

Functional and Taxonomic Traits of the Gut Microbiota in Type 1 Diabetes Children at the Onset: A Metaproteomic Study
 , , , ,
, , , , 
Abstract
1. Introduction
2. Results
2.1. LC-MS/MS Analysis and GM Ecology
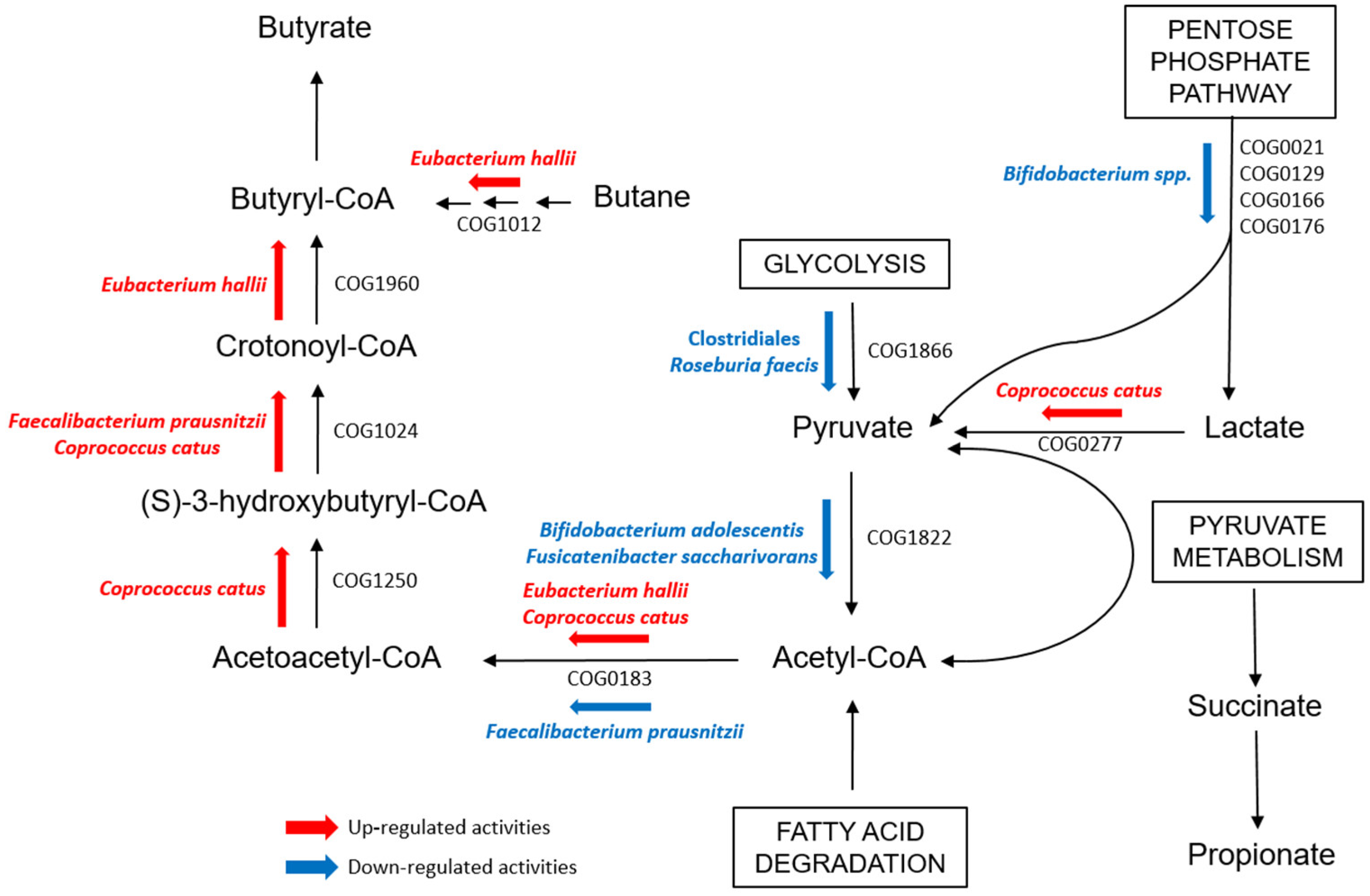
2.2. GM Quantitative Analysis: T1D vs. CTRL vs. SIBL
2.3. GM Quantitative Analysis: T1D Patients’ Classification by Insulin Need
2.4. Human Proteins
3. Discussion
4. Materials and Methods
4.1. Subjects Enrollment and Sample Collection
4.2. Protein Extraction and Digestion
4.3. Chromatography and Mass Spectrometry Analysis
4.4. Bioinformatic Data Processing and Metaproteomic Analysis
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 Diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [PubMed]
- La Marca, V.; Gianchecchi, E.; Fierabracci, A. Type 1 Diabetes and Its Multi-Factorial Pathogenesis: The Putative Role of NK Cells. Int. J. Mol. Sci. 2018, 19, 794. [Google Scholar] [CrossRef] [PubMed]
- Gale, E.A.M. Type 1 Diabetes in the Young: The Harvest of Sorrow Goes On. Diabetologia 2005, 48, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Luopajärvi, K.; Härkönen, T. Early Life Origin of Type 1 Diabetes. Semin. Immunopathol. 2017, 39, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Brugman, S.; Klatter, F.A.; Visser, J.T.J.; Wildeboer-Veloo, A.C.M.; Harmsen, H.J.M.; Rozing, J.; Bos, N.A. Antibiotic Treatment Partially Protects against Type 1 Diabetes in the Bio-Breeding Diabetes-Prone Rat. Is the Gut Flora Involved in the Development of Type 1 Diabetes? Diabetologia 2006, 49, 2015–2018. [Google Scholar] [CrossRef]
- Alam, C.; Bittoun, E.; Bhagwat, D.; Valkonen, S.; Saari, A.; Jaakkola, U.; Eerola, E.; Huovinen, P.; Hänninen, A. Effects of a Germ-Free Environment on Gut Immune Regulation and Diabetes Progression in Non-Obese Diabetic (NOD) Mice. Diabetologia 2011, 54, 1398–1406. [Google Scholar] [CrossRef]
- Serino, M.; Fernandez-Real, J.M.; Garcia-Fuentes, E.; Queipo-Ortuño, M.; Moreno-Navarrete, J.M.; Sanchez, A.; Burcelin, R.; Tinahones, F. The Gut Microbiota Profile Is Associated with Insulin Action in Humans. Acta Diabetol. 2013, 50, 753–761. [Google Scholar] [CrossRef]
- Pearson, J.A.; Wong, S.F.; Wen, L. The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Triplett, E.W. A Model for the Role of Gut Bacteria in the Development of Autoimmunity for Type 1 Diabetes. Diabetologia 2015, 58, 1386–1393. [Google Scholar] [CrossRef]
- Zheng, P.; Li, Z.; Zhou, Z. Gut Microbiome in Type 1 Diabetes: A Comprehensive Review. Diabetes Metab. Res. Rev. 2018, 34, e3043. [Google Scholar] [CrossRef]
