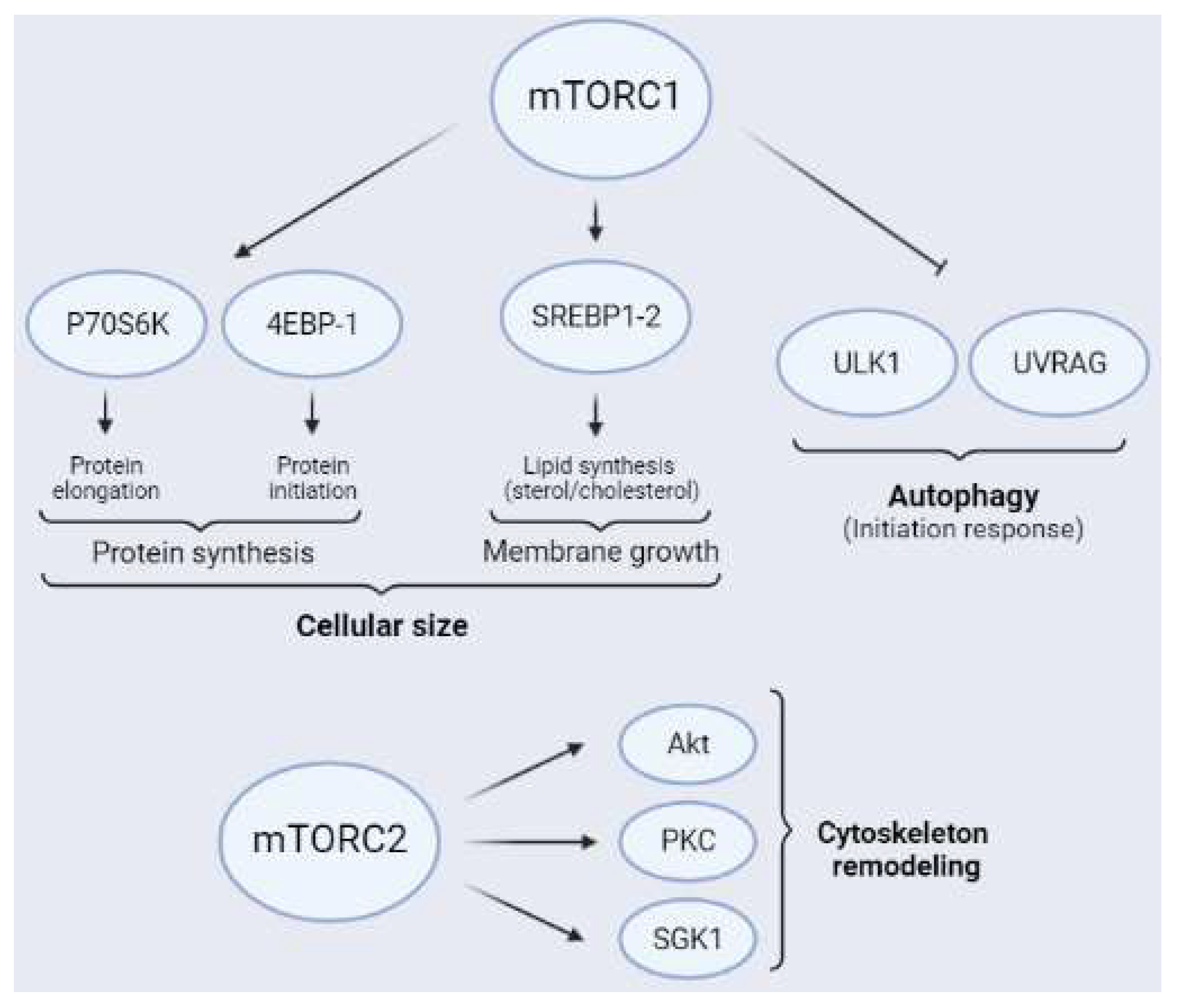
1.3. Downstream Targets of mTORCs
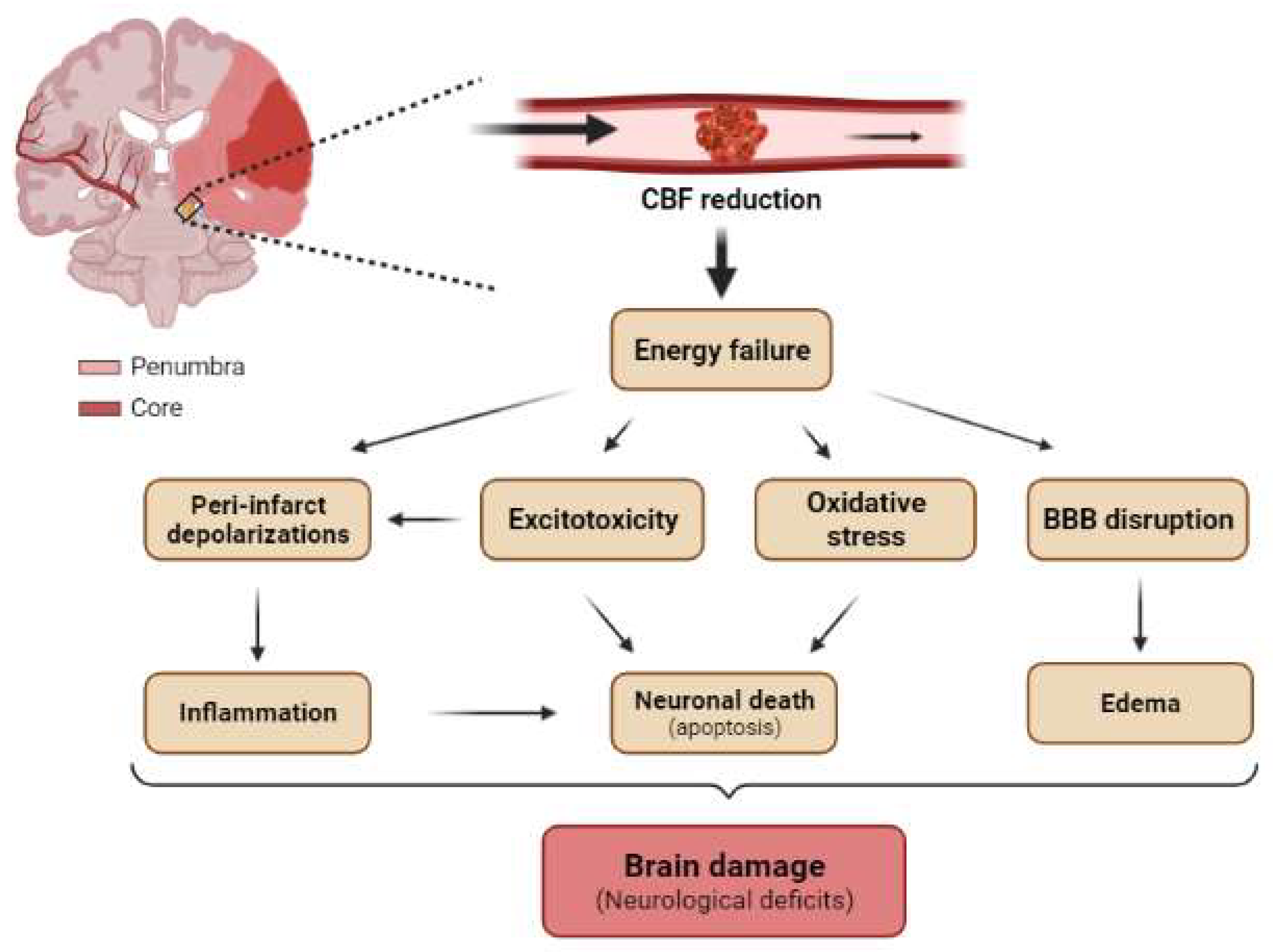
2. mTOR after Cerebral Ischemia
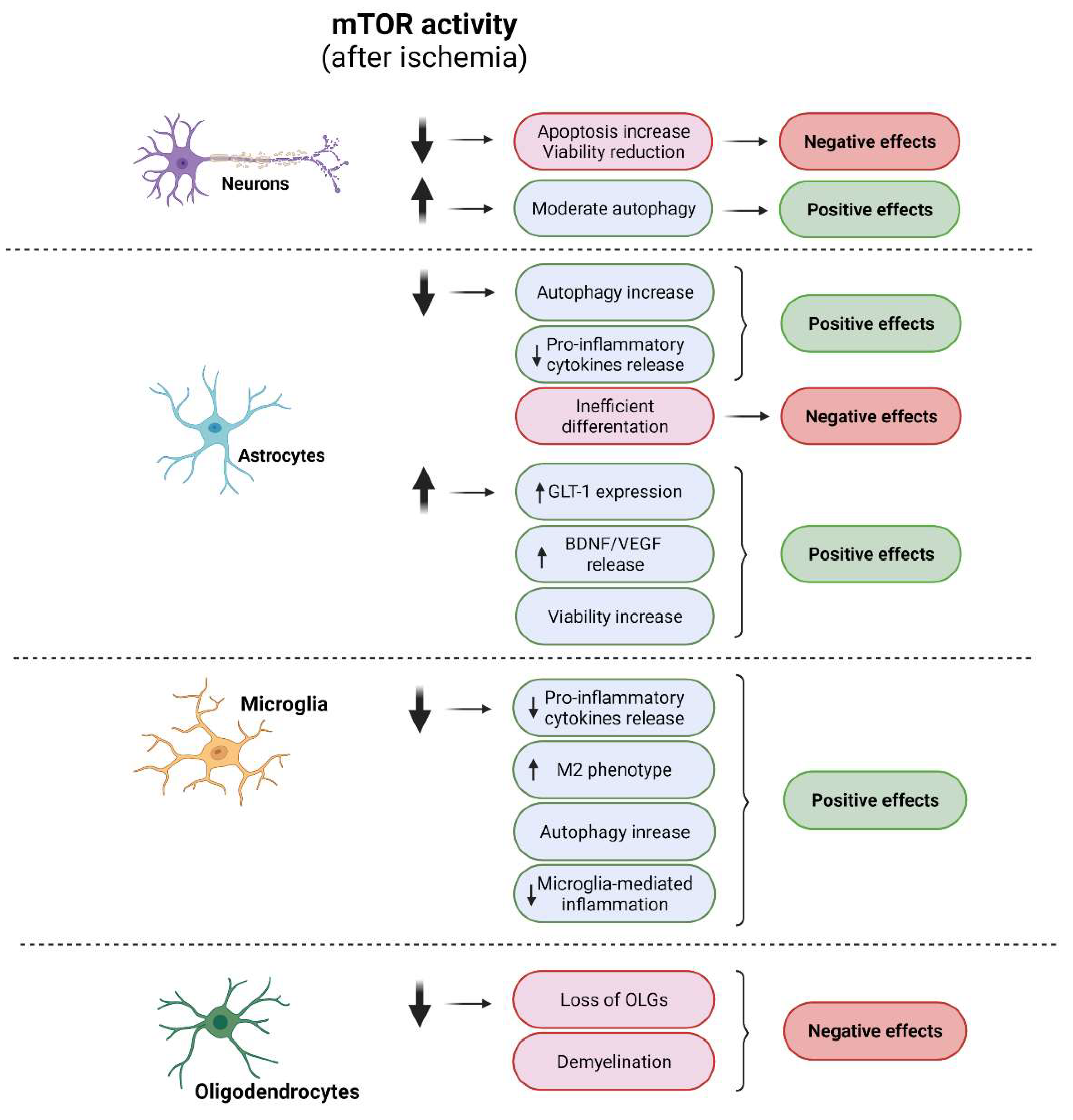
2.1. mTORCs and Neurons
2.2. mTORCs and Glial Cells
2.2.1. Astrocytes
2.2.2. Microglia
2.2.3. Oligodendrocytes
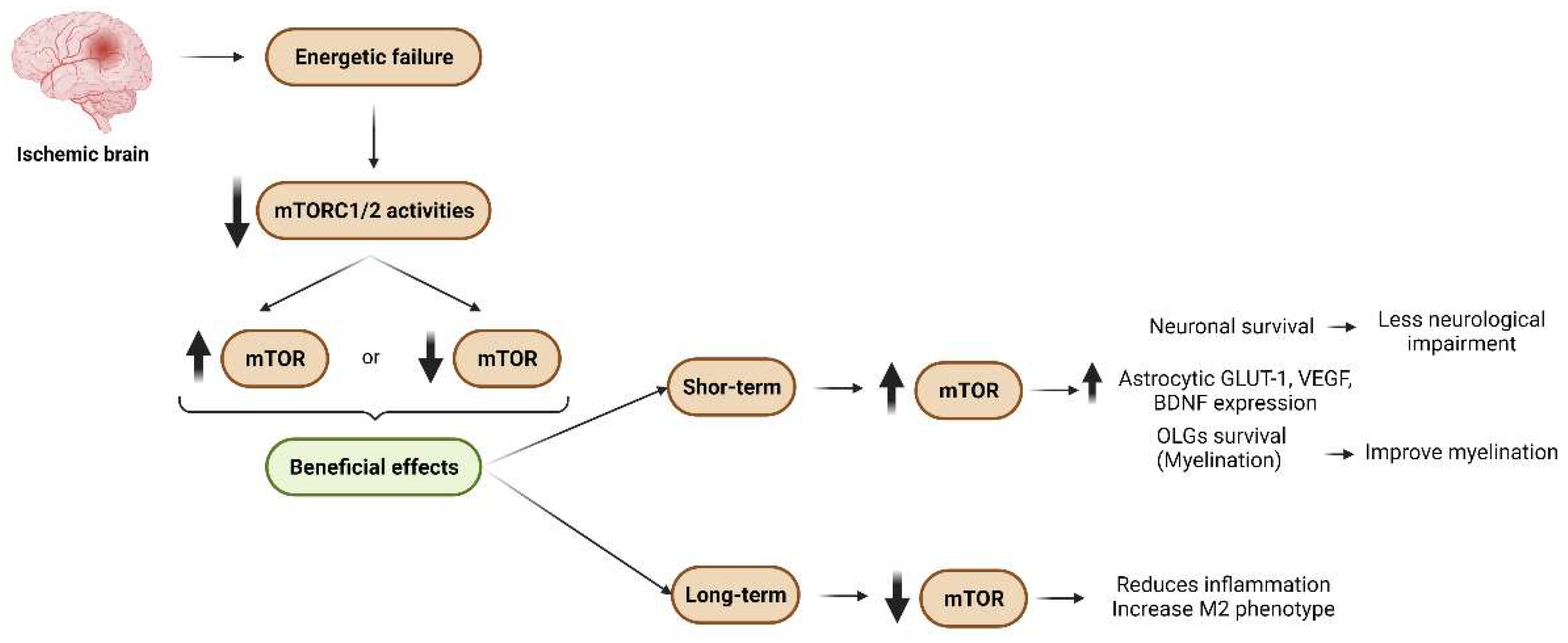
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Dam, T.J.; Zwartkruis, F.J.; Bos, J.L.; Snel, B. Evolution of the TOR pathway. J. Mol. Evol. 2011, 73, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, K.; St-Pierre, M.-K.; Vecchiarelli, H.A.; Bordeleau, M.; Tremblay, M.-È. Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: A story of microglial remodeling. Neurochem. Int. 2021, 145, 104987. [Google Scholar] [CrossRef]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef]
- Moloney, P.B.; Cavalleri, G.L.; Delanty, N. Epilepsy in the mTORopathies: Opportunities for precision medicine. Brain Commun. 2021, 3, fcab222. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Gong, L.; Shu, J.; Chen, X.; Pan, H.; Chen, G.; Bi, Y.; Cui, D.; Li, X.; Liu, D.; Wang, L.; et al. DEPTOR inhibits lung tumorigenesis by inactivating the EGFR-mTOR signals. Cancer Lett. 2021, 519, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Gagné, L.M.; Morin, N.; Lavoie, N.; Bisson, N.; Lambert, J.P.; Mallette, F.A.; Huot, M. Tyrosine phosphorylation of DEPTOR functions as a molecular switch to activate mTOR signaling. J. Biol. Chem. 2021, 297, 101291. [Google Scholar] [CrossRef] [PubMed]
- Wälchli, M.; Berneiser, K.; Mangia, F.; Imseng, S.; Craigie, L.M.; Stuttfeld, E.; Hall, M.N.; Maier, T. Regulation of human mTOR complexes by DEPTOR. Elife 2021, 10, e70781. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Blumhagen, R.; Lao, U.; Kuo, Y.; Edgar, B.A. LST8 regulates cell growth via target-of-rapamycin complex 2 (TORC2). Mol. Cell Biol. 2012, 32, 2203–2213. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Ghomlaghi, M.; Yang, G.; Shin, S.Y.; James, D.E.; Nguyen, L.K. Dynamic modelling of the PI3K/MTOR signalling network uncovers biphasic dependence of mTORC1 activity on the mTORC2 subunit SIN1. PLoS Comput. Biol. 2021, 17, e1008513. [Google Scholar] [CrossRef]
- Sini, P.; James, D.; Chresta, C.; Guichard, S. Simultaneous inhibition of mTORC1 and mTORC2 by mTOR kinase inhibitor AZD8055 induces autophagy and cell death in cancer cells. Autophagy 2010, 6, 553–554. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Perez-Alvarez, M.J.; Villa Gonzalez, M.; Benito-Cuesta, I.; Wandosell, F.G. Role of mTORC1 Controlling Proteostasis after Brain Ischemia. Front. Neurosci. 2018, 12, 60. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Shiota, C.; Woo, J.T.; Lindner, J.; Shelton, K.D.; Magnuson, M.A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 2006, 11, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangloff, Y.G.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell Biol. 2004, 24, 9508–9516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Lee, D.Y. Roles of mTOR Signaling in Brain Development. Exp. Neurobiol. 2015, 24, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B. Spatial regulation of mTORC1 signalling: Beyond the Rag GTPases. Semin. Cell Dev. Biol. 2020, 107, 103–111. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Moten, A.; Lin, H.-K. Akt: A new activation mechanism. Cell Res. 2014, 24, 785–786. [Google Scholar] [CrossRef] [Green Version]
- Thiebaut, A.M.; Buendia, I.; Ginet, V.; Lemarchand, E.; Boudjadja, M.B.; Hommet, Y.; Lebouvier, L.; Lechevallier, C.; Maillasson, M.; Hedou, E.; et al. Thrombolysis by PLAT/tPA increases serum free IGF1 leading to a decrease of deleterious autophagy following brain ischemia. Autophagy 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Muraleedharan, R.; Dasgupta, B. AMPK in the brain: Its roles in glucose and neural metabolism. FEBS J. 2021. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Hong, S.P.; Carlson, M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J. Biol. Chem. 2006, 281, 25336–25343. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef]
