
Hepatitis B and Hepatitis D Viruses: A Comprehensive Update with an Immunological Focus
 , , and
, , and
Abstract
1. Introduction
HBV and HDV Cell Tropism
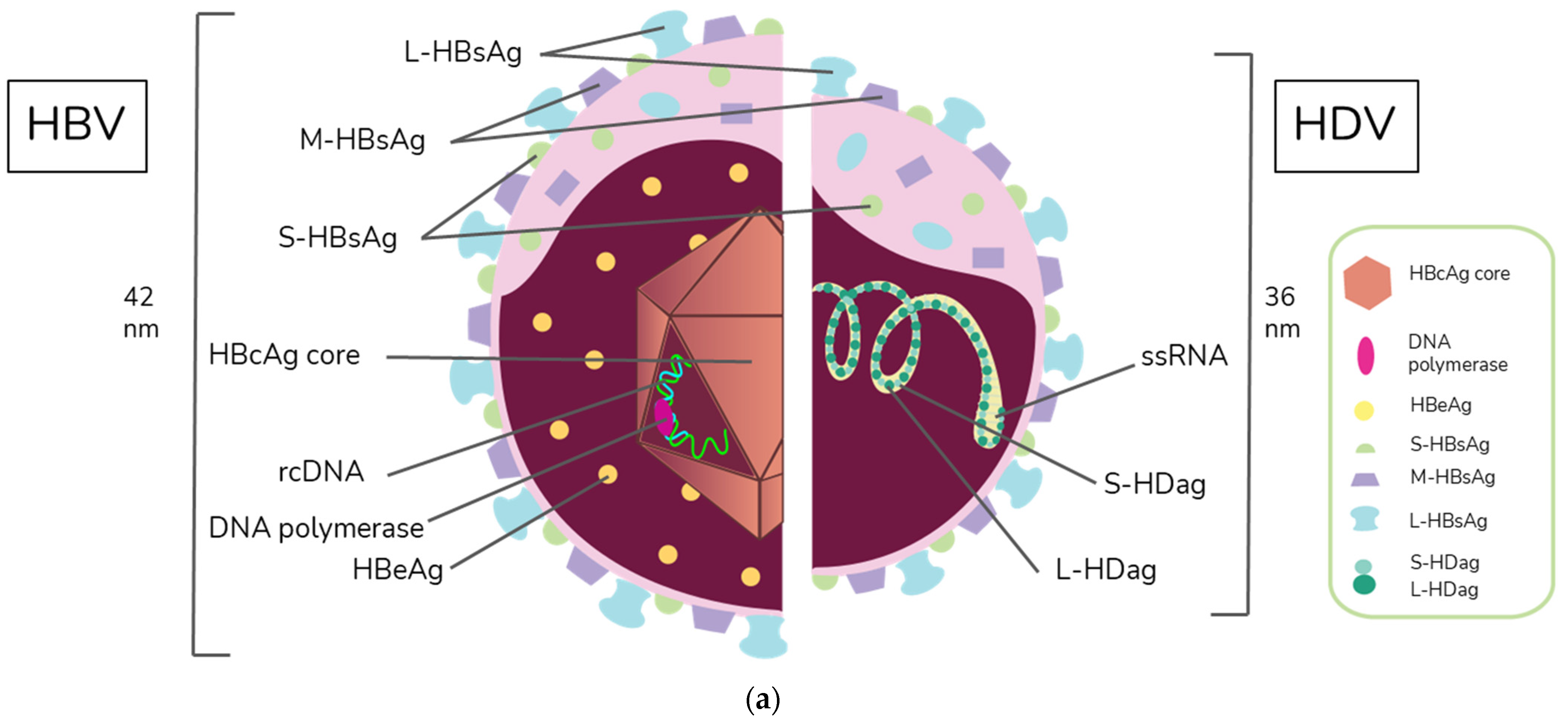
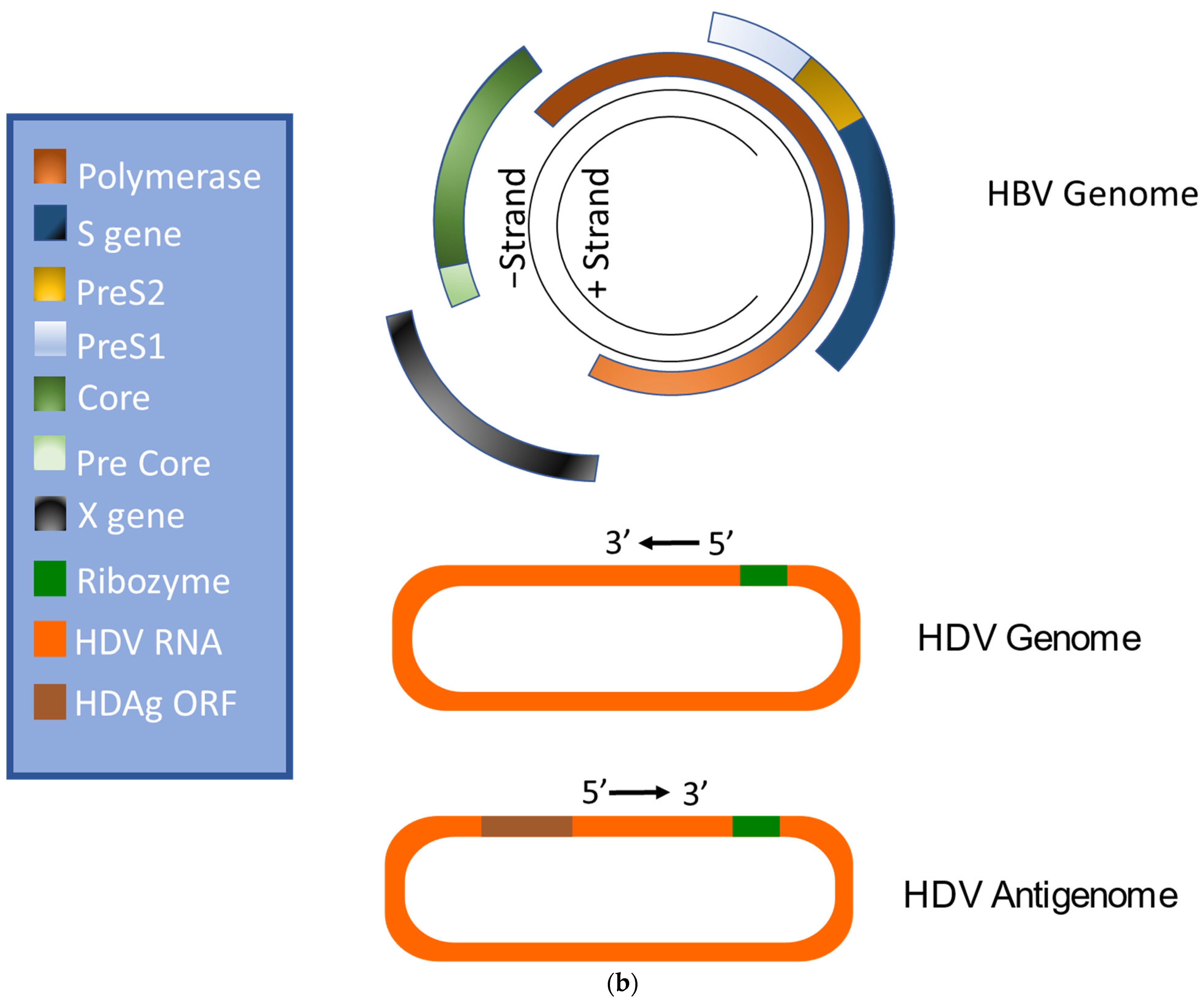
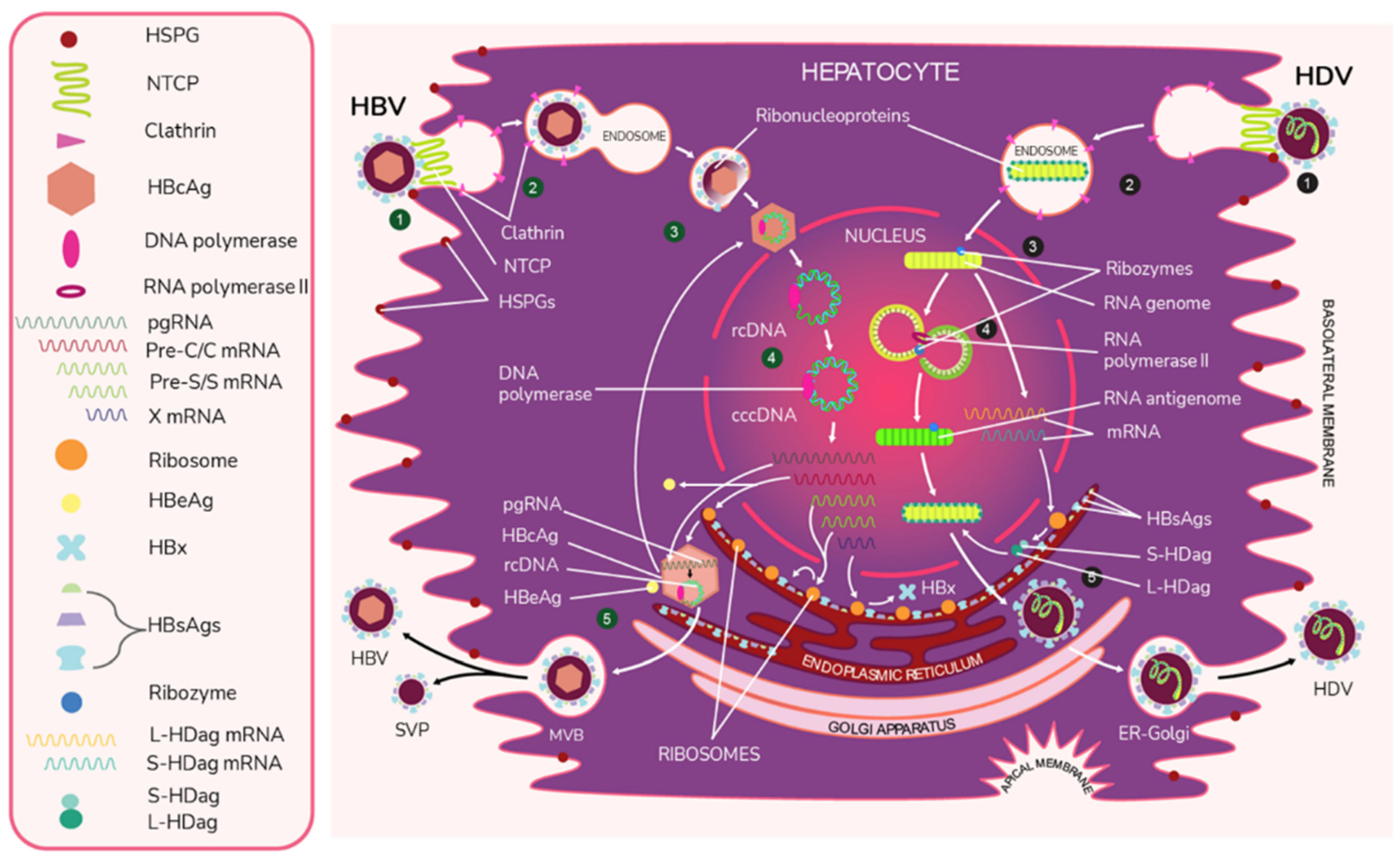
2. Virion Structure and Viral Life Cycle of HBV and HDV
3. Natural Course of HBV and HDV Infection
3.1. HDV RNA and HBV DNA Interplay during Chronic Infection
3.2. Histopathology
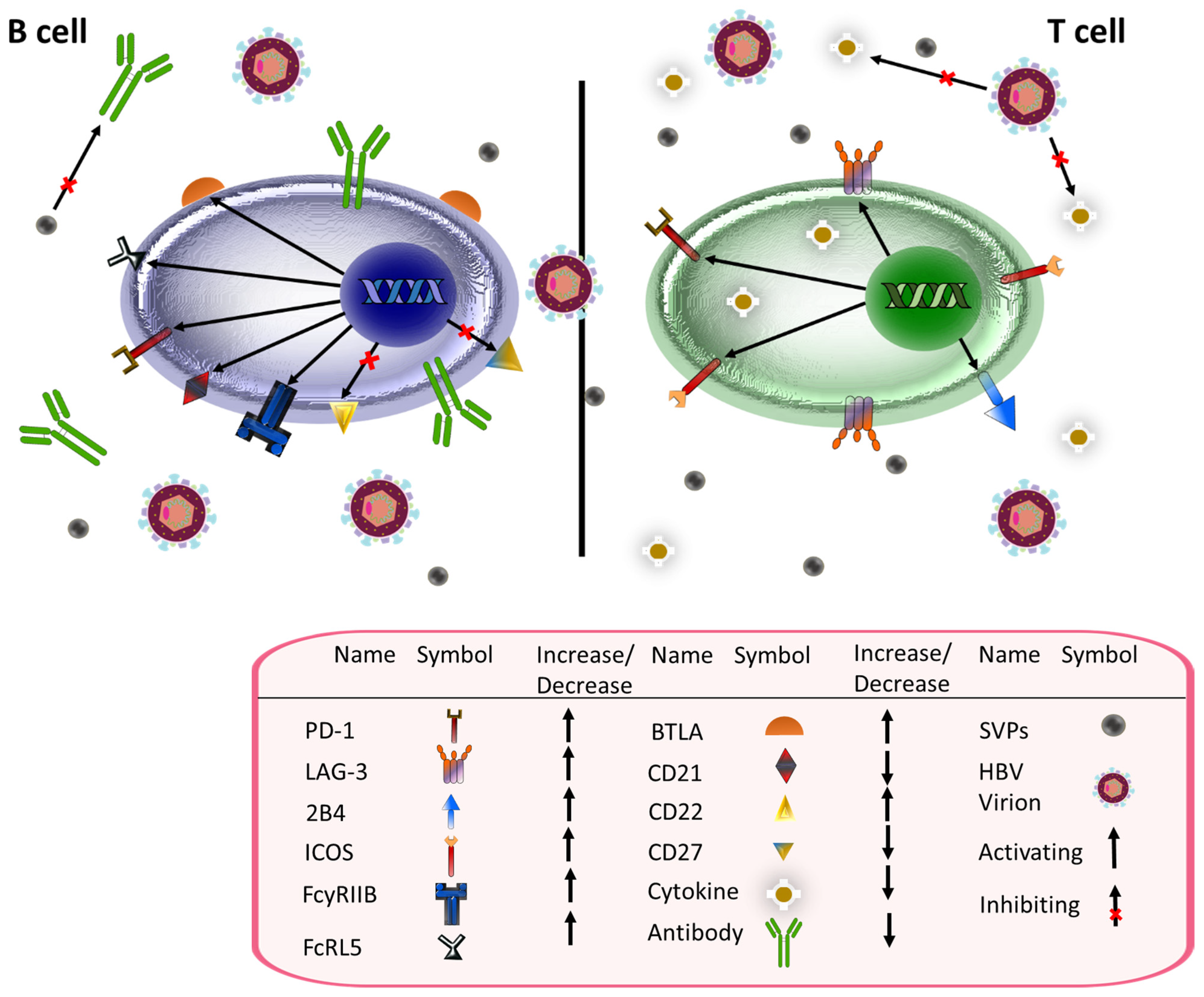
4. HBsAg, CD4+, and CD8+ T Cell-Mediated Immunity
HBsAg and Follicular Helper T Cells
5. HBsAg and B Cell Immunity
6. Antigenicity of HBsAg
7. HBV and Immune Suppression
8. S-HDAg, L-HDAg, and the Immune System
9. Therapies for HBV and HDV
10. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Zu Siederdissen, C.H.; Maasoumy, B.; Cornberg, M. What is new on HBsAg and other diagnostic markers in HBV infection? Best Pract. Res. Clin. Gastroenterol. 2017, 31, 281–289. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b#:~:text=WHO%20estimates%20that%20296%20million,carcinoma%20(primary%20liver%20cancer) (accessed on 13 November 2022).
- MacLachlan, J.H.; Cowie, B.C. Hepatitis B virus epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5, a021410. [Google Scholar] [CrossRef] [PubMed]
- Shao, E.R.; Mboya, I.B.; Gunda, D.W.; Ruhangisa, F.G.; Temu, E.M.; Nkwama, M.L.; Pyuza, J.J.; Kilonzo, K.G.; Lyamuya, F.S.; Maro, V.P. Seroprevalence of hepatitis B virus infection and associated factors among healthcare workers in northern Tanzania. BMC Infect. Dis. 2018, 18, 474. [Google Scholar] [CrossRef]
- Candotti, D.; Allain, J.P. Transfusion-transmitted hepatitis B virus infection. J. Hepatol. 2009, 51, 798–809. [Google Scholar] [CrossRef]
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49, S13–S21. [Google Scholar] [CrossRef]
- Trepo, C.; Guillevin, L. Polyarteritis nodosa and extrahepatic manifestations of HBV infection: The case against autoimmune intervention in pathogenesis. J. Autoimmun. 2001, 16, 269–274. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, C.; Fan, J.; Wang, B.; Li, G. Hepatitis B-related glomerulonephritis and optimization of treatment. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 113–125. [Google Scholar] [CrossRef]
- Mazzaro, C.; Dal Maso, L.; Visentini, M.; Ermacora, A.; Tonizzo, M.; Gattei, V.; Andreone, P. Recent news in the treatment of hepatitis B virus-related cryogobulinemic vasculitis. Minerva Med. 2020, 111, 566–572. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Z. HBV-Induced Immune Imbalance in the Development of HCC. Front. Immunol. 2019, 10, 2048. [Google Scholar] [CrossRef]
- Baig, S.; Alamgir, M. The extrahepatic manifestations of hepatitis B virus. J. Coll. Physicians Surg. Pak. 2008, 18, 451–457. [Google Scholar] [PubMed]
- Kim, G.W.; Imam, H.; Khan, M.; Mir, S.A.; Kim, S.J.; Yoon, S.K.; Hur, W.; Siddiqui, A. HBV-Induced Increased N6 Methyladenosine Modification of PTEN RNA Affects Innate Immunity and Contributes to HCC. Hepatology 2021, 73, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2022, 22, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Hu, K.Q. Rethinking the pathogenesis of hepatitis B virus (HBV) infection. J. Med. Virol. 2015, 87, 1989–1999. [Google Scholar] [CrossRef]
- Engle, R.E.; De Battista, D.; Danoff, E.J.; Nguyen, H.; Chen, Z.; Lusso, P.; Purcell, R.H.; Farci, P. Distinct Cytokine Profiles Correlate with Disease Severity and Outcome in Longitudinal Studies of Acute Hepatitis B Virus and Hepatitis D Virus Infection in Chimpanzees. mBio 2020, 11, e02580-20. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases from 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef]
- Pujol, F.; Jaspe, R.C.; Loureiro, C.L.; Chemin, I. Hepatitis B virus American genotypes: Pathogenic variants? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 825–835. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. Natural history of acute and chronic hepatitis B: The role of HBV genotypes and mutants. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 249–255. [Google Scholar] [CrossRef]
- Tanwar, S.; Dusheiko, G. Is there any value to hepatitis B virus genotype analysis? Curr. Gastroenterol. Rep. 2012, 14, 37–46. [Google Scholar] [CrossRef]
- Tsushima, K.; Tsuge, M.; Hiraga, N.; Uchida, T.; Murakami, E.; Makokha, G.N.; Kurihara, M.; Nomura, M.; Hiyama, Y.; Fujino, H.; et al. Comparison of intracellular responses between HBV genotype A and C infection in human hepatocyte chimeric mice. J. Gastroenterol. 2019, 54, 650–659. [Google Scholar] [CrossRef]
- Ito, K.; Yotsuyanagi, H.; Yatsuhashi, H.; Karino, Y.; Takikawa, Y.; Saito, T.; Arase, Y.; Imazeki, F.; Kurosaki, M.; Umemura, T.; et al. Risk factors for long-term persistence of serum hepatitis B surface antigen following acute hepatitis B virus infection in Japanese adults. Hepatology 2014, 59, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; Hui, A.Y.; Wong, M.L.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, 1494–1498. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Lang, T.; Ryan, K.; Wilson, R.; Skinner, N.A.; Thompson, A.J.; Ahn, S.H.; Weilert, F.; Abbott, W.; Gane, E.; et al. Toll-IL1 receptor-mediated innate immune responses vary across HBV genotype and predict treatment response to pegylated-IFN in HBeAg-positive CHB patients. J. Viral Hepat. 2016, 23, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.M.; Kuo, Y.H.; Wang, J.H.; Hung, C.H.; Hu, T.H.; Lu, S.N.; Chen, C.H. Associations of HBV Genotype B vs. C Infection with Relapse after Cessation of Entecavir or Tenofovir Therapy. Clin. Gastroenterol. Hepatol. 2020, 18, 2989–2997.e2983. [Google Scholar] [CrossRef]
- Negro, F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb. Perspect. Med. 2014, 4, a021550. [Google Scholar] [CrossRef]
- Perez-Vargas, J.; Amirache, F.; Boson, B.; Mialon, C.; Freitas, N.; Sureau, C.; Fusil, F.; Cosset, F.L. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat. Commun. 2019, 10, 2098. [Google Scholar] [CrossRef]
- Ferrante, N.D.; Lo Re, V., 3rd. Epidemiology, Natural History, and Treatment of Hepatitis Delta Virus Infection in HIV/Hepatitis B Virus Coinfection. Curr. HIV/AIDS Rep. 2020, 17, 405–414. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, S.; Ou, X.; Li, S.; Ma, Z.; Wang, W.; Peppelenbosch, M.P.; Liu, J.; Pan, Q. Estimating the Global Prevalence, Disease Progression, and Clinical Outcome of Hepatitis Delta Virus Infection. J. Infect. Dis. 2020, 221, 1677–1687. [Google Scholar] [CrossRef]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef]