- Abdellatif, A.M.; Sarvetnick, N.E. Current Understanding of the Role of Gut Dysbiosis in Type 1 Diabetes. J. Diabetes 2019, 11, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, P.; Hasanzadeh, S.; Tahvildari, A.; Farsi, Y.; Arbabi, M.; Mota, J.F.; Sechi, L.A.; Nasiri, M.J. Is There Any Association between Gut Microbiota and Type 1 Diabetes? A Systematic Review. Gut Pathog. 2019, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Siljander, H.; Honkanen, J.; Knip, M. Microbiome and Type 1 Diabetes. EBioMedicine 2019, 46, 512–521. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, L.; Zhang, S.; Zhao, X.; Gang, X.; Wang, G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front. Endocrinol. 2020, 11, 125. [Google Scholar] [CrossRef]
- Bielka, W.; Przezak, A.; Pawlik, A. The Role of the Gut Microbiota in the Pathogenesis of Diabetes. Int. J. Mol. Sci. 2022, 23, 480. [Google Scholar] [CrossRef] [PubMed]
- Murri, M.; Leiva-Gea, I.; Gomez-Zumaquero, J.M.; Tinahones, F.; Cardona, F.; Soriguer, F.; Queipo-Ortuño, M. Gut Microbiota in Children with Type 1 Diabetes Differs from That in Healthy Children: A Case-Control Study. BMC Med. 2013, 11, 46. [Google Scholar] [CrossRef]
- Pellegrini, S.; Sordi, V.; Bolla, A.M.; Saita, D.; Ferrarese, R.; Canducci, F.; Clementi, M.; Invernizzi, F.; Mariani, A.; Bonfanti, R.; et al. Duodenal Mucosa of Patients with Type 1 Diabetes Shows Distinctive Inflammatory Profile and Microbiota. J. Clin. Endocrinol. Metab. 2017, 102, 1468–1477. [Google Scholar] [CrossRef]
- Traversi, D.; Rabbone, I.; Ignaccolo, M.G.; Carletto, G.; Racca, I.; Vallini, C.; Andriolo, V.; Cadario, F.; Savastio, S.; Siquilini, R.; et al. Gut Microbiota Diversity and T1DM Onset: Preliminary Data of a Case-Control Study. Hum. Microbiome J. 2017, 5, 11–13. [Google Scholar] [CrossRef]
- Del Chierico, F.; Conta, G.; Matteoli, M.C.; Fierabracci, A.; Reddel, S.; Macari, G.; Gardini, S.; Guarrasi, V.; Levi Mortera, S.; Marzano, V.; et al. Gut Microbiota Functional Traits, Blood PH, and Anti-GAD Antibodies Concur in the Clinical Characterization of T1D at Onset. Int. J. Mol. Sci. 2022, 23, 10256. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Xiao, X.; Zheng, J.; Zhou, H. Metaproteomics: A Strategy to Study the Taxonomy and Functionality of the Gut Microbiota. J. Proteom. 2020, 219, 103737. [Google Scholar] [CrossRef]
- Calabrese, F.M.; Porrelli, A.; Vacca, M.; Comte, B.; Nimptsch, K.; Pinart, M.; Pischon, T.; Pujos-Guillot, E.; De Angelis, M. Metaproteomics Approach and Pathway Modulation in Obesity and Diabetes: A Narrative Review. Nutrients 2022, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, M. Metaproteomics: Much More than Measuring Gene Expression in Microbial Communities. mSystem 2019, 4, e0011519. [Google Scholar] [CrossRef] [PubMed]