- Seong, M.; Lee, J.; Kang, H. Hypoxia-induced regulation of mTOR signaling by miR-7 targeting REDD1. J. Cell Biochem. 2019, 120, 4523–4532. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.Y.; Hagen, T. mTORC1 dependent regulation of REDD1 protein stability. PLoS ONE 2013, 8, e63970. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Park, J.H.; Cho, J.H.; Ahn, J.H.; Yan, B.C.; Lee, J.C.; Shin, M.C.; Cheon, S.H.; Cho, Y.S.; Cho, J.H.; et al. Changes and expressions of Redd1 in neurons and glial cells in the gerbil hippocampus proper following transient global cerebral ischemia. J. Neurol. Sci. 2014, 344, 43–50. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Inoki, K. Rags connect mTOR and autophagy. Small GTPases 2012, 3, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Kimball, S.R.; Jefferson, L.S. Molecular mechanisms through which amino acids mediate signaling through the mammalian target of rapamycin. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 39–44. [Google Scholar] [CrossRef]
- Takei, N.; Furukawa, K.; Hanyu, O.; Sone, H.; Nawa, H. A possible link between BDNF and mTOR in control of food intake. Front. Psychol. 2014, 5, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, Y.; Kakiya, N.; Witters, L.A.; Oshiro, N.; Shirao, T.; Nawa, H.; Takei, N. AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signaling in neurons. J. Neurochem. 2013, 127, 66–77. [Google Scholar] [CrossRef]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A Cellular Regulator Interface in Health and Disease. Cells 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Böhm, R.; Imseng, S.; Jakob, R.P.; Hall, M.N.; Maier, T.; Hiller, S. The dynamic mechanism of 4E-BP1 recognition and phosphorylation by mTORC1. Mol. Cell 2021, 81, 2403–2416.e5. [Google Scholar] [CrossRef]
- Batool, A.; Majeed, S.T.; Aashaq, S.; Majeed, R.; Shah, G.; Nazir, N.; Andrabi, K.I. Eukaryotic Initiation Factor 4E (eIF4E) sequestration mediates 4E-BP1 response to rapamycin. Int. J. Biol. Macromol. 2019, 125, 651–659. [Google Scholar] [CrossRef]
- Ayuso, M.I.; Hernández-Jiménez, M.; Martín, M.E.; Salinas, M.; Alcázar, A. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. J. Biol. Chem. 2010, 285, 34355–34363. [Google Scholar] [CrossRef] [Green Version]
- Ayuso, M.I.; Martinez-Alonso, E.; Salvador, N.; Bonova, P.; Regidor, I.; Alcázar, A. Dissociation of eIF4E-binding protein 2 (4E-BP2) from eIF4E independent of Thr37/Thr46 phosphorylation in the ischemic stress response. PLoS ONE 2015, 10, e0121958. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Katsu-Jiménez, Y.; Alves, R.M.P.; Giménez-Cassina, A. Food for thought: Impact of metabolism on neuronal excitability. Exp. Cell Res. 2017, 360, 41–46. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Furukawa, K.; Hamamura, K.; Furukawa, K. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: Induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011, 71, 4989–4997. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W.; Sabatini, D.M. A Central role for mTOR in lipid homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Schüle, M.; Butto, T.; Dewi, S.; Schlichtholz, L.; Strand, S.; Gerber, S.; Endres, K.; Schweiger, S.; Winter, J. mTOR Driven Gene Transcription Is Required for Cholesterol Production in Neurons of the Developing Cerebral Cortex. Int. J. Mol. Sci. 2021, 22, 6034. [Google Scholar] [CrossRef]
