
Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives

 ,
,  and
and
Abstract
1. Introduction
2. Cancer, a Non-Healing Wound
3. Stromal Cells Are Recruited to the Battlefield by Cancer Cells
4. CAFs Biomarkers: A Mixed Bag
4.1. Cytoskeleton and Cytoplasmic Proteins
4.2. ECM-Related Components
4.3. Receptors and Membrane-Bound Proteins
4.4. Growth Factors and Cytokines
5. Fibroblasts: Induced and Inductors
6. CAFs and Tumour Immunity
7. Tumour Stroma and Therapeutic Resistance
8. Therapeutic Strategies
9. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-AZA | 5-Azacytidine |
| α-CTLA-4 | Alpha-Cytotoxic T-Lymphocyte-associated Protein 4 |
| α-PD-L1 | Alpha-Programmed death-ligand 1 |
| αSMA | Alpha Smooth Muscle Actin |
| AKT | Serine/Threonine Kinase |
| AP-1 | Activator protein 1 |
| Arg2 | Arginase 2 |
| Bcl-2 | B-Cell Lymphoma 2 |
| BM | Bone Marrow |
| BMP2 | Bone Morphogenetic Protein 2 |
| CAFs | Cancer-associated Fibroblasts |
| CAR | Chimeric Antigen Receptor |
| CAV-1 | Caveolin-1 |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| CCR2 | C-C Motif Chemokine Receptor 2 |
| Chi3L1 | Chitinase-3-like protein 1 |
| COXs | Cyclooxygenases |
| CRC | Colorectal Carcinoma |
| CTGF | Connective Tissue Growth Factor |
| CXCL | Chemokine (C-X-C motif) Ligand |
| CXCR | C-X-C Motif Chemokine Receptor |
| DNA | Deoxyribonucleic Acid |
| DTR | Diphtheria Toxin Receptor |
| E2 | 17β-Estradiol |
| ECM | Extracellular Matrix |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| EMAP II | Endothelial Monocyte Activating Polypeptide II |
| EMT | Epithelial-to-mesenchymal Transition |
| EndMT | Endothelial-to-mesenchymal Transition |
| EP 1-4 | E-type prostanoid receptors 1-4 |
| EPCs | Endothelial Progenitor Cells |
| ERK | Extracellular Signal-regulated Kinase |
| FACS | Fluorescence-activated Cell Sorting |
| FAFs | Fibrosis-associated Fibroblasts |
| FAK | Focal Adhesion Kinase |
| FAPα | Fibroblast Activation Protein-α |
| FASL | Fas Ligand |
| FGF | Fibroblast Growth Factor |
| FLI1 | Friend Leukemia Integration 1 Transcription Factor |
| FSP-1 | Fibroblast-specific Protein 1 |
| G1 | 1-(4-(6-bromobenzo [1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)-Ethanone |
| GEM | Gemcitabine |
| GFRs | Growth Factor Receptors |
| GLI | Glioma-associated Oncogene |
| GPER | G-Protein-coupled Estrogen Receptor |
| HA | Hyaluronic acid |
| HCC | Hepatocellular Carcinoma |
| HGF | Hepatocyte Growth Factor |
| HGSOC | High-grade Serous Ovarian Cancer |
| HIF1-α | Hypoxia Inducible Factor 1-alpha |
| HLA | Human Leukocyte Antigen |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| HPCs | Hematopoietic Progenitor Cells |
| HSF1 | Heat Shock Factor 1 |
| iCAFs | Inflammatory-like Cancer-associated Fibroblasts |
| ICAM | Intercellular Adhesion Molecule 1 |
| IDH3 | Isocitrate Dehydrogenase 3 |
| IDO1 | Indoleamine 2,3-Dioxygenase 1 |
| IGF | Insulin-like Growth Factor |
| IGFR | Insulin Growth Factor Receptor |
| IL | Interleukin |
| JAK/STAT | Janus kinase-Signal Transducer and Activator of Transcription |
| KLF5 | Kruppel-like Factor 5 |
| LCC | Lewis Lung Cancer |
| LPA | Lipoprotein A |
| MAF | Metastasis-associated Fibroblasts |
| MAPK | Mitogen-activated Protein Kinase |
| MDSCs | Myeloid-derived Suppressor Cells |
| MEK | Mitogen-activated Protein Kinase |
| MET | Methionine |
| MMPs | Matrix metalloproteinases |
| MSCs | Mesenchymal Stem Cells |
| mTOR | Mammalian Target of Rapamycin |
| myCAFs | Myofibroblast-like Cancer-associated Fibroblasts |
| NAFs | Normal Activated Fibroblasts |
| NF-κβ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve Growth Factor |
| NK | Natural Killer |
| NSCLC | Non-Small Cell Lung Carcinoma |
| OPN | Osteopontin |
| PD-L | Programmed Cell Death-ligand |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PDGF | Platelet-derived Growth Factor |
| PDGF-BB | Platelet-derived Growth Factor-BB |
| PDK1 | 3-Phosphoinositide protein kinase-1 |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphoinositide-3-kinase |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| POSTN | Periostin |
| PSCs | Pancreatic Stellate Cells |
| PTCH | Protein Patched Homolog |
| PTEN | Phosphatase and Tensin Homolog |
| ROCK | Rho-associated Protein Kinase |
| ROS | Reactive Oxygen Species |
| RNI | Reactive Nitrogen intermediates |
| SDF-1 | Stromal Cell-Derived Factor 1 |
| SHH | Sonic Hedgehog |
| Smo | Smoothened |
| SOCS1 | Suppressor Of Cytokine Signaling 1 |
| Sox2 | SRY-Box Transcription Factor 2 |
| SPARC | Secreted Protein Acidic and Cysteine Rich |
| SRF | Serum Response Factor |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAMs | Tumour-associated Macrophages |
| TCF | Ternary Complex Factor |
| TGF-β | Transforming Growth Factor beta |
| Tie1/2 | Tyrosine Kinase with Immunoglobulin-like and EGF-like Domains 1/2 |
| TIMP-1 | Tissue Inhibitor of Metalloproteinase-1 |
| TME | Tumour Microenvironment |
| TN-C | Tenascin-C |
| TNF | Tumor Necrosis Factor |
| VCAM | Vascular Cellular Adhesion Molecule |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor |
| VIM | Vimentin |
| Vit | Vitamine |
| WFDC1 | WAP Four-Disulfide Core Domain 1 |
| YAP1 | Yes-associated Protein-1 |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.; Bhakta, N.; Kessenbrock, K.; Prummel, K.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and Translational Impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed]
- Östman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth–bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.; Egeblad, M.; Evans, R.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Loeffler, M.; Krüger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef]
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, I.A.; Desmoulière, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenes. Tissue Repair 2012, 5, S5. [Google Scholar] [CrossRef]
- Eyden, B. Translational Medicine: The myofibroblast: Phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J. Cell. Mol. Med. 2007, 12, 22–37. [Google Scholar] [CrossRef]
- Rognoni, E.; Pisco, A.O.; Hiratsuka, T.; Sipilä, K.H.; Belmonte, J.M.; Mobasseri, S.A.; Philippeos, C.; Dilão, R.; Watt, F.M. Fibroblast state switching orchestrates dermal maturation and wound healing. Mol. Syst. Biol. 2018, 14, e8174. [Google Scholar] [CrossRef]
- Ko, U.H.; Choi, J.; Choung, J.; Moon, S.; Shin, J.H. Physicochemically tuned myofibroblasts for wound healing strategy. Sci. Rep. 2019, 9, 16070. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Yam, N.; Chen, C.; Elawad, K.; Hu, B.; Chen, Y. Force-dependent extracellular matrix remodeling by early-stage cancer cells alters diffusion and induces carcinoma-associated fibroblasts. Biomaterials 2020, 234, 119756. [Google Scholar] [CrossRef] [PubMed]
- Desmoulière, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar] [PubMed]
- Kankuri, E.; Cholujova, D.; Comajova, M.; Vaheri, A.; Bizik, J. Induction of hepatocyte growth factor/scatter factor by fibroblast clustering directly promotes tumor cell invasiveness. Cancer Res. 2005, 65, 9914–9922. [Google Scholar] [CrossRef]
- Trent, J.T.; Kirsner, R.S. Wounds and Malignancy. Adv. Ski. Wound Care 2003, 16, 31–34. [Google Scholar] [CrossRef]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Drev, D.; Bileck, A.; Erdem, Z.N.; Mohr, T.; Timelthaler, G.; Beer, A.; Gerner, C.; Marian, B. Proteomic profiling identifies markers for inflammation-related tumor–fibroblast interaction. Clin. Proteom. 2017, 14, 33. [Google Scholar] [CrossRef]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef]
- Kawanishi, S.; Hiraku, Y.; Pinlaor, S.; Ma, N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol. Chem. 2006, 387, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2020, 33, 127–148. [Google Scholar] [CrossRef]
- National Cancer Institute. Treating Cancer by Reducing Tumor-Related Inflammation. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2022/reducing-inflammation-to-treat-cancer (accessed on 25 September 2022).