- Chen, H.Y.; Shen, D.T.; Ji, D.Z.; Han, P.C.; Zhang, W.M.; Ma, J.F.; Chen, W.S.; Goyal, H.; Pan, S.; Xu, H.G. Prevalence and burden of hepatitis D virus infection in the global population: A systematic review and meta-analysis. Gut 2019, 68, 512–521. [Google Scholar] [CrossRef]
- Niro, G.A.; Ferro, A.; Cicerchia, F.; Brascugli, I.; Durazzo, M. Hepatitis delta virus: From infection to new therapeutic strategies. World J. Gastroenterol. 2021, 27, 3530–3542. [Google Scholar] [CrossRef] [PubMed]
- Sellier, P.O.; Maylin, S.; Brichler, S.; Bercot, B.; Lopes, A.; Chopin, D.; Pogliaghi, M.; Munier, A.L.; Delcey, V.; Simoneau, G.; et al. Hepatitis B Virus-Hepatitis D Virus mother-to-child co-transmission: A retrospective study in a developed country. Liver Int. 2018, 38, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Urban, S. Interplay between Hepatitis D Virus and the Interferon Response. Viruses 2020, 12, 1334. [Google Scholar] [CrossRef] [PubMed]
- Roulot, D.; Brichler, S.; Layese, R.; BenAbdesselam, Z.; Zoulim, F.; Thibault, V.; Scholtes, C.; Roche, B.; Castelnau, C.; Poynard, T.; et al. Origin, HDV genotype and persistent viremia determine outcome and treatment response in patients with chronic hepatitis delta. J. Hepatol. 2020, 73, 1046–1062. [Google Scholar] [CrossRef] [PubMed]
- Tseligka, E.D.; Clement, S.; Negro, F. HDV Pathogenesis: Unravelling Ariadne’s Thread. Viruses 2021, 13, 778. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.; Wick, M.; White, H.; Perrillo, R. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology 1993, 18, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Colpitts, C.C.; Schuster, C.; Zeisel, M.B.; Baumert, T.F. Cell Culture Models for the Investigation of Hepatitis B and D Virus Infection. Viruses 2016, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 3, e00049. [Google Scholar] [CrossRef]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014, 443, 808–813. [Google Scholar] [CrossRef]
- Mason, W.S. Animal models and the molecular biology of hepadnavirus infection. Cold Spring Harb. Perspect. Med. 2015, 5, a021352. [Google Scholar] [CrossRef]
- Giersch, K.; Dandri, M. In Vivo Models of HDV Infection: Is Humanizing NTCP Enough? Viruses 2021, 13, 588. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Marion, P.L.; Nakai, H.; Meuse, L.; Cullen, J.M.; Bordier, B.B.; Schwall, R.; Greenberg, H.B.; Glenn, J.S.; Kay, M.A. Sustained survival of human hepatocytes in mice: A model for in vivo infection with human hepatitis B and hepatitis delta viruses. Nat. Med. 2000, 6, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Broering, R.; Li, X.; Zhang, X.; Liu, J.; Yang, D.; Lu, M. In Vivo Mouse Models for Hepatitis B Virus Infection and Their Application. Front. Immunol. 2021, 12, 766534. [Google Scholar] [CrossRef] [PubMed]
- Bonamassa, B.; Hai, L.; Liu, D. Hydrodynamic gene delivery and its applications in pharmaceutical research. Pharm. Res. 2011, 28, 694–701. [Google Scholar] [CrossRef]
- Chang, J.; Sigal, L.J.; Lerro, A.; Taylor, J. Replication of the human hepatitis delta virus genome Is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J. Virol. 2001, 75, 3469–3473. [Google Scholar] [CrossRef]
- Liu, Y.; Maya, S.; PLoSs, A. Animal Models of Hepatitis B Virus Infection-Success, Challenges, and Future Directions. Viruses 2021, 13, 777. [Google Scholar] [CrossRef]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [CrossRef]
- Selzer, L.; Zlotnick, A. Assembly and Release of Hepatitis B Virus. Cold Spring Harb. Perspect. Med. 2015, 5, a021394. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antiviral Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Seeger, C.; Zoulim, F.; Masson, W.S. Hepadnaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2185–2221. [Google Scholar]
- Wei, L.; PLoss, A. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat. Commun. 2021, 12, 1591. [Google Scholar] [CrossRef]
- Been, M.D. HDV ribozymes. Curr. Top Microbiol. Immunol. 2006, 307, 47–65. [Google Scholar] [CrossRef]
- Mentha, N.; Clement, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef]
- Perez-Vargas, J.; Teppa, E.; Amirache, F.; Boson, B.; de Oliveira, R.P.; Combet, C.; Bockmann, A.; Fusil, F.; Freitas, N.; Carbone, A.; et al. A fusion peptide in preS1 and the human protein disulfide isomerase ERp57 are involved in hepatitis B virus membrane fusion process. Elife 2021, 10, e64507. [Google Scholar] [CrossRef]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef]
- Wei, L.; PLoss, A. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA formation. Nat. Microbiol. 2020, 5, 715–726. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Lin, N.; Ye, A.; Lin, J.; Liu, C.; Huang, J.; Fu, Y.; Wu, S.; Xu, S.; Wang, L.; Ou, Q. Diagnostic Value of Detection of Pregenomic RNA in Sera of Hepatitis B Virus-Infected Patients with Different Clinical Outcomes. J. Clin. Microbiol. 2020, 58, e01275-19. [Google Scholar] [CrossRef]
- Revill, P.A.; Locarnini, S.A. New perspectives on the hepatitis B virus life cycle in the human liver. J. Clin. Investig. 2016, 126, 833–836. [Google Scholar] [CrossRef]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef]
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef]
- Taylor, J.M.; Purcell, R.H.; Farci, P. Hepatitis D (Delta) Virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2222–2241. [Google Scholar]
- Urban, S.; Neumann-Haefelin, C.; Lampertico, P. Hepatitis D virus in 2021: Virology, immunology and new treatment approaches for a difficult-to-treat disease. Gut 2021, 70, 1782–1794. [Google Scholar] [CrossRef]
- Lo, K.; Hwang, S.B.; Duncan, R.; Trousdale, M.; Lai, M.M. Characterization of mRNA for hepatitis delta antigen: Exclusion of the full-length antigenomic RNA as an mRNA. Virology 1998, 250, 94–105. [Google Scholar] [CrossRef]
- Hartwig, D.; Schutte, C.; Warnecke, J.; Dorn, I.; Hennig, H.; Kirchner, H.; Schlenke, P. The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J. Viral Hepat. 2006, 13, 150–157. [Google Scholar] [CrossRef]
- Dziri, S.; Rodriguez, C.; Gerber, A.; Brichler, S.; Alloui, C.; Roulot, D.; Deny, P.; Pawlotsky, J.M.; Gordien, E.; Le Gal, F. Variable In Vivo Hepatitis D Virus (HDV) RNA Editing Rates According to the HDV Genotype. Viruses 2021, 13, 1572. [Google Scholar] [CrossRef]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Rizzetto, M. The adventure of delta. Liver Int. 2016, 36 (Suppl. S1), 135–140. [Google Scholar] [CrossRef]