- Norrmén, C.; Figlia, G.; Lebrun-Julien, F.; Pereira, J.A.; Trötzmüller, M.; Köfeler, H.C.; Rantanen, V.; Wessig, C.; van Deijk, A.L.; Smit, A.B.; et al. mTORC1 controls PNS myelination along the mTORC1-RXRγ-SREBP-lipid biosynthesis axis in Schwann cells. Cell Rep. 2014, 9, 646–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiebaut, A.M.; Hedou, E.; Marciniak, S.J.; Vivien, D.; Roussel, B.D. Proteostasis During Cerebral Ischemia. Front. Neurosci. 2019, 13, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahsan, A.; Liu, M.; Zheng, Y.; Yan, W.; Pan, L.; Li, Y.; Ma, S.; Zhang, X.; Cao, M.; Wu, Z.; et al. Natural compounds modulate the autophagy with potential implication of stroke. Acta Pharm. Sin. B 2021, 11, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Zhang, J.; Lin, C.; Zhang, L.; Liu, B.; Ouyang, L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm. Sin. B 2020, 10, 569–581. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Cheng, Q.; Chen, C. The Role of Autophagy in the Pathogenesis of Ischemic Stroke. Curr. Neuropharmacol. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, T.; Kuczmik, W.; Basta-Kaim, A.; Gabryel, B. Rapamycin induces of protective autophagy in vascular endothelial cells exposed to oxygen-glucose deprivation. Brain Res. 2014, 1553, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, H.; Kai, J.; Zhu, F.; Dong, J.; Xu, Z.; Wong, M.; Zeng, L.H. Rapamycin prevents cerebral stroke by modulating apoptosis and autophagy in penumbra in rats. Ann. Clin. Transl. Neurol. 2018, 5, 138–146. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Chen, X.; Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment. Int. J. Mol. Med. 2022, 49, 5070. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, S.; Dai, Q.; Zheng, J.; Liu, C.; Li, S.; Li, J. IL-17A-Mediated Excessive Autophagy Aggravated Neuronal Ischemic Injuries via Src-PP2B-mTOR Pathway. Front. Immunol. 2019, 10, 2952. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Sheng, M. The growing role of mTOR in neuronal development and plasticity. Mol. Neurobiol. 2006, 34, 205–219. [Google Scholar] [CrossRef]
- Nie, D.; Di Nardo, A.; Han, J.M.; Baharanyi, H.; Kramvis, I.; Huynh, T.; Dabora, S.; Codeluppi, S.; Pandolfi, P.P.; Pasquale, E.B.; et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 2010, 13, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Belin, S.; He, Z. Signaling regulations of neuronal regenerative ability. Curr. Opin. Neurobiol. 2014, 27, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Angliker, N.; Rüegg, M.A. In vivo evidence for mTORC2-mediated actin cytoskeleton rearrangement in neurons. Bioarchitecture 2013, 3, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhu, P.J.; Zhang, S.; Zhou, H.; Stoica, L.; Galiano, M.; Krnjević, K.; Roman, G.; Costa-Mattioli, M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci. 2013, 16, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Hernández, I.H.; Villa-González, M.; Martín, G.; Soto, M.; Pérez-Álvarez, M.J. Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury. Cells 2021, 10, 1639. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Ikhlas, M.; Atherton, N.S. Vascular Reperfusion Injury. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Richard Green, A.; Odergren, T.; Ashwood, T. Animal models of stroke: Do they have value for discovering neuroprotective agents? Trends Pharmacol. Sci. 2003, 24, 402–408. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Hossaini Nasr, S.; Huang, X. Nanotechnology for Targeted Therapy of Atherosclerosis. Front. Pharmacol. 2021, 12, 755569. [Google Scholar] [CrossRef]
- Pérez-Álvarez, M.J.; Maza Mdel, C.; Anton, M.; Ordoñez, L.; Wandosell, F. Post-ischemic estradiol treatment reduced glial response and triggers distinct cortical and hippocampal signaling in a rat model of cerebral ischemia. J. Neuroinflammation 2012, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Perez-Alvarez, M.J.; Mateos, L.; Alonso, A.; Wandosell, F. Estradiol and Progesterone Administration After pMCAO Stimulates the Neurological Recovery and Reduces the Detrimental Effect of Ischemia Mainly in Hippocampus. Mol. Neurobiol. 2015, 52, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Mateos, L.; Perez-Alvarez, M.J.; Wandosell, F. Angiotensin II type-2 receptor stimulation induces neuronal VEGF synthesis after cerebral ischemia. Biochim. Biophys. Acta 2016, 1862, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Yao, Q.; Li, H.H. The rationale of targeting mammalian target of rapamycin for ischemic stroke. Cell. Signal. 2013, 25, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.W.; Wu, D.P.; Liang, H.F.; Tang, G.Y.; Fan, C.L.; Shi, L.; Ye, W.C.; Li, M.M. Total Saponins of Panax notoginseng Activate Akt/mTOR Pathway and Exhibit Neuroprotection in vitro and in vivo against Ischemic Damage. Chin. J. Integr. Med. 2021, 1–9. [Google Scholar] [CrossRef]