- Smith, A.K.; Conneely, K.N.; Pace, T.W.; Mister, D.; Felger, J.C.; Kilaru, V.; Akel, M.J.; Vertino, P.M.; Miller, A.H.; Torres, M.A. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav. Immun. 2014, 38, 227–236. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Wang, H.M.; Hung, C.H.; Lu, S.N.; Chen, C.H.; Lee, C.M.; Hu, T.H.; Wang, J.H. Liver stiffness measurement as an alternative to fibrotic stage in risk assessment of hepatocellular carcinoma incidence for chronic hepatitis C patients. Liver Int. 2013, 33, 756–761. [Google Scholar] [CrossRef]
- Sangiovanni, A.; del Ninno, E.; Fasani, P.; de Fazio, C.; Ronchi, G.; Romeo, R.; Morabito, A.; de Franchis, R.; Colombo, M. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology 2004, 126, 1005–1014. [Google Scholar] [CrossRef]
- Trimis, G.; Chatzistamou, I.; Politi, K.; Kiaris, H.; Papavassiliou, A.G. Expression of p21 waf1/Cip1 in stromal fibroblasts of primary breast tumors. Hum. Mol. Genet. 2008, 17, 3596–3600. [Google Scholar] [CrossRef]
- Hu, M.; Peluffo, G.; Chen, H.; Gelman, R.; Schnitt, S.; Polyak, K. Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. 2009, 106, 3372–3377. [Google Scholar] [CrossRef]
- Linares, J.; Marín-Jiménez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and functions of CAFS secretome during cancer progression and therapy. Front. Cell Dev. Biol. 2021, 8, 621070. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.-W.; Che, Z.M.; Kim, J.; Kim, K.; Kim, K.-Y.; Williams, D.; Kim, J. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: A pivotal role of CCL7. Int. J. Cancer 2009, 127, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Yamamoto, Y.; Sakamoto, N.; Shimomura, I.; Kogure, A.; Kumazaki, M.; Yokoi, A.; Yashiro, M.; Kiyono, T.; Yanagihara, K.; et al. Cancer extracellular vesicles contribute to stromal heterogeneity by inducing chemokines in cancer-associated fibroblasts. Oncogene 2019, 38, 5566–5579. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014, 345, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Watt, D.M.; Morton, J.P. Heterogeneity in pancreatic cancer fibroblasts—tgfβ as a master regulator? Cancers 2021, 13, 4984. [Google Scholar] [CrossRef]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef]
- Brennen, W.N.; Denmeade, S.R.; Isaacs, J.T. Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr.-Relat. Cancer 2013, 20, R269–R290. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2018, 9, 282–301. [Google Scholar] [CrossRef]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin. Cancer Res. 2018, 25, 2194–2205. [Google Scholar] [CrossRef]
- Busch, S.; Acar, A.; Magnusson, Y.; Gregersson, P.; Rydén, L.; Landberg, G. TGF-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene 2013, 34, 27–38. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.-M.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin αvβ3 engagement and PI3K/Pakt/NFΚB signaling. Hepatology 2012, 55, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2013, 33, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef]
- Álvarez-Teijeiro, S.; García-Inclán, C.; Villaronga, M.; Casado, P.; Hermida-Prado, F.; Granda-Díaz, R.; Rodrigo, J.; Calvo, F.; del-Río-Ibisate, N.; Gandarillas, A.; et al. Factors secreted by cancer-associated fibroblasts that sustain cancer stem properties in head and neck squamous carcinoma cells as potential therapeutic targets. Cancers 2018, 10, 334. [Google Scholar] [CrossRef]
- Fordyce, C.; Fessenden, T.; Pickering, C.; Jung, J.; Singla, V.; Berman, H.; Tlsty, T. DNA damage drives an activin A–dependent induction of cyclooxygenase-2 in premalignant cells and lesions. Cancer Prev. Res. 2009, 3, 190–201. [Google Scholar] [CrossRef]
- Guo, X.; Oshima, H.; Kitmura, T.; Taketo, M.M.; Oshima, M. Stromal Fibroblasts Activated by Tumor Cells Promote Angiogenesis in Mouse Gastric Cancer. J. Biol. Chem. 2008, 283, 19864–19871. [Google Scholar] [CrossRef]
- Kellermann, M.G.; Sobral, L.M.; da Silva, S.D.; Zecchin, K.G.; Graner, E.; Lopes, M.A.; Kowalski, L.P.; Coletta, R.D. Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: Induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation. Oral Oncol. 2008, 44, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1–TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2014, 34, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, Y.; Wu, Q.; Xie, L.; Barwick, B.; Fu, C.; Li, X.; Wu, D.; Xia, S.; Chen, J.; et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat. Commun. 2021, 12, 1714. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, B.; Xiang, L.; Xia, S.; Kucuk, O.; Deng, X.; Boise, L.H.; Dong, J.T. TGF-β causes Docetaxel resistance in Prostate Cancer via the induction of Bcl-2 by acetylated KLF5 and Protein Stabilization. Theranostics 2020, 10, 7656–7670. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Li, Y.; Zhao, J.; Li, Q.; Yang, B.; Wang, Y.; Zhu, Z.; Sun, H.; Zhai, Z. Transforming growth factor-β1 contributes to oxaliplatin resistance in colorectal cancer via epithelial to mesenchymal transition. Oncol. Lett. 2017, 14, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Edward, K.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF-β signalling attenuates tumour response to PD-L1 checkpoint blockade by contributing to retention of T cells in the peritumoural stroma. Ann. Oncol. 2017, 28, xi30. [Google Scholar] [CrossRef]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef]
- Yeh, H.-W.; Lee, S.-S.; Chang, C.-Y.; Lang, Y.-D.; Jou, Y.-S. A new switch for TGFΒ in cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. A Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesismTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Philippeos, C.; Telerman, S.B.; Oules, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J. Investig. Derm. 2018, 138, 811–825. [Google Scholar] [CrossRef]
- Primac, I.; Maquoi, E.; Blacher, S.; Heljasvaara, R.; Van Deun, J.; Smeland, H.Y.; Canale, A.; Louis, T.; Stuhr, L.; Sounni, N.E.; et al. Stromal integrin alpha11 regulates PDGFR-beta signaling and promotes breast cancer progression. J. Clin. Investig. 2019, 130, 4609–4628. [Google Scholar] [CrossRef]
- Haubeiss, S.; Schmid, J.O.; Mürdter, T.E.; Sonnenberg, M.; Friedel, G.; van der Kuip, H.; Aulitzky, W.E. Dasatinib reverses cancer-associated fibroblasts (CAFs) from primary lung carcinomas to a phenotype comparable to that of normal fibroblasts. Mol. Cancer 2010, 9, 168. [Google Scholar] [CrossRef]
- Djurec, M.; Graña, O.; Lee, A.; Troulé, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martín-Díaz, L.; Burdiel, M.; et al. SAA3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef]
- Wilson, M.W.; Goldsmith, Z.; Coppess, W.; Gao, B.; McEwen, M.; Irvine, A.; Brennan, R.C.; Morales, V.M. PDGF-PDGFR Signaling Sustain Angiogenesis in an Autocrine and Paracrine Fashion in Retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2016, 57, 12. [Google Scholar]