- Goodrum, G.; Pelchat, M. Insight into the Contribution and Disruption of Host Processes during HDV Replication. Viruses 2018, 11, 21. [Google Scholar] [CrossRef]
- Filipovska, J.; Konarska, M.M. Specific HDV RNA-templated transcription by pol II in vitro. RNA 2000, 6, 41–54. [Google Scholar] [CrossRef]
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J. Virol. 2008, 82, 1118–1127. [Google Scholar] [CrossRef]
- Greco-Stewart, V.S.; Schissel, E.; Pelchat, M. The hepatitis delta virus RNA genome interacts with the human RNA polymerases I and III. Virology 2009, 386, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Macnaughton, T.; Gao, L.; Lai, M.M. RNA-templated replication of hepatitis delta virus: Genomic and antigenomic RNAs associate with different nuclear bodies. J. Virol. 2006, 80, 6478–6486. [Google Scholar] [CrossRef]
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990, 64, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef]
- Jayan, G.C.; Casey, J.L. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of ADAR1 but not ADAR2 expression. J. Virol. 2002, 76, 12399–12404. [Google Scholar] [CrossRef]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta virus large antigen. Science 1992, 256, 1331–1333. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Clin. Liver Dis. 2018, 12, 33–34. [Google Scholar] [CrossRef]
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, S45–S55. [Google Scholar] [CrossRef]
- Shapiro, C.N. Epidemiology of hepatitis B. Pediatr. Infect. Dis. J. 1993, 12, 433–437. [Google Scholar] [CrossRef]
- van Santen, D.K.; Boyd, A.; Bruisten, S.; Sonder, G.J.; Prins, M.; van Houdt, R. Frequent delayed spontaneous seroclearance of hepatitis B virus after incident HBV infection among adult high-risk groups. J. Viral Hepat. 2020, 27, 81–87. [Google Scholar] [CrossRef]
- Veronese, P.; Dodi, I.; Esposito, S.; Indolfi, G. Prevention of vertical transmission of hepatitis B virus infection. World J. Gastroenterol. 2021, 27, 4182–4193. [Google Scholar] [CrossRef]
- Stevens, C.E.; Toy, P.T.; Tong, M.J.; Taylor, P.E.; Vyas, G.N.; Nair, P.V.; Gudavalli, M.; Krugman, S. Perinatal hepatitis B virus transmission in the United States. Prevention by passive-active immunization. JAMA 1985, 253, 1740–1745. [Google Scholar] [CrossRef]
- Taylor, J.M. Infection by Hepatitis Delta Virus. Viruses 2020, 12, 648. [Google Scholar] [CrossRef]
- Sagnelli, C.; Sagnelli, E.; Russo, A.; Pisaturo, M.; Occhiello, L.; Coppola, N. HBV/HDV Co-Infection: Epidemiological and Clinical Changes, Recent Knowledge and Future Challenges. Life 2021, 11, 169. [Google Scholar] [CrossRef]
- Romeo, R. Hepatitis Delta: Natural history and outcome. Clin. Liver Dis. 2013, 2, 235–236. [Google Scholar] [CrossRef]
- Botelho-Souza, L.F.; Vasconcelos, M.P.A.; Dos Santos, A.O.; Salcedo, J.M.V.; Vieira, D.S. Hepatitis delta: Virological and clinical aspects. Virol. J. 2017, 14, 177. [Google Scholar] [CrossRef]
- Masood, U.; John, S. Hepatitis D. In StatPearls; NCBI Bookshelf: Bethesda, MD, USA, 2021. [Google Scholar]
- Hsieh, T.H.; Liu, C.J.; Chen, D.S.; Chen, P.J. Natural course and treatment of hepatitis D virus infection. J. Formos. Med. Assoc. 2006, 105, 869–881. [Google Scholar] [CrossRef]
- Genesca, J.; Jardi, R.; Buti, M.; Vives, L.; Prat, S.; Esteban, J.I.; Esteban, R.; Guardia, J. Hepatitis B virus replication in acute hepatitis B, acute hepatitis B virus-hepatitis delta virus coinfection and acute hepatitis delta superinfection. Hepatology 1987, 7, 569–572. [Google Scholar] [CrossRef]
- Giersch, K.; Dandri, M. Hepatitis B and Delta Virus: Advances on Studies about Interactions between the Two Viruses and the Infected Hepatocyte. J. Clin. Transl. Hepatol. 2015, 3, 220–229. [Google Scholar] [CrossRef]
- Schaper, M.; Rodriguez-Frias, F.; Jardi, R.; Tabernero, D.; Homs, M.; Ruiz, G.; Quer, J.; Esteban, R.; Buti, M. Quantitative longitudinal evaluations of hepatitis delta virus RNA and hepatitis B virus DNA shows a dynamic, complex replicative profile in chronic hepatitis B and D. J. Hepatol. 2010, 52, 658–664. [Google Scholar] [CrossRef]
- Braga, W.S.; de Oliveira, C.M.; de Araujo, J.R.; Mda, C.C.; Rocha, J.M.; Gimaque, J.B.; Silva, M.L.; Vasconcelos, H.L.; Ramasawmy, R.; Parana, R. Chronic HDV/HBV co-infection: Predictors of disease stage---A case series of HDV-3 patients. J. Hepatol. 2014, 61, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Mhlanga, A.; Zakh, R.; Churkin, A.; Reinharz, V.; Glenn, J.S.; Etzion, O.; Cotler, S.J.; Yurdaydin, C.; Barash, D.; Dahari, H. Modeling the Interplay between HDV and HBV in Chronic HDV/HBV Patients. Mathematics 2022, 10, 3917. [Google Scholar] [CrossRef]
- Yurdaydin, C.; Keskin, O.; Kalkan, C.; Karakaya, F.; Caliskan, A.; Karatayli, E.; Karatayli, S.; Bozdayi, A.M.; Koh, C.; Heller, T.; et al. Optimizing lonafarnib treatment for the management of chronic delta hepatitis: The LOWR HDV-1 study. Hepatology 2018, 67, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Shekhtman, L.; Cotler, S.J.; Hershkovich, L.; Uprichard, S.L.; Bazinet, M.; Pantea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Krawczyk, A.; et al. Modelling hepatitis D virus RNA and HBsAg dynamics during nucleic acid polymer monotherapy suggest rapid turnover of HBsAg. Sci. Rep. 2020, 10, 7837. [Google Scholar] [CrossRef] [PubMed]
- Guedj, J.; Rotman, Y.; Cotler, S.J.; Koh, C.; Schmid, P.; Albrecht, J.; Haynes-Williams, V.; Liang, T.J.; Hoofnagle, J.H.; Heller, T.; et al. Understanding early serum hepatitis D virus and hepatitis B surface antigen kinetics during pegylated interferon-alpha therapy via mathematical modeling. Hepatology 2014, 60, 1902–1910. [Google Scholar] [CrossRef]
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: A proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect. Dis. 2015, 15, 1167–1174. [Google Scholar] [CrossRef]
- Castelnau, C.; Le Gal, F.; Ripault, M.P.; Gordien, E.; Martinot-Peignoux, M.; Boyer, N.; Pham, B.N.; Maylin, S.; Bedossa, P.; Deny, P.; et al. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: Relevance of quantitative RT-PCR for follow-up. Hepatology 2006, 44, 728–735. [Google Scholar] [CrossRef]
- Manesis, E.K.; Schina, M.; Le Gal, F.; Agelopoulou, O.; Papaioannou, C.; Kalligeros, C.; Arseniou, V.; Manolakopoulos, S.; Hadziyannis, E.S.; Gault, E.; et al. Quantitative analysis of hepatitis D virus RNA and hepatitis B surface antigen serum levels in chronic delta hepatitis improves treatment monitoring. Antivir. Ther. 2007, 12, 381–388. [Google Scholar] [CrossRef]