- Lei, Y.; Jin, X.; Sun, M.; Ji, Z. miR-129-5p Ameliorates Ischemic Brain Injury by Binding to SIAH1 and Activating the mTOR Signaling Pathway. J. Mol. Neurosci. 2021, 71, 1761–1771. [Google Scholar] [CrossRef]
- Chi, O.Z.; Liu, X.; Cofano, S.; Patel, N.; Jacinto, E.; Weiss, H.R. Rapalink-1 Increased Infarct Size in Early Cerebral Ischemia-Reperfusion With Increased Blood-Brain Barrier Disruption. Front. Physiol. 2021, 12, 706528. [Google Scholar] [CrossRef]
- Lee, J.M.; Grabb, M.C.; Zipfel, G.J.; Choi, D.W. Brain tissue responses to ischemia. J. Clin. Investig. 2000, 106, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Sharma, U.; Jagannathan, N.R.; Gupta, Y.K. Rapamycin ameliorates brain metabolites alterations after transient focal ischemia in rats. Eur. J. Pharmacol. 2015, 757, 28–33. [Google Scholar] [CrossRef]
- Luo, C.; Li, Q.; Gao, Y.; Shen, X.; Ma, L.; Wu, Q.; Wang, Z.; Zhang, M.; Zhao, Z.; Chen, X.; et al. Poloxamer 188 Attenuates Cerebral Hypoxia/Ischemia Injury in Parallel with Preventing Mitochondrial Membrane Permeabilization and Autophagic Activation. J. Mol. Neurosci. 2015, 56, 988–998. [Google Scholar] [CrossRef]
- Yin, L.; Ye, S.; Chen, Z.; Zeng, Y. Rapamycin preconditioning attenuates transient focal cerebral ischemia/reperfusion injury in mice. Int. J. Neurosci. 2012, 122, 748–756. [Google Scholar] [CrossRef]
- Wang, C.; Niu, F.; Ren, N.; Wang, X.; Zhong, H.; Zhu, J.; Li, B. Hyperbaric Oxygen Improves Cerebral Ischemia/Reperfusion Injury in Rats Probably via Inhibition of Autophagy Triggered by the Downregulation of Hypoxia-Inducing Factor-1 Alpha. Biomed Res. Int. 2021, 2021, 6615685. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Peng, A.; Sun, H.; Zhuang, Y.; Yu, M.; Wang, Q.; Wang, J. LncRNA AC136007.2 alleviates cerebral ischemic-reperfusion injury by suppressing autophagy. Aging 2021, 13, 19587–19597. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ji, Y.; Ren, J.; Liu, H.; Khanna, R.; Sun, L. Inhibition of autophagy by CRMP2-derived peptide ST2-104 (R9-CBD3) via a CaMKKβ/AMPK/mTOR pathway contributes to ischemic postconditioning-induced neuroprotection against cerebral ischemia-reperfusion injury. Mol. Brain 2021, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Xia, Q.; Chu, K.T.; Pan, J.; Sun, L.N.; Zeng, B.; Zhu, Y.J.; Wang, Q.; Wang, K.; Luo, B.Y. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: A widely used inhibitor of autophagy. J. Neuropathol. Exp. Neurol. 2011, 70, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Beard, D.J.; Hadley, G.; Thurley, N.; Howells, D.W.; Sutherland, B.A.; Buchan, A.M. The effect of rapamycin treatment on cerebral ischemia: A systematic review and meta-analysis of animal model studies. Int. J. Stroke 2019, 14, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Jadaun, K.S.; Sharma, A.; Siddiqui, E.M.; Mehan, S. Targeting abnormal PI3K/AKT/mTOR signaling in intracerebral hemorrhage: A systematic review on potential drug targets and influences of signaling modulators on other neurological disorders. Curr. Rev. Clin. Exp. Pharmacol. 2021, 16, 1–18. [Google Scholar] [CrossRef]
- Cespedes, A.; Villa, M.; Benito-Cuesta, I.; Perez-Alvarez, M.J.; Ordoñez, L.; Wandosell, F. Energy-Sensing Pathways in Ischemia: The Counterbalance Between AMPK and mTORC. Curr. Pharm. Des. 2019, 25, 4763–4770. [Google Scholar] [CrossRef]
- Fletcher, L.; Evans, T.M.; Watts, L.T.; Jimenez, D.F.; Digicaylioglu, M. Rapamycin treatment improves neuron viability in an in vitro model of stroke. PLoS ONE 2013, 8, e68281. [Google Scholar] [CrossRef] [Green Version]