- French, W.J.; Creemers, E.E.; Tallquist, M.D. Platelet-derived growth factor receptors direct vascular development independent of vascular smooth muscle cell function. Mol. Cell. Biol. 2008, 28, 5646–5657. [Google Scholar] [CrossRef]
- Levéen, P.; Pekny, M.; Gebre-Medhin, S.; Swolin, B.; Larsson, E.; Betsholtz, C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994, 8, 1875–1887. [Google Scholar] [CrossRef]
- Tejada, M.L.; Yu, L.; Dong, J.; Jung, K.; Meng, G.; Peale, F.V.; Frantz, G.D.; Hall, L.; Liang, X.; Gerber, H.P.; et al. Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin. Cancer Res. 2006, 12, 2676–2688. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Du, Q.; Jing, X.; He, X.; Wu, J.; Zhang, Y.; Morikawa, H.; Nakamura, M.; et al. Therapeutic paradigm of dual targeting VEGF and PDGF for effectively treating FGF-2 off-target tumors. Nat. Commun. 2020, 11, 3704. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Wagner, A.J.; Kindler, H.; Gelderblom, H.; Schöffski, P.; Bauer, S.; Hohenberger, P.; Kopp, H.G.; Lopez-Martin, J.A.; Peeters, M.; Reichardt, P.; et al. A phase II study of a human anti-PDGFRα monoclonal antibody (olaratumab, IMC-3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 541–546. [Google Scholar] [CrossRef]
- Wang, M.; Han, J.; Marcar, L.; Black, J.; Liu, Q.; Li, X.; Nagulapalli, K.; Sequist, L.V.; Mak, R.H.; Benes, C.H.; et al. Radiation resistance in kras-mutated lung cancer is enabled by stem-like properties mediated by an osteopontin–EGFR pathway. Cancer Res. 2017, 77, 2018–2028. [Google Scholar] [CrossRef]
- Ishibashi, M.; Neri, S.; Hashimoto, H.; Miyashita, T.; Yoshida, T.; Nakamura, Y.; Udagawa, H.; Kirita, K.; Matsumoto, S.; Umemura, S.; et al. CD200-positive cancer associated fibroblasts augment the sensitivity of Epidermal Growth Factor Receptor mutation-positive lung adenocarcinomas to EGFR Tyrosine kinase inhibitors. Sci. Rep. 2017, 7, 46662. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to Stromal Fibroblasts Induces Resistance of Lung Cancer to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef]
- Zhang, X.H.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef]
- Chen, Y.J.; Lin, C.Y.; Pan, S.H.; Chou, H.Y.; Chen, Y.J.; Chang, G.C.; Chu, W.C.; Lee, Y.M.; Lee, J.Y.; Lee, P.J.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed]
- Klarmann, G.J.; Hurt, E.M.; Mathews, L.A.; Zhang, X.; Duhagon, M.A.; Mistree, T.; Thomas, S.B.; Farrar, W.L. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin. Exp. Metastasis 2009, 26, 433–446. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.; Cook, M.; et al. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.C.; Tchou, J.; et al. Dickkopf-3 links HSF1 and yap/taz signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef]
- Moskovits, N.; Kalinkovich, A.; Bar, J.; Lapidot, T.; Oren, M. p53 Attenuates Cancer Cell Migration and Invasion through Repression of SDF-1/CXCL12 Expression in Stromal Fibroblasts. Cancer Res. 2006, 66, 10671–10676. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4 + stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J.; Huang, H.; Ye, M.; Li, X.; Wu, R.; Liu, H.; Song, Y. Metastasis-associated fibroblasts: An emerging target for metastatic cancer. Biomark. Res. 2021, 9, 47. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Eckert, F.; Schilbach, K.; Klumpp, L.; Bardoscia, L.; Sezgin, E.C.; Schwab, M.; Zips, D.; Huber, S.M. Potential role of CXCR4 targeting in the context of radiotherapy and immunotherapy of cancer. Front. Immunol. 2018, 9, 3018. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.P.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, N.; Yang, A.-G.; Sanders, D.E.; Strube, R.W.; Chen, S.-Y. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. 2004, 12, 84–89. [Google Scholar] [CrossRef]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef]
- Rhim, A.; Oberstein, P.; Thomas, D.; Mirek, E.; Palermo, C.; Sastra, S.; Dekleva, E.; Saunders, T.; Becerra, C.; Tattersall, I.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Chang, P.H.; Hwang-Verslues, W.W.; Chang, Y.C.; Chen, C.C.; Hsiao, M.; Jeng, Y.M.; Chang, K.-J.; Lee, E.Y.-H.; Shew, J.-Y.; Lee, W.-H.; et al. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/beta-catenin pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef]
- Marlow, R.; Strickland, P.; Lee, J.S.; Wu, X.; Pebenito, M.; Binnewies, M.; Le, E.K.; Moran, A.; Macias, H.; Cardiff, R.D.; et al. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008, 68, 7819–7827. [Google Scholar] [CrossRef]
- Alpízar-Alpízar, W.; Laerum, O.D.; Christensen, I.J.; Ovrebo, K.; Skarstein, A.; Høyer-Hansen, G.; Ploug, M.; Illemann, M. Tissue inhibitor of metalloproteinase-1 is confined to tumor-associated myofibroblasts and is increased with progression in gastric adenocarcinoma. J. Histochem. Cytochem. 2016, 64, 483–494. [Google Scholar] [CrossRef]
- Pape, J.; Magdeldin, T.; Stamati, K.; Nyga, A.; Loizidou, M.; Emberton, M.; Cheema, U. Cancer associated fibroblasts mediate cancer progression and remodel the tumouroid stroma. Br. J. Cancer 2020, 123, 1178–1190. [Google Scholar] [CrossRef]
- Duch, P.; Díaz-Valdivia, N.; Ikemori, R.; Gabasa, M.; Radisky, E.S.; Arshakyan, M.; Gea-Sorlí, S.; Mateu-Bosch, A.; Bragado, P.; Carrasco, J.L.; et al. Aberrant TIMP-1 overexpression in tumor-associated fibroblasts drives tumor progression through CD63 in lung adenocarcinoma. Matrix Biol. 2022, 111, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Hara, M.; Takahashi, H.; Shiga, K.; Hirokawa, T.; Maeda, Y.; Yanagita, T.; Ando, N.; Takasu, K.; Suzuki, T.; et al. Cancer cell-induced tissue inhibitor of metalloproteinase-1 secretion by cancer-associated fibroblasts promotes cancer cell migration. Oncol. Rep. 2022, 47, 112. [Google Scholar] [CrossRef] [PubMed]
- El-Tanani, M.K.; Campbell, F.C.; Crowe, P.; Erwin, P.; Harkin, D.P.; Pharoah, P.; Ponder, B.; Rudland, P.S. BRCA1 suppresses osteopontin-mediated breast cancer. J. Biol. Chem. 2006, 281, 26587–26601. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, K.; Morine, Y.; Miyazaki, K.; Yamada, S.; Saito, Y.; Nishi, M.; Tokunaga, T.; Ikemoto, T.; Imura, S.; Shimada, M. The interaction between cancer associated fibroblasts and tumor associated macrophages via the osteopontin pathway in the tumor microenvironment of hepatocellular carcinoma. Oncotarget 2021, 12, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Muchlińska, A.; Nagel, A.; Popęda, M.; Szade, J.; Niemira, M.; Zieliński, J.; Skokowski, J.; Bednarz-Knoll, N.; Żaczek, A.J. Alpha-smooth muscle actin-positive cancer-associated fibroblasts secreting osteopontin promote growth of luminal breast cancer. Cell. Mol. Biol. Lett. 2022, 27, 45. [Google Scholar] [CrossRef]
- Wu, M.; Schneider, D.J.; Mayes, M.D.; Assassi, S.; Arnett, F.C.; Tan, F.K.; Blackburn, M.R.; Agarwal, S.K. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J. Investig. Dermatol. 2012, 132, 1605–1614. [Google Scholar] [CrossRef]