- Schiebenhoefer, H.; Van Den Bossche, T.; Fuchs, S.; Renard, B.Y.; Muth, T.; Martens, L. Challenges and Promise at the Interface of Metaproteomics and Genomics: An Overview of Recent Progress in Metaproteogenomic Data Analysis. Expert Rev. Proteom. 2019, 16, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Sajulga, R.; Easterly, C.W.; Riffle, M.; Mesuere, B.; Muth, T.; Metha, S.; Kumar, P.; Johnson, J.; Gruening, B.A.; Schiebenhoefer, H.; et al. Survey of Metaproteomics Software Tools for Functional Microbiome Analysis. PLoS ONE 2020, 15, e0241503. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Manghina, V.; Fraumene, C.; Palomba, A.; Abbondio, M.; Deligios, M.; Silverman, M.; Uzzau, S. Metaproteogenomics Reveals Taxonomic and Functional Changes between Cecal and Fecal Microbiota in Mouse. Front. Microbiol. 2017, 8, 391. [Google Scholar] [CrossRef]
- Harjutsalo, V.; Podar, T.; Tuomilehto, J. Cumulative Incidence of Type 1 Diabetes in 10,168 Siblings of Finnish Young-Onset Type 1 Diabetic Patients. Diabetes 2005, 54, 563–569. [Google Scholar] [CrossRef]
- Mrena, S.; Virtanen, S.M.; Laippala, P.; Kulmala, P.; Hannila, M.-L.; Akerblom, H.K.; Knip, M. Models for Predicting Type 1 Diabetes in Siblings of Affected Children. Diabetes Care 2006, 29, 662–667. [Google Scholar] [CrossRef]
- Hagopian, W.; Erlich, H.; Lernmark, A.; Rewers, M.; Ziegler, A.-G.; Simell, O.; Akolkar, B.; Vogt, R.J.; Blair, A.; Ilonen, J.; et al. The Environmental Determinants of Diabetes in the Young (TEDDY): Genetic Criteria and International Diabetes Risk Screening of 421 000 Infants. Pediatr. Diabetes 2011, 12, 733–743. [Google Scholar] [CrossRef]
- Dedrick, S.; Sundaresh, B.; Huang, Q.; Brady, C.; Yoo, T.; Cronin, C.; Rudnicki, C.; Flood, M.; Momeni, B.; Ludvigsson, J.; et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front. Endocrinol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Pinto, E.; Anselmo, M.; Calha, M.; Bottrill, A.; Duarte, I.; Andrew, P.W.; Faleiro, M.L. The Intestinal Proteome of Diabetic and Control Children Is Enriched with Different Microbial and Host Proteins. Microbiology 2017, 163, 161–174. [Google Scholar] [CrossRef]