- Arabian, M.; Aboutaleb, N.; Soleimani, M.; Ajami, M.; Habibey, R.; Rezaei, Y.; Pazoki-Toroudi, H. Preconditioning with morphine protects hippocampal CA1 neurons from ischemia-reperfusion injury via activation of the mTOR pathway. Can. J. Physiol. Pharmacol. 2018, 96, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xiong, T.; Qu, Y.; Zhao, F.; Ferriero, D.; Mu, D. mTOR activates hypoxia-inducible factor-1α and inhibits neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Neurosci. Lett. 2012, 507, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Rutkai, I.; Katakam, P.V.; Busija, D.W. The mechanistic target of rapamycin (mTOR) pathway and S6 Kinase mediate diazoxide preconditioning in primary rat cortical neurons. J. Neurochem. 2015, 134, 845–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Yamashita, S.; Hua, Y.; Keep, R.F.; Liu, W.; Xi, G. Thrombin-induced neuronal protection: Role of the mitogen activated protein kinase/ribosomal protein S6 kinase pathway. Brain Res. 2010, 1361, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, M.H.; Qin, C.; Fan, W.H.; Tian, D.S.; Liu, J.L. Fingolimod suppresses neuronal autophagy through the mTOR/p70S6K pathway and alleviates ischemic brain damage in mice. PLoS ONE 2017, 12, e0188748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, M.D.; García-Yébenes, I.; Fradejas, N.; Pérez-Ortiz, J.M.; Mora-Lee, S.; Tranque, P.; Moro, M.; Pende, M.; Calvo, S. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J. Biol. Chem. 2009, 284, 22067–22078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioda, N.; Ishigami, T.; Han, F.; Moriguchi, S.; Shibuya, M.; Iwabuchi, Y.; Fukunaga, K. Activation of phosphatidylinositol 3-kinase/protein kinase B pathway by a vanadyl compound mediates its neuroprotective effect in mouse brain ischemia. Neuroscience 2007, 148, 221–229. [Google Scholar] [CrossRef]
- Xu, X.; Chua, C.C.; Gao, J.; Chua, K.W.; Wang, H.; Hamdy, R.C.; Chua, B.H. Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain Res. 2008, 1227, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Tu, L.; Wang, Y.; Chen, D.; Xiang, P.; Shen, J.; Li, Y.; Wang, S. Protective Effects of Notoginsenoside R1 via Regulation of the PI3K-Akt-mTOR/JNK Pathway in Neonatal Cerebral Hypoxic-Ischemic Brain Injury. Neurochem. Res. 2018, 43, 1210–1226. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.D.; OuYang, Y.P.; Shi, J.G.; Liu, Y.; Yuan, W.; Jia, L.S. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2011, 404, 941–945. [Google Scholar] [CrossRef]
- Fakhri, S.; Iranpanah, A.; Gravandi, M.M.; Moradi, S.Z.; Ranjbari, M.; Majnooni, M.B.; Echeverría, J.; Qi, Y.; Wang, M.; Liao, P.; et al. Natural products attenuate PI3K/Akt/mTOR signaling pathway: A promising strategy in regulating neurodegeneration. Phytomedicine 2021, 91, 153664. [Google Scholar] [CrossRef]
- Hadley, G.; Beard, D.J.; Alexopoulou, Z.; Sutherland, B.A.; Buchan, A.M. Investigation of the novel mTOR inhibitor AZD2014 in neuronal ischemia. Neurosci. Lett. 2019, 706, 223–230. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, R.L.; Li, Y.; Hu, Y.Q.; Xv, H.; Zhu, C.; Wang, X.; Ma, K.T.; Zhao, D. Reverse relationship between autophagy and apoptosis in an in vitro model of cortical neuronal injury. J. Chem. Neuroanat. 2022, 120, 102070. [Google Scholar] [CrossRef]
- Sánchez-Alegría, K.; Flores-León, M.; Avila-Muñoz, E.; Rodríguez-Corona, N.; Arias, C. PI3K Signaling in Neurons: A Central Node for the Control of Multiple Functions. Int. J. Mol. Sci. 2018, 19, 3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Hadley, G.; Beard, D.J.; Couch, Y.; Neuhaus, A.A.; Adriaanse, B.A.; DeLuca, G.C.; Sutherland, B.A.; Buchan, A.M. Rapamycin in ischemic stroke: Old drug, new tricks? J. Cereb. Blood Flow Metab. 2019, 39, 20–35. [Google Scholar] [CrossRef]