- Zachou, K.; Yurdaydin, C.; Drebber, U.; Dalekos, G.N.; Erhardt, A.; Cakaloglu, Y.; Degertekin, H.; Gurel, S.; Zeuzem, S.; Bozkaya, H.; et al. Quantitative HBsAg and HDV-RNA levels in chronic delta hepatitis. Liver Int. 2010, 30, 430–437. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Yurdaydin, C.; Dalekos, G.N.; Erhardt, A.; Cakaloglu, Y.; Degertekin, H.; Gurel, S.; Zeuzem, S.; Zachou, K.; Bozkaya, H.; et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N. Engl. J. Med. 2011, 364, 322–331. [Google Scholar] [CrossRef]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Yurdaydin, C.; Bozkaya, H.; Onder, F.O.; Senturk, H.; Karaaslan, H.; Akdogan, M.; Cetinkaya, H.; Erden, E.; Erkan-Esin, O.; Yalcin, K.; et al. Treatment of chronic delta hepatitis with lamivudine vs. lamivudine + interferon vs. interferon. J. Viral Hepat. 2008, 15, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitch, J.H. Acute Viral Hepatitis. In Scheuer’s Liver Biopsy Interpretation, 10th ed.; Lefkowitch, J.H., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 89–107. [Google Scholar]
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2019, 10, 3112. [Google Scholar] [CrossRef]
- Boltjes, A.; Movita, D.; Boonstra, A.; Woltman, A.M. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J. Hepatol. 2014, 61, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Mani, H.; Kleiner, D.E. Liver biopsy findings in chronic hepatitis B. Hepatology 2009, 49, S61–S71. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M. Patterns of necrosis in liver disease. Clin. Liver Dis. 2017, 10, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; MacSween, R.N.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef]
- Bedossa, P. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. The French METAVIR Cooperative Study Group. Hepatology 1994, 20, 15–20. [Google Scholar] [CrossRef]
- Axley, P.; Mudumbi, S.; Sarker, S.; Kuo, Y.F.; Singal, A.K. Patients with stage 3 compared to stage 4 liver fibrosis have lower frequency of and longer time to liver disease complications. PLoS ONE 2018, 13, e0197117. [Google Scholar] [CrossRef]
- Mohamadnejad, M.; Tavangar, S.M.; Sotoudeh, M.; Kosari, F.; Khosravi, M.; Geramizadeh, B.; Montazeri, G.; Estakhri, A.; Mirnasseri, M.M.; Fazlollahi, A.; et al. Histopathological Study of Chronic Hepatitis B: A Comparative Study of Ishak and METAVIR Scoring Systems. Int. J. Organ Transplant. Med. 2010, 1, 171–176. [Google Scholar] [CrossRef]
- Liao, B.; Zhang, F.; Lin, S.; He, H.; Liu, Y.; Zhang, J.; Xu, Y.; Yi, J.; Chen, Y.; Liu, H.; et al. Epidemiological, clinical and histological characteristics of HBV/HDV co-infection: A retrospective cross-sectional study in Guangdong, China. PLoS ONE 2014, 9, e115888. [Google Scholar] [CrossRef] [PubMed]
- Fallet, B.; Hao, Y.; Florova, M.; Cornille, K.; de Los Aires, A.V.; Zubani, G.G.; Ertuna, Y.I.; Greiff, V.; Menzel, U.; Hammad, K.; et al. Chronic Viral Infection Promotes Efficient Germinal Center B Cell Responses. Cell Rep. 2020, 30, 1013–1026.e1017. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Zheng, X.; Yang, W. The Role of Metabolic Dysfunction in T-Cell Exhaustion During Chronic Viral Infection. Front. Immunol. 2022, 13, 843242. [Google Scholar] [CrossRef] [PubMed]
- Ozga, A.J.; Chow, M.T.; Lopes, M.E.; Servis, R.L.; Di Pilato, M.; Dehio, P.; Lian, J.; Mempel, T.R.; Luster, A.D. CXCL10 chemokine regulates heterogeneity of the CD8(+) T cell response and viral set point during chronic infection. Immunity 2022, 55, 82–97.e88. [Google Scholar] [CrossRef]
- Le Bert, N.; Gill, U.S.; Hong, M.; Kunasegaran, K.; Tan, D.Z.M.; Ahmad, R.; Cheng, Y.; Dutertre, C.A.; Heinecke, A.; Rivino, L.; et al. Effects of Hepatitis B Surface Antigen on Virus-Specific and Global T Cells in Patients with Chronic Hepatitis B Virus infection. Gastroenterology 2020, 159, 652–664. [Google Scholar] [CrossRef]
- Aliabadi, E.; Urbanek-Quaing, M.; Maasoumy, B.; Bremer, B.; Grasshoff, M.; Li, Y.; Niehaus, C.E.; Wedemeyer, H.; Kraft, A.R.M.; Cornberg, M. Impact of HBsAg and HBcrAg levels on phenotype and function of HBV-specific T cells in patients with chronic hepatitis B virus infection. Gut 2021, 71, 2300–2312. [Google Scholar] [CrossRef]
- Muller, L.; Di Benedetto, S.; Pawelec, G. The Immune System and Its Dysregulation with Aging. Subcell. Biochem. 2019, 91, 21–43. [Google Scholar] [CrossRef]
- Zu, J.; Zhuang, G.; Liang, P.; Cui, F.; Wang, F.; Zheng, H.; Liang, X. Estimating age-related incidence of HBsAg seroclearance in chronic hepatitis B virus infections of China by using a dynamic compartmental model. Sci. Rep. 2017, 7, 2912. [Google Scholar] [CrossRef]
- Xiong, S.; Zhu, D.; Liang, B.; Li, M.; Pan, W.; He, J.; Wang, H.; Sutter, K.; Dittmer, U.; Lu, M.; et al. Longitudinal characterization of phenotypic profile of T cells in chronic hepatitis B identifies immune markers associated with HBsAg loss. EBioMedicine 2021, 69, 103464. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4(+) T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Lui, P.P.; Cho, I.; Ali, N. Tissue regulatory T cells. Immunology 2020, 161, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Raziorrouh, B.; Heeg, M.; Kurktschiev, P.; Schraut, W.; Zachoval, R.; Wendtner, C.; Wachtler, M.; Spannagl, M.; Denk, G.; Ulsenheimer, A.; et al. Inhibitory phenotype of HBV-specific CD4+ T-cells is characterized by high PD-1 expression but absent coregulation of multiple inhibitory molecules. PLoS ONE 2014, 9, e105703. [Google Scholar] [CrossRef] [PubMed]
- Ferrando-Martinez, S.; Bennett, A.S.; Lino, E.; Gehring, A.J.; Feld, J.; Janssen, H.L.A.; Robbins, S.H. Functional Exhaustion of HBV-Specific CD8 T Cells Impedes PD-L1 Blockade Efficacy in Chronic HBV Infection. Front. Immunol. 2021, 12, 648420. [Google Scholar] [CrossRef]
- Jiang, D.; Chen, C.; Yan, D.; Zhang, X.; Liu, X.; Yan, D.; Cui, D.; Yang, S. Exhausted phenotype of circulating CD8(+) T cell subsets in hepatitis B virus carriers. BMC Immunol. 2022, 23, 18. [Google Scholar] [CrossRef]
- Heim, K.; Binder, B.; Sagar; Wieland, D.; Hensel, N.; Llewellyn-Lacey, S.; Gostick, E.; Price, D.A.; Emmerich, F.; Vingerhoet, H.; et al. TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut 2020, 70, 1550–1560. [Google Scholar] [CrossRef]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-specific CD8+ T cell function by PD-1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef]
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell Mol. Immunol. 2019, 16, 634–643. [Google Scholar] [CrossRef]