- Gavin, P.G.; Mullaney, J.A.; Loo, D.; Le Cao, K.-A.; Gottlieb, P.A.; Hill, M.M.; Zipris, D.; Hamilton-Williams, E.E. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care 2018, 41, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated Multi-Omics of the Human Gut Microbiome in a Case Study of Familial Type 1 Diabetes. Nat. Microbiol. 2016, 10, 16180. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de los Reyes-Gavilan, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and Their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Gomez-Arango, L.F.; Barrett, H.L.; Wilkinson, S.A.; Callaway, L.K.; McIntyre, H.D.; Morrison, M.; Dekker Nitert, M. Low Dietary Fiber Intake Increases Collinsella Abundance in the Gut Microbiota of Overweight and Obese Pregnant Women. Gut Microbes 2018, 9, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Canaway, S.; Phillips, I.; Betts, P. Pancreatic Exocrine Insufficiency and Type 1 Diabetes Mellitus. Br. J. Nurs. 2000, 9, 2030–2032. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Bruggeman, B.; Campbell-Thompson, M.; Atkinson, M.A.; Haller, M.J.; Schatz, D. Exocrine Pancreas Dysfunction in Type 1 Diabetes. Endocr. Pract. 2020, 26, 1505–1513. [Google Scholar] [CrossRef]
- Higuchi, B.S.; Rodrigues, N.; Gonzaga, M.I.; Cicogna Paiolo, J.C.; Stefanutto, N.; Omimori, W.P.; Pinheiro, D.G.; Brisotti, J.L.; Matheucci Jr, E.; Sammartino Mariano, V.; et al. Intestinal Dysbiosis in Autoimmune Diabetes Is Correlated with Poor Glycemic Control and Increased Interleukin-6: A Pilot Study. Front. Immunol. 2018, 25, 1689. [Google Scholar] [CrossRef]
- Leiva-Gea, I.; Sànchez-Alcoholado, L.; Martin-Tejedor, B.; Castellano-Castillo, D.; Moreno-Indias, I.; Urda-Cardona, A.; Tinahones, F.; Fernàndez-Garcia, J.C.; Queipo Ortuño, M.I. Gut Microbiota Differs in Composition and Functionality Between Children with Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care 2018, 41, 2385–2395. [Google Scholar] [CrossRef]
- Salamon, D.; Sroka-Oleksiak, A.; Kapusta, P.; Szopa, M.; Mrozińska, S.; Ludwig-Słomczyńska, A.H.; Wolkov, P.P.; Bulanda, M.; Klupa, T.; Malecki, M.T.; et al. Characteristics of Gut Microbiota in Adult Patients with Type 1 and Type 2 Diabetes Based on Next-generation Sequencing of the 16S RRNA Gene Fragment. Pol. Arch. Intern. Med. 2018, 128, 336–343. [Google Scholar] [CrossRef]
- Patterson, E.; Marques, T.M.; O’Sullivan, O.; Fitzgerald, P.; Fitzgerald, G.F.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F.; Stanton, C.; Ross, R.P. Streptozotocin-Induced Type-1-Diabetes Disease Onset in Sprague-Dawley Rats Is Associated with an Altered Intestinal Microbiota Composition and Decreased Diversity. Microbiology 2015, 161, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Usta-Gorgun, B.; Yilmaz-Ersan, L. Short-Chain Fatty Acids Production by Bifidobacterium Species in the Presence of Salep. Electron. J. Biotechnol. 2020, 47, 29–35. [Google Scholar] [CrossRef]