- Corallo, C.; Volpi, N.; Franci, D.; Montella, A.; Biagioli, M.; Mariotti, G.; D’Onofrio, F.; Gonnelli, S.; Nuti, R.; Giordano, N. Is osteopontin involved in cutaneous fibroblast activation? its hypothetical role in scleroderma pathogenesis. Int. J. Immunopathol. Pharmacol. 2014, 27, 97–102. [Google Scholar] [CrossRef]
- Butti, R.; Nimma, R.; Kundu, G.; Bulbule, A.; Kumar, T.V.; Gunasekaran, V.P.; Tomar, D.; Kumar, D.; Mane, A.; Gill, S.S.; et al. Tumor-derived osteopontin drives the resident fibroblast to myofibroblast differentiation through Twist1 to promote breast cancer progression. Oncogene 2021, 40, 2002–2017. [Google Scholar] [CrossRef]
- Sharon, Y.; Raz, Y.; Cohen, N.; Ben-Shmuel, A.; Schwartz, H.; Geiger, T.; Erez, N. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 2015, 75, 963–973. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Falasca, M.; Oon, C.E. Cancer-associated fibroblasts: Epigenetic regulation and therapeutic intervention in breast cancer. Cancers 2020, 12, 2949. [Google Scholar] [CrossRef]
- Feng, C.; Kou, L.; Yin, P.; Jing, Y. Excessive activation of IL-33/ST2 in cancer-associated fibroblasts promotes invasion and metastasis in ovarian cancer. Oncol. Lett. 2022, 23, 158. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M.; Sotgia, F.; Lisanti, M.P. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog–GLI signalling in breast cancer cells. Oncotarget 2015, 6, 10728–10745. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Shelton, M.; Anene, C.A.; Nsengimana, J.; Roberts, W.; Newton-Bishop, J.; Boyne, J.R. The role of CAF derived exosomal micrornas in the tumour microenvironment of melanoma. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188456. [Google Scholar] [CrossRef]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef]
- Souma, K.; Shichino, S.; Hashimoto, S.; Ueha, S.; Tsukui, T.; Nakajima, T.; Suzuki, H.I.; Shand, F.H.W.; Inagaki, Y.; Nagase, T.; et al. Lung fibroblasts express a miR-19a-19b-20a sub-cluster to suppress TGF-β-associated fibroblast activation in murine pulmonary fibrosis. Sci. Rep. 2018, 8, 16642. [Google Scholar] [CrossRef]
- Melling, G.E.; Flannery, S.E.; Abidin, S.A.; Clemmens, H.; Prajapati, P.; Hinsley, E.E.; Hunt, S.; Catto, J.W.F.; Della Coletta, R.; Mellone, M.; et al. A miRNA-145/TGF-beta1 negative feedback loop regulates the cancer-associated fibroblast phenotype. Carcinogenesis 2018, 39, 798–807. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, D.; Wang, Y.; Sun, P.; Hou, X.; Larner, J.; Xiong, W.; Mi, J. Mir-21/SMAD 7 signaling determines TGF-β1-induced CAF formation. Sci. Rep. 2013, 3, 2038. [Google Scholar] [CrossRef]
- Han, M.; Wang, F.; Gu, Y.; Pei, X.; Guo, G.; Yu, C.; Li, L.; Zhu, M.; Xiong, Y.; Wang, Y. MicroRNA-21 induces breast cancer cell invasion and migration by suppressing smad7 via EGF and TGF-β pathways. Oncol. Rep. 2016, 35, 73–80. [Google Scholar] [CrossRef]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z.; et al. Melanoma cell-secreted exosomal mir-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/Jak2/Stat3 signaling pathway. J. Exp. Clin. Cancer Res. 2018, 37, 242. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Dror, S.; Sander, L.; Schwartz, H.; Sheinboim, D.; Barzilai, A.; Dishon, Y.; Apcher, S.; Golan, T.; Greenberger, S.; Barshack, I.; et al. Melanoma mirna trafficking controls tumour primary niche formation. Nat. Cell Biol. 2016, 18, 1006–1017. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Fang, P.; Ma, Z.; Wang, X.; Chen, H.; Wang, S.; Meng, F.; Wang, C.; Zhang, E.; et al. Cancer-associated fibroblast-specific lncRNA LINC01614 enhances glutamine uptake in lung adenocarcinoma. J. Hematol. Oncol. 2022, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Ti, W.; Wang, J.; Cheng, Y. The interaction between long non-coding RNAS and cancer-associated fibroblasts in Lung Cancer. Front. Cell Dev. Biol. 2022, 9, 714125. [Google Scholar] [CrossRef]
- Li, Q.; Pan, X.; Wang, X.; Jiao, X.; Zheng, J.; Li, Z.; Huo, Y. Long noncoding RNA MALAT1 promotes cell proliferation through suppressing Mir-205 and promoting SMAD4 expression in osteosarcoma. Oncotarget 2017, 8, 106648–106660. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Luo, H.; Yang, G.; Yu, T.; Luo, S.; Wu, C.; Sun, Y.; Liu, M.; Tu, G. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr.-Relat. Cancer 2014, 21, 355–369. [Google Scholar] [CrossRef]
- Kim, H.J.; Yang, K.; Kim, K.; Lee, Y.J.; Lee, S.; Ahn, S.Y.; Ahn, Y.H.; Kang, J.L. Reprogramming of cancer-associated fibroblasts by apoptotic cancer cells inhibits lung metastasis via notch1-WISP-1 signaling. Cell. Mol. Immunol. 2022, 19, 1373–1391. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of exosomes in crosstalk between cancer-associated fibroblasts and cancer cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Nocquet, L.; Juin, P.P.; Souazé, F. Mitochondria at center of exchanges between cancer cells and cancer-associated fibroblasts during tumor progression. Cancers 2020, 12, 3017. [Google Scholar] [CrossRef] [PubMed]
- Dardare, J.; Witz, A.; Merlin, J.-L.; Gilson, P.; Harlé, A. SMAD4 and the TGFΒ pathway in patients with pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef] [PubMed]
- Bertrand-Chapel, A.; Caligaris, C.; Fenouil, T.; Savary, C.; Aires, S.; Martel, S.; Huchedé, P.; Chassot, C.; Chauvet, V.; Cardot-Ruffino, V.; et al. Smad2/3 mediate oncogenic effects of TGF-β in the absence of SMAD4. Commun. Biol. 2022, 5, 1068. [Google Scholar] [CrossRef] [PubMed]
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C induces phenotypic changes in fibroblasts to myofibroblasts with high contractility through the integrin αvβ1/transforming growth factor β/SMAD signaling axis in human breast cancer. Am. J. Pathol. 2020, 190, 2123–2135. [Google Scholar] [CrossRef]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial–Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Valenti, G.; Quinn, H.M.; Heynen, G.J.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef]
- Diéguez-Hurtado, R.; Kato, K.; Giaimo, B.D.; Nieminen-Kelhä, M.; Arf, H.; Ferrante, F.; Bartkuhn, M.; Zimmermann, T.; Bixel, M.G.; Eilken, H.M.; et al. Loss of the transcription factor RBPJ induces disease-promoting properties in brain pericytes. Nat. Commun. 2019, 10, 2817. [Google Scholar] [CrossRef]
- Trylcova, J.; Busek, P.; Smetana, K., Jr.; Balaziova, E.; Dvorankova, B.; Mifkova, A.; Sedo, A. Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumor Biol. 2015, 36, 5873–5879. [Google Scholar] [CrossRef]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.; Sholl, L.; Ben-Aharon, I.; Beck, A.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.; Whitesell, L.; et al. The Reprogramming of Tumor Stroma by HSF1 Is a Potent Enabler of Malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Stern, M.; Dubois, T.; Sastre-Garau, X.; Delattre, O.; Vincent-Salomon, A.; Mechta-Grigoriou, F. 225 oxidative stress promotes myofibroblast differentiation and tumour spreading. Eur. J. Cancer Suppl. 2010, 8, 124. [Google Scholar] [CrossRef][Green Version]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Erratum: Corrigendum: Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 822. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Schmitt, T.M.; Hulbert, A.; Brockenbrough, J.S.; Nguyen, H.N.; Cuevas, C.; Dotson, A.M.; Tan, X.; Hotes, J.L.; Greenberg, P.D.; et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015, 28, 638–652. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678. [Google Scholar] [CrossRef]