- Schreiner, D.; King, C.G. CD4+ Memory T Cells at Home in the Tissue: Mechanisms for Health and Disease. Front. Immunol. 2018, 9, 2394. [Google Scholar] [CrossRef]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e1024. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Sausen, D.G.; Reed, K.M.; Bhutta, M.S.; Gallo, E.S.; Borenstein, R. Evasion of the Host Immune Response by Betaherpesviruses. Int. J. Mol. Sci. 2021, 22, 7503. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Fernando, K.; Ni, H.; Weissman, D. HIV envelope suppresses CD4+ T cell activation independent of T regulatory cells. J. Immunol. 2008, 180, 5593–5600. [Google Scholar] [CrossRef]
- Kim, J.H.; Ghosh, A.; Ayithan, N.; Romani, S.; Khanam, A.; Park, J.J.; Rijnbrand, R.; Tang, L.; Sofia, M.J.; Kottilil, S.; et al. Circulating serum HBsAg level is a biomarker for HBV-specific T and B cell responses in chronic hepatitis B patients. Sci. Rep. 2020, 10, 1835. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Lu, J.; Wu, J.; Xie, F.; Tian, J.; Tang, X.; Guo, H.; Ma, J.; Xu, P.; Mao, L.; Xu, H.; et al. CD4(+) T Cell-Released Extracellular Vesicles Potentiate the Efficacy of the HBsAg Vaccine by Enhancing B Cell Responses. Adv. Sci. 2019, 6, 1802219. [Google Scholar] [CrossRef]
- Mintz, M.A.; Cyster, J.G. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 2020, 296, 48–61. [Google Scholar] [CrossRef]
- Huang, C. Germinal Center Reaction. Adv. Exp. Med. Biol. 2020, 1254, 47–53. [Google Scholar] [CrossRef]
- Wang, X.; Dong, Q.; Li, Q.; Li, Y.; Zhao, D.; Sun, J.; Fu, J.; Meng, F.; Lin, H.; Luan, J.; et al. Dysregulated Response of Follicular Helper T Cells to Hepatitis B Surface Antigen Promotes HBV Persistence in Mice and Associates with Outcomes of Patients. Gastroenterology 2018, 154, 2222–2236. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef] [PubMed]
- Will, J.P.; Hirani, D.; Thielen, F.; Klein, F.; Vohlen, C.; Dinger, K.; Dotsch, J.; Alejandre Alcazar, M.A. Strain-dependent effects on lung structure, matrix remodeling, and Stat3/Smad2 signaling in C57BL/6N and C57BL/6J mice after neonatal hyperoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R169–R181. [Google Scholar] [CrossRef] [PubMed]
- Fergusson, G.; Ethier, M.; Guevremont, M.; Chretien, C.; Attane, C.; Joly, E.; Fioramonti, X.; Prentki, M.; Poitout, V.; Alquier, T. Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Mol. Metab. 2014, 3, 848–854. [Google Scholar] [CrossRef]
- Sturm, M.; Becker, A.; Schroeder, A.; Bilkei-Gorzo, A.; Zimmer, A. Effect of chronic corticosterone application on depression-like behavior in C57BL/6N and C57BL/6J mice. Genes Brain Behav. 2015, 14, 292–300. [Google Scholar] [CrossRef]
- Huang, L.R.; Wu, H.L.; Chen, P.J.; Chen, D.S. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 17862–17867. [Google Scholar] [CrossRef]
- Jacobi, F.J.; Wild, K.; Smits, M.; Zoldan, K.; Csernalabics, B.; Flecken, T.; Lang, J.; Ehrenmann, P.; Emmerich, F.; Hofmann, M.; et al. OX40 stimulation and PD-L1 blockade synergistically augment HBV-specific CD4 T cells in patients with HBeAg-negative infection. J. Hepatol. 2019, 70, 1103–1113. [Google Scholar] [CrossRef]
- Khanam, A.; Ayithan, N.; Tang, L.; Poonia, B.; Kottilil, S. IL-21-Deficient T Follicular Helper Cells Support B Cell Responses Through IL-27 in Patients With Chronic Hepatitis B. Front. Immunol. 2020, 11, 599648. [Google Scholar] [CrossRef]
- Zotos, D.; Coquet, J.M.; Zhang, Y.; Light, A.; D’Costa, K.; Kallies, A.; Corcoran, L.M.; Godfrey, D.I.; Toellner, K.M.; Smyth, M.J.; et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 2010, 207, 365–378. [Google Scholar] [CrossRef]
- Vijayan, D.; Redzwan, N.M.; Avery, D.T.; Wirasinha, R.C.; Brink, R.; Walters, G.; Adelstein, S.; Kobayashi, M.; Gray, P.; Elliott, M.; et al. IL-27 Directly Enhances Germinal Center B Cell Activity and Potentiates Lupus in Sanroque Mice. J. Immunol. 2016, 197, 3008–3017. [Google Scholar] [CrossRef]
- Ayithan, N.; Tang, L.; Tan, S.K.; Chen, D.; Wallin, J.J.; Fletcher, S.P.; Kottilil, S.; Poonia, B. Follicular Helper T (TFH) Cell Targeting by TLR8 Signaling For Improving HBsAg-Specific B Cell Response In Chronic Hepatitis B Patients. Front. Immunol. 2021, 12, 735913. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, E.; Gao, S.; Xiong, Y.; Lu, M. Toward a Functional Cure for Hepatitis B: The Rationale and Challenges for Therapeutic Targeting of the B Cell Immune Response. Front. Immunol. 2019, 10, 2308. [Google Scholar] [CrossRef] [PubMed]
- Ponde, R.A.A. Expression and detection of anti-HBs antibodies after hepatitis B virus infection or vaccination in the context of protective immunity. Arch. Virol. 2019, 164, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Barcena, R.; Moraleda, G.; Moreno, J.; Martin, M.D.; de Vicente, E.; Nuno, J.; Mateos, M.L.; del Campo, S. Prevention of de novo HBV infection by the presence of anti-HBs in transplanted patients receiving core antibody-positive livers. World J. Gastroenterol. 2006, 12, 2070–2074. [Google Scholar] [CrossRef]
- Jeon, J.W.; Kim, S.M.; Cho, H.; Baek, C.H.; Kim, H.; Shin, S.; Kim, Y.H.; Han, D.J.; Kim, S.B. Presence of Hepatitis B Surface Antibody in Addition to Hepatitis B Core Antibody Confers Protection Against Hepatitis B Virus Infection in Hepatitis B Surface Antigen-negative Patients Undergoing Kidney Transplantation. Transplantation 2018, 102, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Le Bert, N.; Salimzadeh, L.; Gill, U.S.; Dutertre, C.A.; Facchetti, F.; Tan, A.; Hung, M.; Novikov, N.; Lampertico, P.; Fletcher, S.P.; et al. Comparative characterization of B cells specific for HBV nucleocapsid and envelope proteins in patients with chronic hepatitis B. J. Hepatol. 2020, 72, 34–44. [Google Scholar] [CrossRef]
- Burton, A.R.; Pallett, L.J.; McCoy, L.E.; Suveizdyte, K.; Amin, O.E.; Swadling, L.; Alberts, E.; Davidson, B.R.; Kennedy, P.T.; Gill, U.S.; et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J. Clin. Investig. 2018, 128, 4588–4603. [Google Scholar] [CrossRef]
- Portugal, S.; Obeng-Adjei, N.; Moir, S.; Crompton, P.D.; Pierce, S.K. Atypical memory B cells in human chronic infectious diseases: An interim report. Cell Immunol. 2017, 321, 18–25. [Google Scholar] [CrossRef]
- Hehle, V.; Beretta, M.; Bourgine, M.; Ait-Goughoulte, M.; Planchais, C.; Morisse, S.; Vesin, B.; Lorin, V.; Hieu, T.; Stauffer, A.; et al. Potent human broadly neutralizing antibodies to hepatitis B virus from natural controllers. J. Exp. Med. 2020, 217, 10. [Google Scholar] [CrossRef]