- Perez-Alvarez, M.J.; Wandosell, F. Stroke and Neuroinflamation: Role of Sexual Hormones. Curr. Pharm. Des. 2016, 22, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.T.; Zhang, J.R.; Kapupara, K.; Tsai, R.K. mTORC2 activation protects retinal ganglion cells via Akt signaling after autophagy induction in traumatic optic nerve injury. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Erickson, E.K.; DaCosta, A.J.; Mason, S.C.; Blednov, Y.A.; Mayfield, R.D.; Harris, R.A. Cortical astrocytes regulate ethanol consumption and intoxication in mice. Neuropsychopharmacology 2021, 46, 500–508. [Google Scholar] [CrossRef]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2021, 12, 825816. [Google Scholar] [CrossRef]
- Peng, L.; Zhao, Y.; Li, Y.; Zhou, Y.; Li, L.; Lei, S.; Yu, S.; Zhao, Y. Effect of DJ-1 on the neuroprotection of astrocytes subjected to cerebral ischemia/reperfusion injury. J. Mol. Med. 2019, 97, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, Z.W.; Zhao, Y.H.; Liu, B.W.; Liu, N.W.; Ke, C.C.; Tan, H.M. Bone Marrow Stromal Cells Combined With Sodium Ferulate and n-Butylidenephthalide Promote the Effect of Therapeutic Angiogenesis via Advancing Astrocyte-Derived Trophic Factors After Ischemic Stroke. Cell Transplant. 2017, 26, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubern, C.; Camós, S.; Hurtado, O.; Rodríguez, R.; Romera, V.G.; Sobrado, M.; Cañadas, R.; Moro, M.A.; Lizasoain, I.; Serena, J.; et al. Characterization of Gcf2/Lrrfip1 in experimental cerebral ischemia and its role as a modulator of Akt, mTOR and β-catenin signaling pathways. Neuroscience 2014, 268, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.F.; Zhou, L.; Xie, Y.J.; Xu, S.M.; Zhu, J.; Teng, P.; Shao, C.Y.; Wang, Y.; Luo, J.H.; Shen, Y. Upregulation of glutamate transporter GLT-1 by mTOR-Akt-NF-кB cascade in astrocytic oxygen-glucose deprivation. Glia 2013, 61, 1959–1975. [Google Scholar] [CrossRef]
- Huang, W.Y.; Jiang, C.; Ye, H.B.; Jiao, J.T.; Cheng, C.; Huang, J.; Liu, J.; Zhang, R.; Shao, J.F. miR-124 upregulates astrocytic glutamate transporter-1 via the Akt and mTOR signaling pathway post ischemic stroke. Brain Res. Bull. 2019, 149, 231–239. [Google Scholar] [CrossRef]
- Barialai, L.; Strecker, M.I.; Luger, A.L.; Jäger, M.; Bruns, I.; Sittig, A.C.M.; Mildenberger, I.C.; Heller, S.M.; Delaidelli, A.; Lorenz, N.I.; et al. AMPK activation protects astrocytes from hypoxia-induced cell death. Int. J. Mol. Med. 2020, 45, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Shi, X.; Yang, M.; Yang, T.; Li, T.; Chen, J. Mesenchymal stem cells-derived IL-6 activates AMPK/mTOR signaling to inhibit the proliferation of reactive astrocytes induced by hypoxic-ischemic brain damage. Exp. Neurol. 2019, 311, 15–32. [Google Scholar] [CrossRef]
- Zhang, Y.; Miao, J.M. Ginkgolide K promotes astrocyte proliferation and migration after oxygen-glucose deprivation via inducing protective autophagy through the AMPK/mTOR/ULK1 signaling pathway. Eur. J. Pharmacol. 2018, 832, 96–103. [Google Scholar] [CrossRef]
- Zhu, C.; Zhou, Q.; Luo, C.; Chen, Y. Dexmedetomidine Protects Against Oxygen-Glucose Deprivation-Induced Injury Through Inducing Astrocytes Autophagy via TSC2/mTOR Pathway. Neuromolecular Med. 2020, 22, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Li, X.; Liu, S.F.; Qu, W.S.; Wang, W.; Tian, D.S. Inhibition of mTOR pathway restrains astrocyte proliferation, migration and production of inflammatory mediators after oxygen-glucose deprivation and reoxygenation. Neurochem. Int. 2015, 83–84, 9–18. [Google Scholar] [CrossRef]
- Zha, H.; Fan, Y.; Yang, L.; Yin, M.; Miao, W.; He, J.; Wang, Y. Autophagy protects against cerebral ischemic reperfusion injury by inhibiting neuroinflammation. Am. J. Transl. Res. 2021, 13, 4726–4737. [Google Scholar]