- Zeltz, C.; Primac, I.; Erusappan, P.; Alam, J.; Noel, A.; Gullberg, D. Cancer-associated fibroblasts in desmoplastic tumors: Emerging role of integrins. Semin. Cancer Biol. 2020, 62, 166–181. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid. Redox Signal. 2011, 14, 2361–2371. [Google Scholar] [CrossRef]
- Zeltz, C.; Alam, J.; Liu, H.; Erusappan, P.M.; Hoschuetzky, H.; Molven, A.; Parajuli, H.; Cukierman, E.; Costea, D.-E.; Lu, N.; et al. A11β1 integrin is induced in a subset of cancer-associated fibroblasts in desmoplastic tumor stroma and mediates in vitro cell migration. Cancers 2019, 11, 765. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent proinvasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Glod, J.W.; Banerjee, D. Mesenchymal Stem Cells: Flip Side of the Coin. Cancer Res. 2009, 69, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Timaner, M.; Letko-Khait, N.; Kotsofruk, R.; Benguigui, M.; Beyar-Katz, O.; Rachman-Tzemah, C.; Raviv, Z.; Bronshtein, T.; Machluf, M.; Shaked, Y. Therapy-educated mesenchymal stem cells enrich for tumor-initiating cells. Cancer Res. 2018, 78, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chu, Y.; Huang, Z.; Cai, J.; Wang, Z. The metastatic phenotype shift toward myofibroblast of adipose-derived mesenchymal stem cells promotes ovarian cancer progression. Carcinogenesis 2019, 41, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Ishii, G.; Sangai, T.; Oda, T.; Aoyagi, Y.; Hasebe, T.; Kanomata, N.; Endoh, Y.; Okumura, C.; Okuhara, Y.; Magae, J.; et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003, 309, 232–240. [Google Scholar] [CrossRef]
- Worthley, D.L.; Ruszkiewicz, A.; Davies, R.; Moore, S.; Nivison-Smith, I.; Bik To, L.; Browett, P.; Western, R.; Durrant, S.; So, J.; et al. Human gastrointestinal neoplasia-associated myofibroblasts can develop from bone marrow-derived cells following allogeneic stem cell transplantation. Stem Cells 2009, 27, 1463–1468. [Google Scholar] [CrossRef]
- Lapshyn, H.; Bolm, L.; Kohler, I.; Werner, M.; Billmann, F.G.; Bausch, D.; Hopt, U.T.; Makowiec, F.; Wittel, U.A.; Keck, T.; et al. Histopathological tumor invasion of the mesenterico-portal vein is characterized by aggressive biology and stromal fibroblast activation. HPB 2017, 19, 67–74. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2019, 146, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Brosh, R.; Buganim, Y.; Ezra, O.; Goldstein, I.; Solomon, H.; Kogan, I.; Goldfinger, N.; Klocker, H.; Rotter, V. Modulated expression of WFDC1 during carcinogenesis and cellular senescence. Carcinogenesis 2008, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Berdiel-Acer, M.; Sanz-Pamplona, R.; Calon, A.; Cuadras, D.; Berenguer, A.; Sanjuan, X.; Paules, M.J.; Salazar, R.; Moreno, V.; Batlle, E.; et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol. Oncol. 2014, 8, 1290–1305. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Feng, H.Z.; Wei, H.; Cala, S.; Jin, J.P. Up-regulation of alpha-smooth muscle actin in cardiomyocytes from non-hypertrophic and non-failing transgenic mouse hearts expressing N-terminal truncated cardiac troponin I. FEBS Open Bio 2013, 4, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Deciphering cancer fibroblasts. J. Exp. Med. 2018, 215, 2967–2968. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Sun, C.; Liang, Z.; Li, H.; Chen, L.; Luo, H.; Zhang, H.; Ding, P.; Sun, X.; Qin, Z.; et al. FSP1+ fibroblast subpopulation is essential for the maintenance and regeneration of medullary thymic epithelial cells. Sci. Rep. 2015, 5, 14871. [Google Scholar] [CrossRef] [PubMed]
- Österreicher, C.H.; Penz-Österreicher, M.; Grivennikov, S.I.; Guma, M.; Koltsova, E.K.; Datz, C.; Sasik, R.; Hardiman, G.; Karin, M.; Brenner, D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 2010, 108, 308–313. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, Y.; Liang, A.; Jia, L.; Du, J. FSP-1 silencing in bone marrow cells suppresses neointima formation in vein graft. Circ. Res. 2012, 110, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1)Gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindström, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-β–slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3873–3883. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin is required for lung adenocarcinoma metastasis via heterotypic tumor cell–cancer-associated fibroblast interactions during collective invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Chiquet, M. Tenascins: Regulation and putative functions during pathological stress. J. Pathol. 2003, 200, 488–499. [Google Scholar] [CrossRef]
- Lundin, M.; Nordling, S.; Lundin, J.; Haglund, C. Tenascin-C expression and its prognostic significance in colorectal cancer. Oncology 2007, 72, 403–409. [Google Scholar] [CrossRef]
- Tsunoda, T.; Inada, H.; Kalembeyi, I.; Imanaka-Yoshida, K.; Sakakibara, M.; Okada, R.; Katsuta, K.; Sakakura, T.; Majima, Y.; Yoshida, T. Involvement of large tenascin-C splice variants in breast cancer progression. Am. J. Pathol. 2003, 162, 1857–1867. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Silvers, C.R.; Messing, E.M.; Miyamoto, H.; Lee, Y.-F. Tenascin-C expression in the lymph node pre-metastatic niche in muscle-invasive bladder cancer. Br. J. Cancer 2021, 125, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Sonongbua, J.; Siritungyong, S.; Thongchot, S.; Kamolhan, T.; Utispan, K.; Thuwajit, P.; Pongpaibul, A.; Wongkham, S.; Thuwajit, C. Periostin induces epithelial-to-mesenchymal transition via the integrin α5β1/twist-2 axis in Cholangiocarcinoma. Oncol. Rep. 2020, 43, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.A.; Damon, B.; Mironov, V.; Kasyanov, V.; Ramamurthi, A.; Moreno-Rodriguez, R.; Trusk, T.; Potts, J.D.; Goodwin, R.L.; Davis, J.; et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 2007, 101, 695–711. [Google Scholar] [CrossRef]
- Walker, J.T.; McLeod, K.; Kim, S.; Conway, S.J.; Hamilton, D.W. Periostin as a multifunctional modulator of the wound healing response. Cell Tissue Res. 2016, 365, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.W. Functional role of Periostin in development and wound repair: Implications for connective tissue disease. J. Cell Commun. Signal. 2008, 2, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Doldi, V.; Lecchi, M.; Ljevar, S.; Colecchia, M.; Campi, E.; Centonze, G.; Marenghi, C.; Rancati, T.; Miceli, R.; Verderio, P.; et al. Potential of the stromal matricellular protein periostin as a biomarker to improve risk assessment in prostate cancer. Int. J. Mol. Sci. 2022, 23, 7987. [Google Scholar] [CrossRef]