- Salimzadeh, L.; Le Bert, N.; Dutertre, C.A.; Gill, U.S.; Newell, E.W.; Frey, C.; Hung, M.; Novikov, N.; Fletcher, S.; Kennedy, P.T.; et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J. Clin. Investig. 2018, 128, 4573–4587. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Rydell, G.E.; Prakash, K.; Norder, H.; Lindh, M. Hepatitis B surface antigen on subviral particles reduces the neutralizing effect of anti-HBs antibodies on hepatitis B viral particles in vitro. Virology 2017, 509, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.F. Detecting hepatitis B surface antigen mutants. Emerg. Infect. Dis. 2006, 12, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Romano, L.; Paladini, S.; Galli, C.; Raimondo, G.; Pollicino, T.; Zanetti, A.R. Hepatitis B vaccination. Hum. Vaccin. Immunother. 2015, 11, 53–57. [Google Scholar] [CrossRef]
- Maillard, P.; Pillot, J. At least three epitopes are recognized by the human repertoire in the hepatitis B virus group a antigen inducing protection; possible consequences for seroprevention and serodiagnosis. Res. Virol. 1998, 149, 153–161. [Google Scholar] [CrossRef]
- Golsaz-Shirazi, F.; Mohammadi, H.; Amiri, M.M.; Khoshnoodi, J.; Kardar, G.A.; Jeddi-Tehrani, M.; Shokri, F. Localization of immunodominant epitopes within the "a" determinant of hepatitis B surface antigen using monoclonal antibodies. Arch. Virol. 2016, 161, 2765–2772. [Google Scholar] [CrossRef]
- Kaymakoglu, S.; Baran, B.; Onel, D.; Badur, S.; Atamer, T.; Akyuz, F. Acute hepatitis B due to immune-escape mutations in a naturally immune patient. Acta Gastroenterol. Belg. 2014, 77, 262–265. [Google Scholar]
- Cooreman, M.P.; Leroux-Roels, G.; Paulij, W.P. Vaccine- and hepatitis B immune globulin-induced escape mutations of hepatitis B virus surface antigen. J. Biomed. Sci. 2001, 8, 237–247. [Google Scholar] [CrossRef]
- Hossain, M.G.; Mahmud, M.M.; Nazir, K.; Ueda, K. PreS1 Mutations Alter the Large HBsAg Antigenicity of a Hepatitis B Virus Strain Isolated in Bangladesh. Int. J. Mol. Sci. 2020, 21, 546. [Google Scholar] [CrossRef]
- Mangold, C.M.; Unckell, F.; Werr, M.; Streeck, R.E. Secretion and antigenicity of hepatitis B virus small envelope proteins lacking cysteines in the major antigenic region. Virology 1995, 211, 535–543. [Google Scholar] [CrossRef]
- Fedorowicz-Stronska, O.; Kapusta, J.; Czyz, M.; Kaczmarek, M.; Pniewski, T. Immunogenicity of parenterally delivered plant-derived small and medium surface antigens of hepatitis B virus. Plant Cell Rep. 2016, 35, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Chun, T.W.; Fauci, A.S. Pathogenic mechanisms of HIV disease. Annu. Rev. Pathol. 2011, 6, 223–248. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Joshua, J.M.; Mathews, S.M. Chemotherapy-induced neutropenia among breast Cancer patients in a tertiary care hospital: Risk and consequences. J. Oncol. Pharm. Pract. 2022, 10781552221074004. [Google Scholar] [CrossRef]
- Kochhar, G.S.; Mohan, B.P.; Khan, S.R.; Chandan, S.; Kassab, L.L.; Ponnada, S.; Desai, A.; Caldera, F.; Dulai, P.S.; Farraye, F.A. Hepatitis-B Vaccine Response in Inflammatory Bowel Disease Patients: A Systematic Review and Meta-analysis. Inflamm. Bowel Dis. 2021, 27, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Feuerherd, M.K.F. Altered HBV-Specific T-Cell Immunity in HBV/HIV Co-Infection and Chronic Hepatitis B. Ph.D. Thesis, Technische Universität München, München, Germany, 2021. [Google Scholar]
- Loomba, R.; Liang, T.J. Hepatitis B Reactivation Associated With Immune Suppressive and Biological Modifier Therapies: Current Concepts, Management Strategies, and Future Directions. Gastroenterology 2017, 152, 1297–1309. [Google Scholar] [CrossRef]
- Zhou, K.; Terrault, N. Management of hepatitis B in special populations. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 311–320. [Google Scholar] [CrossRef]
- Sarmati, L.; Malagnino, V. HBV Infection in HIV-Driven Immune Suppression. Viruses 2019, 11, 1077. [Google Scholar] [CrossRef]
- Chihota, B.V.; Wandeler, G.; Chilengi, R.; Mulenga, L.; Chung, R.T.; Bhattacharya, D.; Egger, M.; Vinikoor, M.J. High Rates of Hepatitis B Virus (HBV) Functional Cure Among Human Immunodeficiency Virus-HBV Coinfected Patients on Antiretroviral Therapy in Zambia. J. Infect. Dis. 2020, 221, 218–222. [Google Scholar] [CrossRef]
- Yeo, Y.H.; Ho, H.J.; Yang, H.I.; Tseng, T.C.; Hosaka, T.; Trinh, H.N.; Kwak, M.S.; Park, Y.M.; Fung, J.Y.Y.; Buti, M.; et al. Factors Associated With Rates of HBsAg Seroclearance in Adults With Chronic HBV Infection: A Systematic Review and Meta-analysis. Gastroenterology 2019, 156, 635–646.e639. [Google Scholar] [CrossRef]
- Lok, A.S.; Zoulim, F.; Dusheiko, G.; Ghany, M.G. Hepatitis B cure: From discovery to regulatory approval. Hepatology 2017, 66, 1296–1313. [Google Scholar] [CrossRef]
- Gantner, P.; Cotte, L.; Allavena, C.; Bani-Sadr, F.; Huleux, T.; Duvivier, C.; Valantin, M.A.; Jacomet, C.; Joly, V.; Cheret, A.; et al. Higher rates of HBsAg clearance with tenofovir-containing therapy in HBV/HIV co-infection. PLoS ONE 2019, 14, e0215464. [Google Scholar] [CrossRef] [PubMed]
- Jaroszewicz, J.; Reiberger, T.; Meyer-Olson, D.; Mauss, S.; Vogel, M.; Ingiliz, P.; Payer, B.A.; Stoll, M.; Manns, M.P.; Schmidt, R.E.; et al. Hepatitis B surface antigen concentrations in patients with HIV/HBV co-infection. PLoS ONE 2012, 7, e43143. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Li, N.; Chen, X.; Zhang, T.; Li, H.; Li, W.; Huang, X.; Liu, Z.; Zhang, Y.; Wu, H. Acute HIV infection is beneficial for controlling chronic hepatitis B. Clin. Infect. Dis. 2015, 60, 128–134. [Google Scholar] [CrossRef][Green Version]
- Song, T.; Li, L.; Su, B.; Liu, L.; Liu, Y.; Yang, X.; Zhang, Q.; Guo, N.; Zhang, T.; Sun, G.; et al. NKG2C+ natural killer cell function improves the control of HBV replication in individuals with acute HIV infection coinfected with HBV. Medicine 2020, 99, e20073. [Google Scholar] [CrossRef]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Duchler, M.; Offterdinger, M.; Holzmuller, H.; Lipp, J.; Chu, C.T.; Aschauer, B.; Bach, F.H.; Hofer, E. NKG2-C is a receptor on human natural killer cells that recognizes structures on K562 target cells. Eur. J. Immunol. 1995, 25, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.C.; Nguyen, T.H.O.; Harpur, C.M.; Stankovic, S.; Kanagarajah, A.R.; Koutsakos, M.; Saunders, P.M.; Cai, Z.; Gray, J.A.; Widjaja, J.M.L.; et al. Natural killer cell receptors regulate responses of HLA-E-restricted T cells. Sci. Immunol. 2021, 6, eabe9057. [Google Scholar] [CrossRef]