- Rodriguez-Vargas, J.M.; Martin-Hernandez, K.; Wang, W.; Kunath, N.; Suganthan, R.; Amé, J.C.; Oliver, F.J.; Ye, J.; Bjørås, M.; Dantzer, F. Parp3 promotes astrocytic differentiation through a tight regulation of Nox4-induced ROS and mTorc2 activation. Cell Death Dis. 2020, 11, 954. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Marsh, B.J.; Stevens, S.L.; Hunter, B.; Stenzel-Poore, M.P. Inflammation and the emerging role of the toll-like receptor system in acute brain ischemia. Stroke 2009, 40, S34–S37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Yrjänheikki, J.; Tikka, T.; Keinänen, R.; Goldsteins, G.; Chan, P.H.; Koistinaho, J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc. Natl. Acad. Sci. USA 1999, 96, 13496–13500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Gaojian, T.; Dingfei, Q.; Linwei, L.; Xiaowei, W.; Zheng, Z.; Wei, L.; Tong, Z.; Benxiang, N.; Yanning, Q.; Wei, Z.; et al. Parthenolide promotes the repair of spinal cord injury by modulating M1/M2 polarization via the NF-κB and STAT 1/3 signaling pathway. Cell Death Discov. 2020, 6, 97. [Google Scholar] [CrossRef]
- Xue, Y.; Nie, D.; Wang, L.J.; Qiu, H.C.; Ma, L.; Dong, M.X.; Tu, W.J.; Zhao, J. Microglial Polarization: Novel Therapeutic Strategy against Ischemic Stroke. Aging Dis. 2021, 12, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Li, D.; Wang, C.; Yao, Y.; Chen, L.; Liu, G.; Zhang, R.; Liu, Q.; Shi, F.-D.; Hao, J. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 3388–3399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Deng, S.; Zhang, Z.; Gu, Y.; Xia, S.; Bao, X.; Cao, X.; Xu, Y. 6-Gingerol attenuates microglia-mediated neuroinflammation and ischemic brain injuries through Akt-mTOR-STAT3 signaling pathway. Eur. J. Pharmacol. 2020, 883, 173294. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Q.; Zhang, X. C1q/tumor necrosis factor-related protein-1 attenuates microglia autophagy and inflammatory response by regulating the Akt/mTOR pathway. Life Sci. 2020, 256, 117992. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Li, W.; Song, Y.; Li, Z.; Tang, Y.; Zhang, Z.; Yang, G.-Y. Sestrin2 regulates microglia polarization through mTOR-mediated autophagic flux to attenuate inflammation during experimental brain ischemia. J. Neuroinflammation 2020, 17, 329. [Google Scholar] [CrossRef]
- Ruan, C.; Guo, H.; Gao, J.; Wang, Y.; Liu, Z.; Yan, J.; Li, X.; Lv, H. Neuroprotective effects of metformin on cerebral ischemia-reperfusion injury by regulating PI3K/Akt pathway. Brain Behav. 2021, 11, e2335. [Google Scholar] [CrossRef]
- Li, S.; Hua, X.; Zheng, M.; Wu, J.; Ma, Z.; Xing, X.; Ma, J.; Zhang, J.; Shan, C.; Xu, J. PLXNA2 knockdown promotes M2 microglia polarization through mTOR/STAT3 signaling to improve functional recovery in rats after cerebral ischemia/reperfusion injury. Exp. Neurol. 2021, 346, 113854. [Google Scholar] [CrossRef]
- Xin, W.; Chan, J.R. Myelin plasticity: Sculpting circuits in learning and memory. Nat. Rev. Neurosci. 2020, 21, 682–694. [Google Scholar] [CrossRef]
- Pfeiffer, S.E.; Warrington, A.E.; Bansal, R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993, 3, 191–197. [Google Scholar] [CrossRef]
- Lebrun-Julien, F.; Bachmann, L.; Norrmén, C.; Trötzmüller, M.; Köfeler, H.; Rüegg, M.A.; Hall, M.N.; Suter, U. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J. Neurosci. 2014, 34, 8432–8448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardiola-Diaz, H.M.; Ishii, A.; Bansal, R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia 2012, 60, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Zhou, L.; Du, X.X.; Ji, Y.; Xu, J.; Tian, J.; Jiang, W.; Zou, Y.; Yu, S.; Gan, L.; et al. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 2011, 20, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, S.E.; McLane, L.E.; Bercury, K.K.; Macklin, W.B.; Wood, T.L. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci. 2014, 34, 4453–4465. [Google Scholar] [CrossRef] [PubMed]
- Figlia, G.; Gerber, D.; Suter, U. Myelination and mTOR. Glia 2018, 66, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef]
- Carson, R.P.; Kelm, N.D.; West, K.L.; Does, M.D.; Fu, C.; Weaver, G.; McBrier, E.; Parker, B.; Grier, M.D.; Ess, K.C. Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann. Clin. Transl. Neurol. 2015, 2, 1041–1054. [Google Scholar] [CrossRef]