- Mishra, S.; Wang, S.; Nagpal, R.; Miller, B.; Singh, R.; Taraphder, S.; Yadav, H. Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives. Microorganisms 2019, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut Microbiome Metagenomics Analysis Suggests a Functional Model for the Development of Autoimmunity for Type 1 Diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef]
- Ganesan, K.; Chung, S.K.; Vanamala, J.; Xu, B. Causal Relationship between Diet-Induced Gut Microbiota Changes and Diabetes: A Novel Strategy to Transplant Faecalibacterium Prausnitzii in Preventing Diabetes. Int. J. Mol. Sci. 2018, 19, 3720. [Google Scholar] [CrossRef]
- Mariño, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect against Type 1 Diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef]
- Kim, C.H. Microbiota or Short-Chain Fatty Acids: Which Regulates Diabetes? Cell. Mol. Immunol. 2018, 15, 88–91. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Ardissone, A.; Dias, R.; Simell, V.; Leonard, M.T.; Kemppainen, K.M.; Drew, J.C.; Schatz, D.; Atkinson, M.A.; Kolaczkowski, B.; et al. Bacteroides Dorei Dominates Gut Microbiome Prior to Autoimmunity in Finnish Children at High Risk for Type 1 Diabetes. Front. Microbiol. 2014, 5, 678. [Google Scholar] [CrossRef]
- Soyucen, E.; Glucan, A.; Aktuglu-Zeybek, A.C.; Onal, H.; Kiykim, E.; Aydin, A. Differences in the Gut Microbiota of Healthy Children and Those with Type 1 Diabetes. Pediatr. Int. 2014, 56, 336–343. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Duran, C.; Martinez-Lopez, R.; Zapico, I.; Perez, E.; Romeu, E.; Arroyo, J.; Hernaez, M.L.; Pitarch, A.; Monteoliva, L.; Gil, C. Distinct Human Gut Microbial Taxonomic Signatures Uncovered with Different Sample Processing and Microbial Cell Disruption Methods for Metaproteomic Analysis. Front. Microbiol. 2021, 12, 618566. [Google Scholar] [CrossRef] [PubMed]
- Confer, A.W.; Ayalew, S. The OmpA Family of Proteins: Roles in Bacterial Pathogenesis and Immunity. Vet. Microbiol. 2013, 3, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Biassoni, R.; Di Marco, E.; Squillario, M.; Barla, A.; Piccolo, G.; Ugolotti, E.; Gatti, C.; Minuto, N.; Patti, G.; Maghnie, M.; et al. Gut Microbiota in T1DM-Onset Pediatric Patients: Machine-Learning Algorithms to Classify Microorganisms as Disease Linked. J. Clin. Endocrinol. Metab. 2020, 105, e3114–e3126. [Google Scholar] [CrossRef]
- Marzano, V.; Pane, S.; Foglietta, G.; Levi Mortera, S.; Vernocchi, P.; Onetti Muda, A.; Putignani, L. Mass Spectrometry Based-Proteomic Analysis of Anisakis Spp.: A Preliminary Study towards a New Diagnostic Tool. Genes 2020, 11, 693. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Cheng, K.; Ning, Z.; Zhang, X.; Li, L.; Liao, B.; Mayne, J.; Stintzi, A.; Figeys, D. MetaLab: An Automated Pipeline for Metaproteomic Data Analysis. Microbiome 2017, 5, 157. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTRL | T1D | SIBL | |