- Chang, Y.; Jeong, S.W.; Jang, J.Y. Hepatitis B Virus Reactivation Associated With Therapeutic Interventions. Front. Med. 2021, 8, 770124. [Google Scholar] [CrossRef]
- Kusumoto, S.; Arcaini, L.; Hong, X.; Jin, J.; Kim, W.S.; Kwong, Y.L.; Peters, M.G.; Tanaka, Y.; Zelenetz, A.D.; Kuriki, H.; et al. Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 2019, 133, 137–146. [Google Scholar] [CrossRef]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef]
- Sehn, L.H.; Martelli, M.; Trneny, M.; Liu, W.; Bolen, C.R.; Knapp, A.; Sahin, D.; Sellam, G.; Vitolo, U. A randomized, open-label, Phase III study of obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-Cell lymphoma: Final analysis of GOYA. J. Hematol. Oncol. 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Chen, C.; Fang, W.; Cai, X.; Zhang, X.; Zhao, M.; Zhang, B.; Jiang, W.; Lin, Z.; et al. Hepatitis B virus reactivation in cancer patients with positive Hepatitis B surface antigen undergoing PD-1 inhibition. J. Immunother. Cancer 2019, 7, 322. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Altstetter, S.M.; Protzer, U. Innate immune recognition and modulation in hepatitis D virus infection. World J. Gastroenterol. 2020, 26, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Landahl, J.; Bockmann, J.H.; Scheurich, C.; Ackermann, C.; Matzat, V.; Heide, J.; Nuurei, T.; D’Antonio, G.; von Felden, J.; Sette, A.; et al. Detection of a Broad Range of Low-Level Major Histocompatibility Complex Class II-Restricted, Hepatitis Delta Virus (HDV)-Specific T-Cell Responses Regardless of Clinical Status. J. Infect. Dis. 2019, 219, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Szeto, C.; Gras, S. The pockets guide to HLA class I molecules. Biochem. Soc. Trans. 2021, 49, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Karimzadeh, H.; Kiraithe, M.M.; Kosinska, A.D.; Glaser, M.; Fiedler, M.; Oberhardt, V.; Alizei, E.S.; Hofmann, M.; Mok, J.Y.; Nguyen, M.; et al. Amino Acid Substitutions within HLA-B*27-Restricted T Cell Epitopes Prevent Recognition by Hepatitis Delta Virus-Specific CD8(+) T Cells. J. Virol. 2018, 92, e01891-17. [Google Scholar] [CrossRef]
- Schirdewahn, T.; Grabowski, J.; Sekyere, S.O.; Bremer, B.; Wranke, A.; Lunemann, S.; Schlaphoff, V.; Kirschner, J.; Hardtke, S.; Manns, M.P.; et al. The Third Signal Cytokine Interleukin 12 Rather Than Immune Checkpoint Inhibitors Contributes to the Functional Restoration of Hepatitis D Virus-Specific T Cells. J. Infect. Dis. 2017, 215, 139–149. [Google Scholar] [CrossRef]
- Karimzadeh, H.; Kiraithe, M.M.; Oberhardt, V.; Alizei, E.S.; Bockmann, J.; Zur Wiesch, J.S.; Budeus, B.; Hoffmann, D.; Wedemeyer, H.; Cornberg, M.; et al. Mutations in Hepatitis D Virus Allow It to Escape Detection by CD8(+) T Cells and Evolve at the Population Level. Gastroenterology 2019, 156, 1820–1833. [Google Scholar] [CrossRef]
- Kefalakes, H.; Koh, C.; Sidney, J.; Amanakis, G.; Sette, A.; Heller, T.; Rehermann, B. Hepatitis D Virus-Specific CD8(+) T Cells Have a Memory-Like Phenotype Associated With Viral Immune Escape in Patients With Chronic Hepatitis D Virus Infection. Gastroenterology 2019, 156, 1805–1819.e1809. [Google Scholar] [CrossRef]
- Mauch, C.; Grimm, C.; Meckel, S.; Wands, J.R.; Blum, H.E.; Roggendorf, M.; Geissler, M. Induction of cytotoxic T lymphocyte responses against hepatitis delta virus antigens which protect against tumor formation in mice. Vaccine 2001, 20, 170–180. [Google Scholar] [CrossRef]
- Fiedler, M.; Lu, M.; Siegel, F.; Whipple, J.; Roggendorf, M. Immunization of woodchucks (Marmota monax) with hepatitis delta virus DNA vaccine. Vaccine 2001, 19, 4618–4626. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Roggendorf, M. Vaccination against hepatitis delta virus infection: Studies in the woodchuck (Marmota monax) model. Intervirology 2001, 44, 154–161. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Combating Hepatitis B and C to Reach Elimination by 2030: Advocacy Brief. Available online: https://apps.who.int/iris/handle/10665/206453 (accessed on 18 October 2021).
- Center for Drug Evaluation and Research. Chronic Hepatitis B Virus Infection: Developing Drugs for Treatment Guidance for Industry. 2022. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chronic-hepatitis-b-virus-infection-developing-drugs-treatment (accessed on 12 November 2022).
- Nayagam, S.; Thursz, M.; Sicuri, E.; Conteh, L.; Wiktor, S.; Low-Beer, D.; Hallett, T.B. Requirements for global elimination of hepatitis B: A modelling study. Lancet Infect. Dis. 2016, 16, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Table 1. Recommended Child and Adolescent Immunization Schedule for Ages 18 Years or Younger, United States. 2021. Available online: https://www.cdc.gov/vaccines/schedules/hcp/imz/child-adolescent.html (accessed on 23 October 2021).
- Centers for Disease Control and Prevention. Hepatitis B Vaccine Information Statement. Available online: https://www.cdc.gov/vaccines/hcp/vis/vis-statements/hep-b.html (accessed on 23 October 2021).
- Stevens, C.E.; Taylor, P.E.; Tong, M.J.; Toy, P.T.; Vyas, G.N.; Nair, P.V.; Weissman, J.Y.; Krugman, S. Yeast-recombinant hepatitis B vaccine. Efficacy with hepatitis B immune globulin in prevention of perinatal hepatitis B virus transmission. JAMA 1987, 257, 2612–2616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhou, X.; Zhou, Y.H. Hepatitis B vaccine development and implementation. Hum. Vaccin. Immunother. 2020, 16, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Hodgens, A.; Marathi, R. Hepatitis B Vaccine. In StatPearls; NCBI Bookshelf: Bethesda, MD, USA, 2021. [Google Scholar]
- Aspinall, E.J.; Hawkins, G.; Fraser, A.; Hutchinson, S.J.; Goldberg, D. Hepatitis B prevention, diagnosis, treatment and care: A review. Occup. Med. 2011, 61, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Postexposure Prophylaxis Treatment of Hepatitis B (HBV). Available online: https://www.cdc.gov/hepatitis/hbv/pep.htm (accessed on 28 October 2021).
- European Association for the Study of the Liver. Electronic address, e.e.e.; European Association for the Study of the, L. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Hepatitis B Questions and Answers for Health Professionals. Available online: https://www.cdc.gov/hepatitis/hbv/hbvfaq.htm (accessed on 1 November 2021).
- Centers for Disease Control and Prevention. Hepatitis D-Faqs and Laboratory Testing Requests. Available online: https://www.cdc.gov/hepatitis/hdv/index.htm (accessed on 1 November 2021).