- Ruan, K.; Bao, S.; Ouyang, G. The multifaceted role of Periostin in tumorigenesis. Cell. Mol. Life Sci. 2009, 66, 2219–2230. [Google Scholar] [CrossRef]
- Morra, L.; Moch, H. Periostin expression and epithelial-mesenchymal transition in cancer: A review and an update. Virchows Arch. 2011, 459, 465–475. [Google Scholar] [CrossRef]
- Qin, X.; Yan, M.; Zhang, J.; Wang, X.; Shen, Z.; Lv, Z.; Li, Z.; Wei, W.; Chen, W. TGFΒ3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci. Rep. 2016, 6, 20587. [Google Scholar] [CrossRef]
- Wei, W.F.; Chen, X.J.; Liang, L.J.; Yu, L.; Wu, X.G.; Zhou, C.F.; Wang, Z.C.; Fan, L.S.; Hu, Z.; Liang, L.; et al. Periostin + cancer-associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Mol. Oncol. 2020, 15, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Kanno, A.; Satoh, K.; Masamune, A.; Hirota, M.; Kimura, K.; Umino, J.; Hamada, S.; Satoh, A.; Egawa, S.; Motoi, F.; et al. Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int. J. Cancer 2008, 122, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Fotsitzoudis, C.; Koulouridi, A.; Messaritakis, I.; Konstantinidis, T.; Gouvas, N.; Tsiaoussis, J.; Souglakos, J. Cancer-associated fibroblasts: The origin, biological characteristics and role in cancer—A glance on colorectal cancer. Cancers 2022, 14, 4394. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, T.P.; Stuart, M.P.; Oosting, J.; Tollenaar, R.A.; Sier, C.F.; Mesker, W.E. Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer 2019, 19, 284. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, C.; Song, Q.; Zhu, M.; Xu, Y.; Xiao, M.; Zheng, W. Identifying cancer-associated fibroblasts as emerging targets for hepatocellular carcinoma. Cell Biosci. 2020, 10, 127. [Google Scholar] [CrossRef]
- Henry, L.R.; Lee, H.-O.; Lee, J.S.; Klein-Szanto, A.; Watts, P.; Ross, E.A.; Chen, W.-T.; Cheng, J.D. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin. Cancer Res. 2007, 13, 1736–1741. [Google Scholar] [CrossRef]
- Xue, Y.; Beyer, G.; Mayerle, J. FAPα in pancreatic stellate cells upregulated by TGFβ1: A novel insight into pancreatic cancer progress. Ann. Transl. Med. 2020, 8, 910. [Google Scholar] [CrossRef]
- Hoos, A. Development of immuno-oncology drugs—from CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef]
- Xin, L.; Gao, J.; Zheng, Z.; Chen, Y.; Lv, S.; Zhao, Z.; Yu, C.; Yang, X.; Zhang, R. Fibroblast activation protein-α as a target in the bench-to-bedside diagnosis and treatment of tumors: A narrative review. Front. Oncol. 2021, 11, 648187. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yi, S.; Liu, W.; Jia, C.; Wang, G.; Hua, X.; Tai, Y.; Zhang, Q.; Chen, G. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med. Oncol. 2013, 30, 663. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and cancer: Insight into tumor progression and immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Bozyk, P.D.; Moore, B.B. Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. American Journal of Respiratory Cell and Molecular Biology. Am. J. Respir. Cell Mol. Biol. 2011, 45, 445–452. [Google Scholar] [CrossRef]
- Odaka, T.; Kobayashi, K.; Takahashi, K.; Nakamura, H.; Matsuoka, T. Effect of prostaglandin E2on urokinase-type plasminogen activator production by human Lung Fibroblasts. Scand. J. Clin. Lab. Investig. 2009, 69, 225–233. [Google Scholar] [CrossRef]
- Ma, X.; Aoki, T.; Tsuruyama, T.; Narumiya, S. Definition of prostaglandin E2–EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth. Cancer Res. 2015, 75, 2822–2832. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019, 79, 5367–5382. [Google Scholar] [CrossRef]
- Liu, C.; Mak, M. Fibroblast-mediated uncaging of cancer cells and dynamic evolution of the physical microenvironment. Sci. Rep. 2022, 12, 791. [Google Scholar] [CrossRef]
- Bauer, G. Central Signaling Elements of Intercellular Reactive Oxygen/Nitrogen Species-dependent Induction of Apoptosis in Malignant Cells. Anticancer Res. 2017, 37, 499–514. [Google Scholar] [CrossRef]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef]
- Chen, S.; Giannakou, A.; Wyman, S.; Gruzas, J.; Golas, J.; Zhong, W.; Loreth, C.; Sridharan, L.; Yamin, T.T.; Damelin, M.; et al. Cancer-associated fibroblasts suppress SOX2-induced dysplasia in a lung squamous cancer coculture. Proc. Natl. Acad. Sci. USA 2018, 115, E11671–E11680. [Google Scholar] [CrossRef] [PubMed]
- Mintz, B.; Illmensee, K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1975, 72, 3585–3589. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.G.P.; Shearer, M.; O’neill, C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Weinberg, R.A.; Ariza, A. Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6389–6393. [Google Scholar] [CrossRef]
- Ressler, S.J.; Dang, T.D.; Wu, S.M.; Tse, D.Y.; Gilbert, B.E.; Vyakarnam, A.; Yang, F.; Schauer, I.G.; Barron, D.A.; Rowley, D.R. WFDC1 is a key modulator of inflammatory and wound repair responses. Am. J. Pathol. 2014, 184, 2951–2964. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Teng, F.; Tian, W.Y.; Wang, Y.M.; Zhang, Y.F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016, 9, 8. [Google Scholar] [CrossRef]
- Allen, M.; Jones, L.J. Jekyll and Hyde: The role of the microenvironment on the progression of cancer. J. Pathol. 2011, 223, 162–176. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef]
- Lederle, W.; Hartenstein, B.; Meides, A.; Kunzelmann, H.; Werb, Z.; Angel, P.; Mueller, M.M. MMP13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis 2009, 31, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef] [PubMed]
- Leeming, D.J.; Koizumi, M.; Qvist, P.; Barkholt, V.; Zhang, C.; Henriksen, K.; Byrjalsen, I.; Karsdal, M.A. Serum N-Terminal Propeptide of Collagen Type I is Associated with the Number of Bone Metastases in Breast and Prostate Cancer and Correlates to Other Bone Related Markers. Biomark. Cancer 2011, 3, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Stanisavljevic, J.; Loubat-Casanovas, J.; Herrera, M.; Luque, T.; Peña, R.; Lluch, A.; Albanell, J.; Bonilla, F.; Rovira, A.; Peña, C.; et al. Snail1-Expressing Fibroblasts in the Tumor Microenvironment Display Mechanical Properties That Support Metastasis. Cancer Res. 2014, 75, 284–295. [Google Scholar] [CrossRef]
- Łabędź, N.; Stachowicz-Suhs, M.; Psurski, M.; Anisiewicz, A.; Banach, J.; Piotrowska, A.; Dzięgiel, P.; Maciejczyk, A.; Matkowski, R.; Wietrzyk, J. Modulation of fibroblast activity via vitamin D3 is dependent on tumor type—Studies on mouse mammary gland cancer. Cancers 2022, 14, 4585. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Pang, N.; Li, J.; Sun, A.; Yang, Z.; Cheng, S.; Qi, X.-R. Prior anti-cafs break down the CAFS barrier and improve accumulation of docetaxel micelles in tumor. Int. J. Nanomed. 2018, 13, 5971–5990. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial–mesenchymal plasticity. Life Sci. Alliance 2019, 2, e201900425. [Google Scholar] [CrossRef]