- Ercan, E.; Han, J.M.; Di Nardo, A.; Winden, K.; Han, M.J.; Hoyo, L.; Saffari, A.; Leask, A.; Geschwind, D.H.; Sahin, M. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J. Exp. Med. 2017, 214, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Carson, R.P.; Fu, C.; Winzenburger, P.; Ess, K.C. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum. Mol. Genet. 2013, 22, 140–152. [Google Scholar] [CrossRef]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, W.A.; Gangoli, N.; Gokina, P.; Kim, H.A.; Covey, M.; Levison, S.W.; Wood, T.L. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J. Neurosci. 2009, 29, 6367–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hu, X.; Leak, R.K.; Shi, Y.; An, C.; Suenaga, J.; Chen, J.; Gao, Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp. Neurol. 2015, 272, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Liu, Z.; Buller, B.; Li, Y.; Golembieski, W.; Gan, X.; Wang, F.; Lu, M.; Ali, M.M.; Zhang, Z.G.; et al. MiR-17-92 enriched exosomes derived from multipotent mesenchymal stromal cells enhance axon-myelin remodeling and motor electrophysiological recovery after stroke. J. Cereb. Blood Flow Metab. 2021, 41, 1131–1144. [Google Scholar] [CrossRef]
- Villa Gonzalez, M.; Pérez-Álvarez, M.J. A 3R-Tau-mediated mechanism in oligodendrocytes: Could it be the key for neuroprotection after stroke? Neural Regen. Res. 2021, 16, 2401–2402. [Google Scholar] [CrossRef]
- Villa González, M.; Vallés-Saiz, L.; Hernández, I.H.; Avila, J.; Hernández, F.; Pérez-Alvarez, M.J. Focal cerebral ischemia induces changes in oligodendrocytic tau isoforms in the damaged area. Glia 2020, 68, 2471–2485. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, F.; Wang, P.; Mana, L.; Sheng, N.; Huang, S. Study on myelin injury of AD mice treated with Shenzhiling oral liquid in the PI3K/Akt-mTOR pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 2058738420923907. [Google Scholar] [CrossRef]
- Ueno, Y.; Koike, M.; Shimada, Y.; Shimura, H.; Hira, K.; Tanaka, R.; Uchiyama, Y.; Hattori, N.; Urabe, T. L-carnitine enhances axonal plasticity and improves white-matter lesions after chronic hypoperfusion in rat brain. J. Cereb. Blood Flow Metab. 2015, 35, 382–391. [Google Scholar] [CrossRef] [Green Version]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The Endocannabinoid System and Oligodendrocytes in Health and Disease. Front. Neurosci. 2018, 12, 733. [Google Scholar] [CrossRef] [Green Version]
- Arai, K.; Lo, E.H. Wiring and plumbing: Oligodendrocyte precursors and angiogenesis in the oligovascular niche. J. Cereb. Blood Flow Metab. 2021, 41, 2132–2133. [Google Scholar] [CrossRef]
- Maki, T.; Morancho, A.; Martinez-San Segundo, P.; Hayakawa, K.; Takase, H.; Liang, A.C.; Gabriel-Salazar, M.; Medina-Gutiérrez, E.; Washida, K.; Montaner, J.; et al. Endothelial Progenitor Cell Secretome and Oligovascular Repair in a Mouse Model of Prolonged Cerebral Hypoperfusion. Stroke 2018, 49, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wang, L.; Shi, S.; Yang, C.; Jiang, W.; Luan, Z.; Liu, L.; Yao, R. Oligogenesis in the “oligovascular unit” involves PI3K/AKT/mTOR signaling in hypoxic-ischemic neonatal mice. Brain Res. Bull. 2020, 155, 81–91. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa-González, M.; Martín-López, G.; Pérez-Álvarez, M.J. Dysregulation of mTOR Signaling after Brain Ischemia. Int. J. Mol. Sci. 2022, 23, 2814. https://doi.org/10.3390/ijms23052814
Villa-González M, Martín-López G, Pérez-Álvarez MJ. Dysregulation of mTOR Signaling after Brain Ischemia. International Journal of Molecular Sciences. 2022; 23(5):2814. https://doi.org/10.3390/ijms23052814
Chicago/Turabian StyleVilla-González, Mario, Gerardo Martín-López, and María José Pérez-Álvarez. 2022. "Dysregulation of mTOR Signaling after Brain Ischemia" International Journal of Molecular Sciences 23, no. 5: 2814. https://doi.org/10.3390/ijms23052814
APA StyleVilla-González, M., Martín-López, G., & Pérez-Álvarez, M. J. (2022). Dysregulation of mTOR Signaling after Brain Ischemia. International Journal of Molecular Sciences, 23(5), 2814. https://doi.org/10.3390/ijms23052814