- Pierra Rouviere, C.; Dousson, C.B.; Tavis, J.E. HBV replication inhibitors. Antiviral Res. 2020, 179, 104815. [Google Scholar] [CrossRef]
- Yang, S.C.; Lee, C.M.; Hu, T.H.; Wang, J.H.; Lu, S.N.; Hung, C.H.; Changchien, C.S.; Chen, C.H. Virological response to entecavir reduces the risk of liver disease progression in nucleos(t)ide analogue-experienced HBV-infected patients with prior resistant mutants. J. Antimicrob. Chemother. 2013, 68, 2154–2163. [Google Scholar] [CrossRef]
- Papatheodoridis, G.V.; Chan, H.L.; Hansen, B.E.; Janssen, H.L.; Lampertico, P. Risk of hepatocellular carcinoma in chronic hepatitis B: Assessment and modification with current antiviral therapy. J. Hepatol. 2015, 62, 956–967. [Google Scholar] [CrossRef]
- Dandri, M.; Petersen, J. cccDNA Maintenance in Chronic Hepatitis B-Targeting the Matrix of Viral Replication. Infect. Drug Resist. 2020, 13, 3873–3886. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.Y.; Wong, V.W.; Wong, G.L.; Yip, T.C. Towards HBV functional cure-do we have a crystal ball for that? Clin. Mol. Hepatol. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Dusheiko, G. Will we need novel combinations to cure HBV infection? Liver Int. 2020, 40 (Suppl. S1), 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hershkovich, L.; Shekhtman, L.; Bazinet, M.; Pantea, V.; Placinta, G.; Moscalu, I.; Cotler, S.J.; Vaillant, A.; Dahari, H. Rapid monophasic HBsAg decline during NAP-based therapy predicts functional cure. Hepatology 2021, 74 (Suppl. S1), 514A–515A. [Google Scholar]
- Farci, P.; Niro, G.A. Current and Future Management of Chronic Hepatitis D. Gastroenterol. Hepatol. 2018, 14, 342–351. [Google Scholar]
- Deterding, K.; Wedemeyer, H. Beyond Pegylated Interferon-Alpha: New Treatments for Hepatitis Delta. AIDS Rev. 2019, 21, 126–134. [Google Scholar] [CrossRef]
- Yardeni, D.; Heller, T.; Koh, C. Chronic hepatitis D-What is changing? J. Viral Hepat. 2022, 29, 240–251. [Google Scholar] [CrossRef]
- Giersch, K.; Homs, M.; Volz, T.; Helbig, M.; Allweiss, L.; Lohse, A.W.; Petersen, J.; Buti, M.; Pollicino, T.; Sureau, C.; et al. Both interferon alpha and lambda can reduce all intrahepatic HDV infection markers in HBV/HDV infected humanized mice. Sci. Rep. 2017, 7, 3757. [Google Scholar] [CrossRef]
- Abdrakhman, A.; Ashimkhanova, A.; Almawi, W.Y. Effectiveness of pegylated interferon monotherapy in the treatment of chronic hepatitis D virus infection: A meta-analysis. Antiviral Res. 2021, 185, 104995. [Google Scholar] [CrossRef]
- Canini, L.; Koh, C.; Cotler, S.J.; Uprichard, S.L.; Winters, M.A.; Han, M.A.T.; Kleiner, D.E.; Idilman, R.; Yurdaydin, C.; Glenn, J.S.; et al. Pharmacokinetics and pharmacodynamics modeling of lonafarnib in patients with chronic hepatitis delta virus infection. Hepatol. Commun. 2017, 1, 288–292. [Google Scholar] [CrossRef]
- Cardozo-Ojeda, F.; Duehren, S.; Hamid, S.; Lurie, Y.; Gane, E.; Bader, N.; Yardeni, D.; Nevo-Shor, A.; Channa, S.M.; Mawani, M.; et al. Mathematical modeling of HDV RNA kinetics suggests high peginterferon-lambda efficacy in blocking viral production: Insights from the LIMT-1 study. J. Hepatol. 2022, 77 (Suppl. S1), S859. [Google Scholar] [CrossRef]
- Shekhtman, L.; Cotler, S.J.; PLoss, A.; Dahari, H. Mathematical modeling suggests that entry-inhibitor bulevirtide may interfere with hepatitis D virus clearance from circulation. J. Hepatol. 2022, 76, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Bartenschlager, R.; Kubitz, R.; Zoulim, F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014, 147, 48–64. [Google Scholar] [CrossRef]
- Lampertico, P.; Roulot, D.; Wedemeyer, H. Bulevirtide with or without pegIFNalpha for patients with compensated chronic hepatitis delta: From clinical trials to real-world studies. J. Hepatol. 2022, 77, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.; Heller, T.; Glenn, J.S. Pathogenesis of and New Therapies for Hepatitis D. Gastroenterology 2019, 156, 461–476.e461. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, M.; Pantea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Le Gal, F.; Gordien, E.; Krawczyk, A.; et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): A non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 877–889. [Google Scholar] [CrossRef]
- Razavi, H. Global Epidemiology of Viral Hepatitis. Gastroenterol. Clin. N. Am. 2020, 49, 179–189. [Google Scholar] [CrossRef]
- Borenstein, R.; Hanson, B.A.; Markosyan, R.M.; Gallo, E.S.; Narasipura, S.D.; Bhutta, M.; Shechter, O.; Lurain, N.S.; Cohen, F.S.; Al-Harthi, L.; et al. Ginkgolic acid inhibits fusion of enveloped viruses. Sci. Rep. 2020, 10, 4746. [Google Scholar] [CrossRef]
- Bhutta, M.S.; Shechter, O.; Gallo, E.S.; Martin, S.D.; Jones, E.; Doncel, G.F.; Borenstein, R. Ginkgolic Acid Inhibits Herpes Simplex Virus Type 1 Skin Infection and Prevents Zosteriform Spread in Mice. Viruses 2021, 13, 86. [Google Scholar] [CrossRef]
- Bhutta, M.S.; Sausen, D.G.; Gallo, E.S.; Dahari, H.; Doncel, G.F.; Borenstein, R. Ginkgolic Acid Inhibits Coronavirus Strain 229E Infection of Human Epithelial Lung Cells. Pharmaceuticals 2021, 14, 980. [Google Scholar] [CrossRef]
- Liu, J.; Liang, W.; Jing, W.; Liu, M. Countdown to 2030: Eliminating hepatitis B disease, China. Bull. World Health Organ. 2019, 97, 230–238. [Google Scholar] [CrossRef]
- Woodring, J.; Pastore, R.; Brink, A.; Ishikawa, N.; Takashima, Y.; Tohme, R.A. Progress Toward Hepatitis B Control and Elimination of Mother-to-Child Transmission of Hepatitis B Virus-Western Pacific Region, 2005-2017. MMWR Morb. Mortal Wkly. Rep. 2019, 68, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Khetsuriani, N.; Mosina, L.; Van Damme, P.; Mozalevskis, A.; Datta, S.; Tohme, R.A. Progress Toward Hepatitis B Control-World Health Organization European Region, 2016-2019. MMWR Morb. Mortal Wkly. Rep. 2021, 70, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Breakwell, L.; Tevi-Benissan, C.; Childs, L.; Mihigo, R.; Tohme, R. The status of hepatitis B control in the African region. Pan. Afr. Med. J. 2017, 27, 17. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study|Viral Profile | HDV Dominant | HBV Dominant | HDV/HBV Equivalent * | HBeAg (+/−) | HDV RNA Assay ** | Reference |
|---|---|---|---|---|---|---|
| Individual Patient Data | ||||||
| Yurdaydin et al., 2018 | 17/20 | 0/20 | 3/20 | 3 (+) 12 (−) 5 (N/A) | In-house PCR. (LLoQ) 70 IU/mL and (LLoD) 50 IU/mL | [97] |
| Shekhtman et al., 2020 | 12/12 | 0/12 | 0/12 | 12 (−) | Robogene MK I LLoQ 3.26 log U/mL | [98] |
| Schaper et al., 2010 | 13/25 | 6/25 | 6/25 | 5 (+) 20 (−) | rt-RT-PCR in the LightCyclerTM system | [94] |
| Guedj et al., 2014 | 11/12 | 0/12 | 1/12 | 9 (−) 3 (+) | real-time PCR (external) (LLOQ): 100 (GE/mL) | [99] |
| Koh et al., 2015 | 8/9 | 0/9 | 1/9 | 9 (−) | qRT-PCR LLoD 70 IU/mL | [100] |
| Patient Data (Cohort Mean) | ||||||
| Castelnau et al., 2006 | 14 | NA | NA | All negative | RT-PCR 100 copies/mL | [101] |
| Manesis et al., 2007 | 53 | NA | NA | All negative | PCR TaqMan Universal 100 copies/mL of serum and the linearity of quantification ranged from 103 to 109 copies/mL. | [102] |
| Zachou et al., 2009 *** | 73 | NA | NA | (+) 12 (−) 68 | RT-PCR | [103] |
| Wedemeyer et al., 2011 | 90 | NA | NA | 14 (+) 76 (−) | TaqMan One Step PCR Master Mix LLOD of HDV RNA was 120 copies/mL of serum | [104] |
| Braga et al., 2014 (64 patients–only % given) | 56.3% | 3.1% | 40.6% | All non-reactive | qRT-PCR Sensitivity not listed. | [95] |
| Koh et al., 2015 | 14 | NA | NA | All negative | qRT-PCR LLoD 70 IU/mL | [100] |
| Bogomolov et al., 2016 | 8 | 0 | 16 | All Negative | Amplisense HDV-FL During treatment: qPCR: limit of detection (LOD) 15 copies/ml | [105] |
| Yurdaydin et al., 2007 | 39 | 0 | 0 | (+) 2 (−) 37 | RT-PCR Sensitivity: 100,000 copies/mL | [106] |
| Virus/Viral Component | Effect on Immune System | Reference |
|---|---|---|
| HBV | Increased inhibitory CD8+ receptors including PD-1, LAG-3 as well as inhibition of IFN-γ, IL-2 and TNF-α secretion | [127,140] |
| HBV | Increased frequency of central memory CD4+ T cells | [140] |
| HBV | Inhibition of germinal center formation and Tfh expansion | [146] |
| HBV | Inhibit Tfh secretion of IL-21 | [153] |
| HBV | Increased plasmablasts, plasma cells, regulatory B cells, and CD19+ B cells | [153] |
| HBV | Inhibition of HBsAg-specific B cell maturation | [161,165] |
| HBV | Decreased expression of genes involved in class switching, increased expression of genes in endothelial adhesion, homeostasis, apoptosis | [161] |
| HBV | Decrease antibody production | [161,162] |
| HBV | Immune Escape | [172,173] |
| HBV | Increased atypical memory B cells | [162,165] |
| High HBsAg | Elevated PD-1 on CD4+ T cells, and increased levels of CD4+ T cells with high PD-1 levels | [140] |
| High HBsAg | Fewer T cells secreting multiple types of cytokines | [140] |
| High HBsAg | higher PD-1 expression on EMRA T cells | [140] |
| High HBsAg | Increased expression of Inhibitory receptors (FcRL5, coexpression of 2B4 and PD-1) on CD4+ T cells | [140] |
| High HBsAg | Inhibit antibody efficacy | [167] |
| HBsAg | Increased number of T cells specific for viral envelope | [120] |
| HDV | Lack of B/T cell stimulation | [199,200,203,206,207,208] |
| HDV | Escape mutations | [202,204,205] |
| HDV | HLA and immune response | [202,204] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sausen, D.G.; Shechter, O.; Bietsch, W.; Shi, Z.; Miller, S.M.; Gallo, E.S.; Dahari, H.; Borenstein, R. Hepatitis B and Hepatitis D Viruses: A Comprehensive Update with an Immunological Focus. Int. J. Mol. Sci. 2022, 23, 15973. https://doi.org/10.3390/ijms232415973
Sausen DG, Shechter O, Bietsch W, Shi Z, Miller SM, Gallo ES, Dahari H, Borenstein R. Hepatitis B and Hepatitis D Viruses: A Comprehensive Update with an Immunological Focus. International Journal of Molecular Sciences. 2022; 23(24):15973. https://doi.org/10.3390/ijms232415973
Chicago/Turabian StyleSausen, Daniel G., Oren Shechter, William Bietsch, Zhenzhen Shi, Samantha M. Miller, Elisa S. Gallo, Harel Dahari, and Ronen Borenstein. 2022. "Hepatitis B and Hepatitis D Viruses: A Comprehensive Update with an Immunological Focus" International Journal of Molecular Sciences 23, no. 24: 15973. https://doi.org/10.3390/ijms232415973
APA StyleSausen, D. G., Shechter, O., Bietsch, W., Shi, Z., Miller, S. M., Gallo, E. S., Dahari, H., & Borenstein, R. (2022). Hepatitis B and Hepatitis D Viruses: A Comprehensive Update with an Immunological Focus. International Journal of Molecular Sciences, 23(24), 15973. https://doi.org/10.3390/ijms232415973