- Hao, J.; Zeltz, C.; Pintilie, M.; Li, Q.; Sakashita, S.; Wang, T.; Cabanero, M.; Martins-Filho, S.N.; Wang, D.Y.; Pasko, E.; et al. Characterization of distinct populations of carcinoma-associated fibroblasts from non–small cell lung carcinoma reveals a role for ST8SIA2 in cancer cell invasion. Neoplasia 2019, 21, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. 2016, 15, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanna, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Ao, M.; Brewer, B.M.; Yang, L.; Franco Coronel, O.E.; Hayward, S.W.; Webb, D.J.; Li, D. Stretching fibroblasts remodels fibronectin and alters cancer cell migration. Sci. Rep. 2015, 5, 8334. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Vidal-Vanaclocha, F. The prometastatic microenvironment of the liver. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2008, 1, 113–129. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Liu, X.; Kammertoens, T.; Blankenstein, T.; Qin, Z. Fibroblast-specific protein 1/S100A4-positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res. 2013, 73, 2770–2781. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Remark, R.; Gnjatic, S.; Merad, M. Host tissue determinants of tumour immunity. Nat. Rev. Cancer 2019, 19, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.M.; McCleland, M.; Koeppen, H. Microenvironmental regulation of tumour immunity and response to immunotherapy. J. Pathol. 2021, 254, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of tumor escape from immune system: Role of mesenchymal stromal cells. Immunol. Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Flavell, R.A. ‘Yin–Yang’ functions of transforming growth factor-β and T regulatory cells in immune regulation. Immunol. Rev. 2007, 220, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Olkhanud, P.B.; Baatar, D.; Bodogai, M.; Hakim, F.; Gress, R.; Anderson, R.L.; Deng, J.; Xu, M.; Briest, S.; Biragyn, A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009, 69, 5996–6004. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.-S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein–α. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Kawabata-Iwakawa, R.; Rokudai, S.; Toyoda, M.; Nishiyama, M.; Chikamatsu, K. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1407–1417. [Google Scholar] [CrossRef]
- Tong, C.C.L.; Kao, J.; Sikora, A.G. Recognizing and reversing the immunosuppressive tumor microenvironment of head and neck cancer. Immunol. Res. 2012, 54, 266–274. [Google Scholar] [CrossRef]
- Augsten, M.; Sjöberg, E.; Frings, O.; Vorrink, S.U.; Frijhoff, J.; Olsson, E.; Borg, K.; Östman, A. Cancer-associated fibroblasts expressing CXCL14 rely upon NOS1-derived nitric oxide signaling for their tumor-supporting properties. Cancer Res. 2014, 74, 2999–3010. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 2019, 249, 193–205. [Google Scholar] [CrossRef]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef]
- Chen, L.; Qiu, X.; Wang, X.; He, J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem. Biophys. Res. Commun. 2017, 487, 8–14. [Google Scholar] [CrossRef]
- Harper, J.; Sainson, R.C. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Phillips, M.; Alice, A.; Bambina, S.; Zebertavage, L.; Friedman, D.; Cottam, B.; Newell, P.; et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PLoS ONE 2019, 14, e0211117. [Google Scholar] [CrossRef]
- Zhang, Y.; Ertl, H.C. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. BCR 2014, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Timosenko, E.; Hadjinicolaou, A.V.; Cerundolo, V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immuno Ther. 2017, 9, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Gao, J.; Wang, S.; Ye, N.; Chong, Y.; Huang, Y.; Wang, J.; Li, B.; Yin, W.; Wang, D. Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumor Biol. 2016, 37, 1889–1899. [Google Scholar] [CrossRef]
- Tanaka, H.Y.; Kano, M.R. Stromal barriers within the tumor microenvironment and obstacles to nanomedicine. Cancer Drug Deliv. Syst. Based Tumor Microenviron. 2019, 57–89. [Google Scholar] [CrossRef]
- Edwards, P.; Kang, B.W.; Chau, I. Targeting the stroma in the management of pancreatic cancer. Front. Oncol. 2021, 11, 691185. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Altrock, P.M.; Yoon, N.; Bull, J.A.; Wu, H.; Ruiz-Ramírez, J.; Miroshnychenko, D.; Kimmel, G.J.; Kim, E.; Vander Velde, R.J.; Rejniak, K.; et al. The impact of tumor stromal architecture on therapy response and clinical progression. bioRxiv 2018. [CrossRef]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-Cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar]
- Gallego-Rentero, M.; Gutiérrez-Pérez, M.; Fernández-Guarino, M.; Mascaraque, M.; Portillo-Esnaola, M.; Gilaberte, Y.; Carrasco, E.; Juarranz, Á. TGFΒ1 secreted by cancer-associated fibroblasts as an inductor of resistance to photodynamic therapy in squamous cell carcinoma cells. Cancers 2021, 13, 5613. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.P.J.; Spaargaren, M.; Clevers, H.; Pals, S.T. Hepatocyte growth factor/met and CD44 in colorectal cancer: Partners in tumorigenesis and therapy resistance. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188437. [Google Scholar] [CrossRef] [PubMed]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-associated fibroblasts: Tumorigenicity and targeting for cancer therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Hatada, I.; Nakagama, H.; Ochiya, T. Epigenetic reprogramming using 5-azacytidine promotes an anti-cancer response in pancreatic adenocarcinoma cells. Cell Death Dis. 2018, 9, 468. [Google Scholar] [CrossRef]
- Al-Harbi, B.; Hendrayani, S.F.; Silva, G.; Aboussekhra, A. Let-7b inhibits cancer-promoting effects of breast cancer-associated fibroblasts through IL-8 repression. Oncotarget 2018, 9, 17825–17838. [Google Scholar] [CrossRef]
- Kuba, K.; Matsumoto, K.; Date, K.; Shimura, H.; Tanaka, M.; Nakamura, T. HGF/NK4, a four-kringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res. 2000, 60, 6737–6743. [Google Scholar]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef]
- Sattler, M.; Reddy, M.M.; Hasina, R.; Gangadhar, T.; Salgia, R. The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther. Adv. Med. Oncol. 2011, 3, 171–184. [Google Scholar] [CrossRef]
- Sadiq, A.A.; Salgia, R. MET as a possible target for non–small-cell lung cancer. J. Clin. Oncol. 2013, 31, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Nogueira, P.; Mancino, M.; Fuster, G.; López-Plana, A.; Jauregui, P.; Almendro, V.; Enreig, E.; Menéndez, S.; Rojo, F.; Noguera-Castells, A.; et al. Tumor-associated fibroblasts promote HER2-targeted therapy resistance through FGFR2 activation. Clin. Cancer Res. 2019, 26, 1432–1448. [Google Scholar] [CrossRef] [PubMed]