|---|---|---|---|
| Mean age in years (± SD 1) | 10 ± 3.2 | 10 ± 3.2 | 13 ± 4.4 |
| Gender (male/female) | 14/16 | 22/17 | 6/9 |
| Clinical Parameters | Average | Standard Deviation | Reference Value | Patients with Values Exceeding the Reference Value |
|---|---|---|---|---|
| Age (years) | 9.56 | 3.19 | ≥14 | 4/39 |
| IAA (U/mL) | 7.56 | 4.76 | ≥7 | 16/34 |
| IA2 (U/mL) | 11.78 | 12.79 | ≥1 | 32/37 |
| anti GAD (U/mL) | 36.79 | 45.39 | ≥1 | 28/37 |
| BMI (kg/m2) | 16.44 | 2.95 | ≤14 | 10/39 |
| Blood pH at onset | 7.27 | 0.11 | ≤7.3 | 19/39 |
| Exogenous insulin need (IU/kg BM) | 0.87 | 0.28 | ≥1 | 14/39 |
| C-peptide (ng/mL) | 0.29 | 0.19 | <0.6 | 36/39 |
| HbA1c (%) | 101.13 | 23.46 | >38 | 39/39 |
| CRP (mg/L) | 0.56 | 1.61 | >1 | 4/39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi Mortera, S.; Marzano, V.; Vernocchi, P.; Matteoli, M.C.; Guarrasi, V.; Gardini, S.; Del Chierico, F.; Rapini, N.; Deodati, A.; Fierabracci, A.; et al. Functional and Taxonomic Traits of the Gut Microbiota in Type 1 Diabetes Children at the Onset: A Metaproteomic Study. Int. J. Mol. Sci. 2022, 23, 15982. https://doi.org/10.3390/ijms232415982
Levi Mortera S, Marzano V, Vernocchi P, Matteoli MC, Guarrasi V, Gardini S, Del Chierico F, Rapini N, Deodati A, Fierabracci A, et al. Functional and Taxonomic Traits of the Gut Microbiota in Type 1 Diabetes Children at the Onset: A Metaproteomic Study. International Journal of Molecular Sciences. 2022; 23(24):15982. https://doi.org/10.3390/ijms232415982
Chicago/Turabian StyleLevi Mortera, Stefano, Valeria Marzano, Pamela Vernocchi, Maria Cristina Matteoli, Valerio Guarrasi, Simone Gardini, Federica Del Chierico, Novella Rapini, Annalisa Deodati, Alessandra Fierabracci, and et al. 2022. "Functional and Taxonomic Traits of the Gut Microbiota in Type 1 Diabetes Children at the Onset: A Metaproteomic Study" International Journal of Molecular Sciences 23, no. 24: 15982. https://doi.org/10.3390/ijms232415982
APA StyleLevi Mortera, S., Marzano, V., Vernocchi, P., Matteoli, M. C., Guarrasi, V., Gardini, S., Del Chierico, F., Rapini, N., Deodati, A., Fierabracci, A., Cianfarani, S., & Putignani, L. (2022). Functional and Taxonomic Traits of the Gut Microbiota in Type 1 Diabetes Children at the Onset: A Metaproteomic Study. International Journal of Molecular Sciences, 23(24), 15982. https://doi.org/10.3390/ijms232415982