- Wakelee, H.A.; Gettinger, S.N.; Engelman, J.A.; Janne, P.A.; West, H.J.; Subramaniam, D.S.; Leach, J.W.; Wax, M.B.; Yaron, Y.; Lara, P. A phase IB/II study of XL184 (BMS 907351) with and without erlotinib (E) in patients (PTS) with non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2010, 28 (Suppl. 15), 3017. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Okamoto, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nakagawa, K. MET tyrosine kinase inhibitor Crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J. Thorac. Oncol. 2011, 6, 1624–1631. [Google Scholar] [CrossRef]
- Blumenschein, G.R., Jr.; Mills, G.B.; Gonzalez-Angulo, A.M. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 3287–3296. [Google Scholar] [CrossRef]
- Munasinghe, A.; Malik, K.; Mohamedi, F.; Moaraf, S.; Kocher, H.; Jones, L.; Hill, N.J. Fibronectin acts as a molecular switch to determine SPARC function in pancreatic cancer. Cancer Lett. 2020, 477, 88–96. [Google Scholar] [CrossRef]
- Schwarz, R.E.; Awasthi, N.; Konduri, S.; Caldwell, L.; Cafasso, D.; Schwarz, M.A. Antitumor effects of EMAP II against pancreatic cancer through inhibition of fibronectin-dependent proliferation. Cancer Biol. Ther. 2010, 9, 632–639. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.-P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2, e92232. [Google Scholar] [CrossRef]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef]
- Fang, J.; Xiao, L.; Joo, K.I.; Liu, Y.; Zhang, C.; Liu, S.; Conti, P.S.; Li, Z.; Wang, P. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int. J. Cancer 2016, 138, 1013–1023. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Gerling, M.; Büller, N.V.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergström, Å.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.; et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, H.; Guo, Q.; Zhou, A.; Wang, X.; Li, P.; Zhang, S. Exosomal Sonic Hedgehog derived from cancer-associated fibroblasts promotes proliferation and migration of esophageal squamous cell carcinoma. Cancer Med. 2020, 9, 2500–2513. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Kadhim, S.A.; Jiang, P.; McMannus, C.; Osgood, R.; Printz, M.; Araiza, F.; Feiser, G.; Symons, R.; Anderson, J.; Thompson, C.; et al. Abstract 5392: Synergistic anti-tumor effects of pegylated recombinant human hyaluronidase (pegph20) with Gemcitabine in subcutaneous pancreatic cancer xenograft models. Cancer Res. 2010, 70 (Suppl. 8), 5392. [Google Scholar] [CrossRef]
- Pavlaki, M.; Zucker, S. Matrix metalloproteinase inhibitors (MMPIs): The beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 2003, 22, 177–203. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.; Karamouzis, M.; Papatsoris, A.; Papavassiliou, A. Matrix metalloproteinase inhibitors as anticancer agents. Int. J. Biochem. Cell Biol. 2008, 40, 1156–1168. [Google Scholar] [CrossRef]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
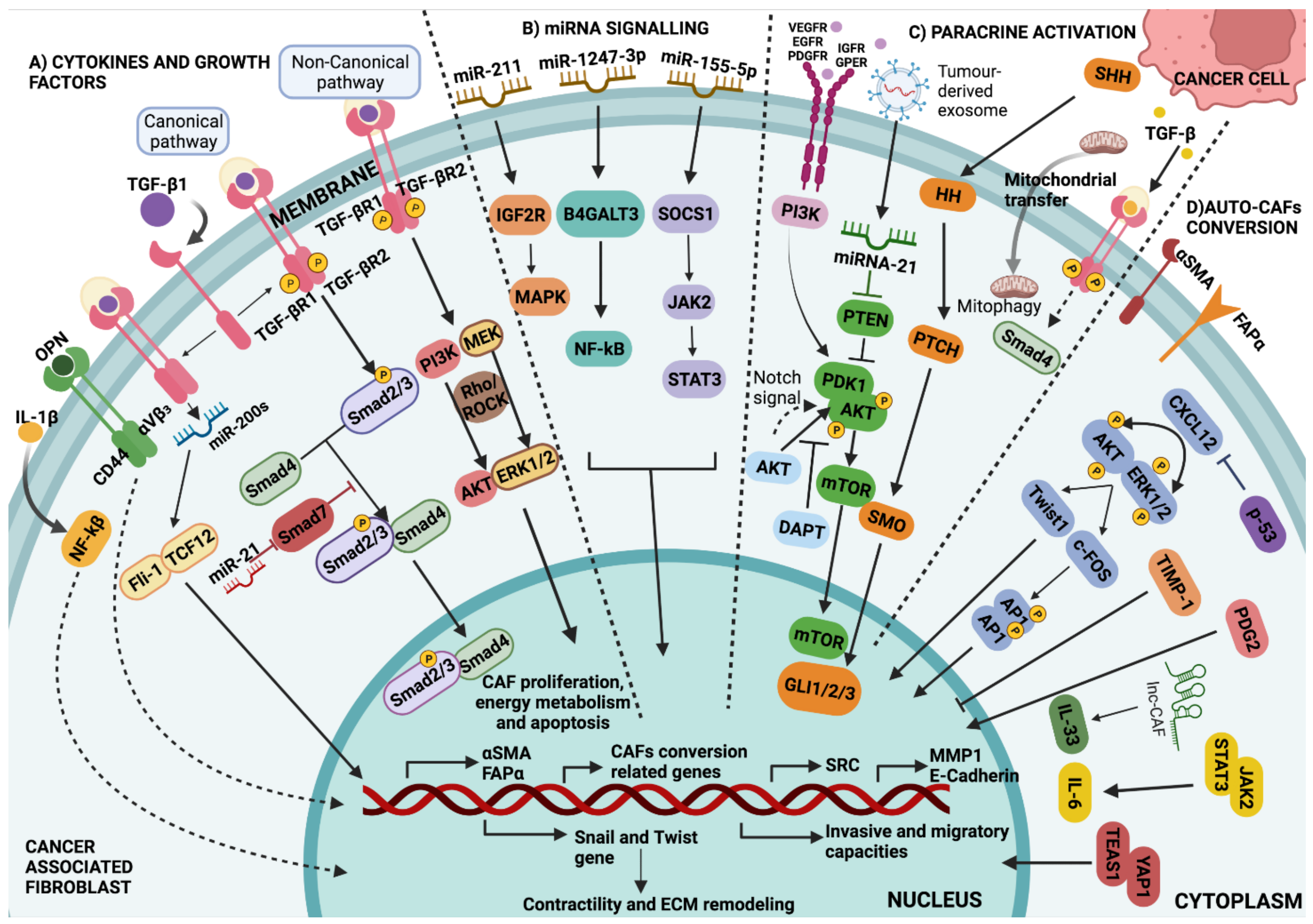{kind=link}
{kind=link}
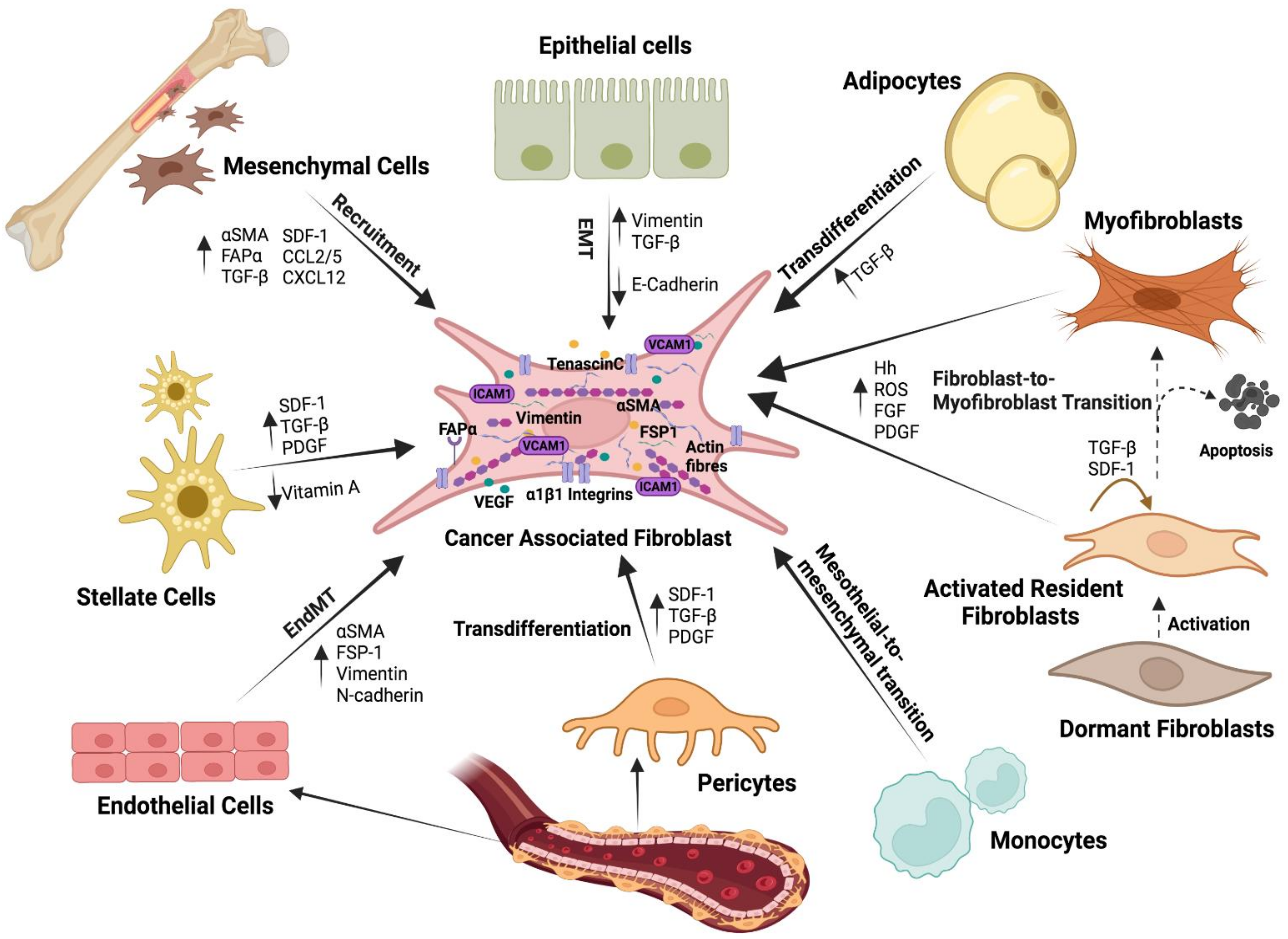{kind=link}
{kind=link}
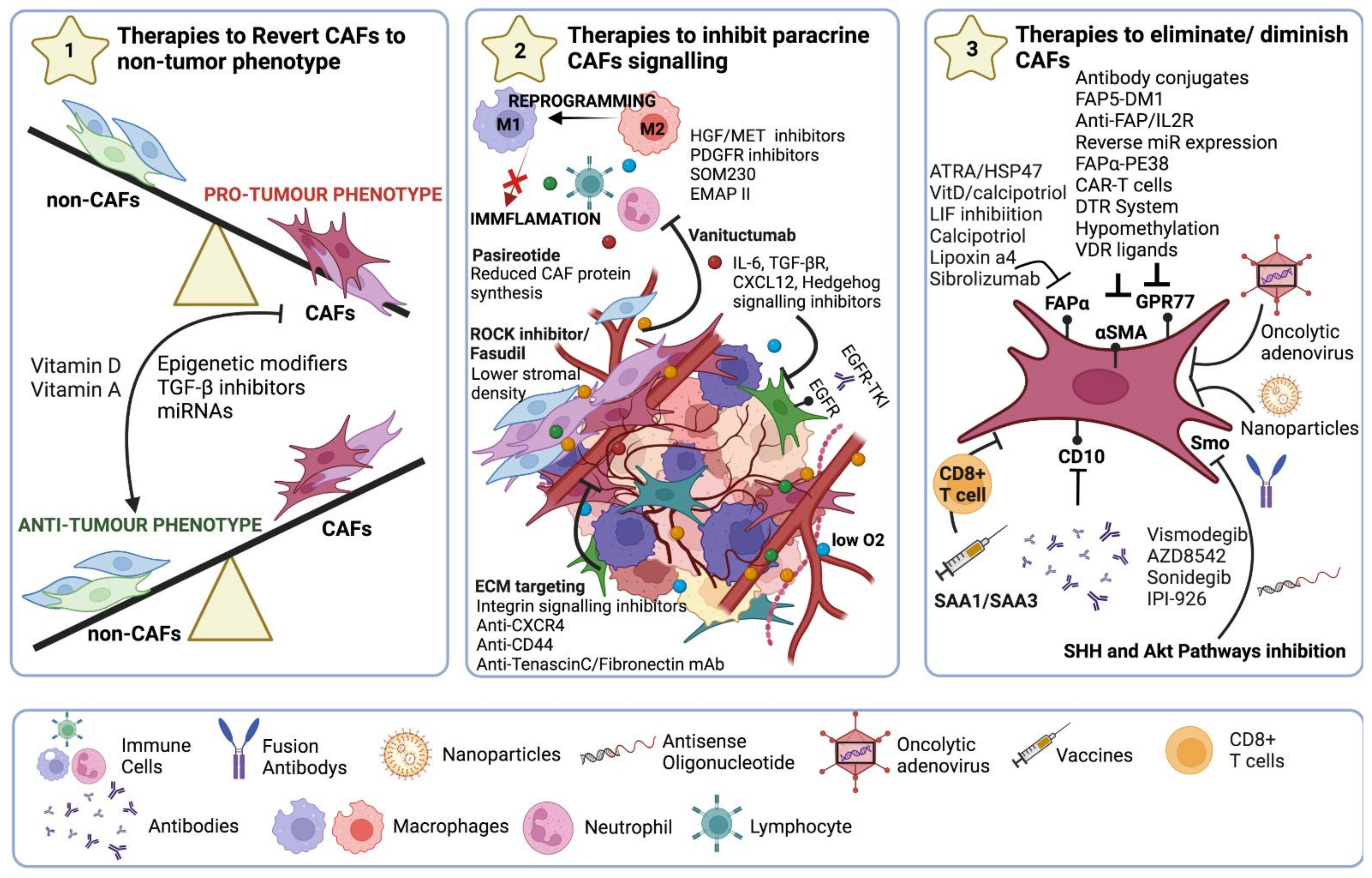{kind=link}
| Factors | Mode of Action | References |
|---|---|---|
| Cytokines | CAF activation/inactivation | [4,22,23,24,32,33,34,35,36,37,38,39,40,41,42,43,44,45] |
| IL-1 | CAF activation: Promotes cell growth and invasion by NF-κB and IL-1β/IL-1R pathway | [8,45,46,47] |
| TGF-beta | CAF activation: Promotes cell proliferation and migration by TGF-β/Smad pathway | [22,37,38,39,40,41,42,48,49,50,51,52,53,54,55,56,57,58,59,60,61] |
| IL-6 | CAF activation: Promotes cell chemoresistance, cancer progression, cell metastasis and chemoresistance and cell adhesion by STAT3/NF-kbeta, JAK2/STAT3, ERK1/2, STAT3/Notch, and STAT3/PD-L1 pathways | [23,24,36,39,46,50,59,62,63,64] |
| PDGF | CAF activation | [39,40,48,49,65,66,67,68,69,70,71,72,73,74,75,76,77,78] |
| EGF | CAF activation | [49,79,80,81] |
| IGF | CAF activation: Promotes cell chemoresistance, cell survival, and cell growth by IGF-/AKT, IGF/IGFR, and PI3K/AKT/mTOR pathways | [49,82,83,84,85] |
| JAK2/STAT3 | CAF activation | [40,86] |
| YAP1/TEAS1 | CAF activation | [87,88] |
| p53 | CAF inactivation | [89] |
| VEGF | CAF activation | [51,75,76,90,91] |
| CXCL12 | CAF activation: Promotes cell metastasis, cell invasion, EMT and cisplatin resistance and cancer progression by PI3K/AKT, TGF-β, Wnt/β-catenin, and CXCL12/CXCR4 pathways | [32,33,34,35,70,83,92,93,94,95,96,97,98,99] |
| TIMP-1 | CAF inactivation | [100,101,102,103] |
| OPN | CAF activation | [44,53,79,104,105,106,107,108,109,110] |
| MAPK | CAF activation | [44] |
| AKT | CAF activation | [44,98] |
| ERK1/2 | CAF activation | [109,110] |
| c-Ski | CAF inactivation | [111] |
| IL-33 | CAF activation: Promotes cell migration and invasion | [112] |
| TNF | CAF activation | [113] |
| SHH | CAF activation | [114] |
| miRNAs | CAF activation/inactivation | [115,116,117,118,119] |
| miR-200s | CAF activation: Invasion, metastasis via transcription factors Fli-1 and TCF12 | [70] |
| miR-21 | CAF activation: miR-21 and Smad7 induce CAF by TGF-β1 signalling regulation. Motility and invasion by MMP inhibitor RECK | [120,121] |
| miR-155-5p | CAF activation: Angiogenesis by SOCS1/JAK2/STAT3 signalling pathway | [122] |
| miR-1247-3p | CAF activation: Stemness, EMT, chemoresistance, and tumorigenicity by IL-6/8; lung metastasis by β1-integrin-NF-κB pathway | [123] |
| miR-211 | CAF activation | [124] |
| lncRNA | CAF activation/inactivation | [125,126,127] |
| LPA | CAF activation | [128] |
| GPE | CAF activation: Promotes cell proliferation by GPER/EGFR/ERK pathway | [129] |
| DAPT | CAF inactivation | [130] |
| Tumour-derived exosome | CAF activation | [131] |
| Mitocondrial transfer | CAF activation | [132] |
| SMAD | CAF activation/inactivation | [121,127,133,134] |
| Smad2/3 | CAF activation | [134,135] |
| Smad4 | CAF activation | [127,133,134] |
| Smad7 | CAF inactivation | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toledo, B.; Picon-Ruiz, M.; Marchal, J.A.; Perán, M. Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 15576. https://doi.org/10.3390/ijms232415576
Toledo B, Picon-Ruiz M, Marchal JA, Perán M. Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives. International Journal of Molecular Sciences. 2022; 23(24):15576. https://doi.org/10.3390/ijms232415576
Chicago/Turabian StyleToledo, Belén, Manuel Picon-Ruiz, Juan Antonio Marchal, and Macarena Perán. 2022. "Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives" International Journal of Molecular Sciences 23, no. 24: 15576. https://doi.org/10.3390/ijms232415576
APA StyleToledo, B., Picon-Ruiz, M., Marchal, J. A., & Perán, M. (2022). Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives. International Journal of Molecular Sciences, 23(24), 15576. https://doi.org/10.3390/ijms232415576