
IL-1RAP, a Key Therapeutic Target in Cancer
 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Description of the IL-1 Axis & Focus on IL-1RAP
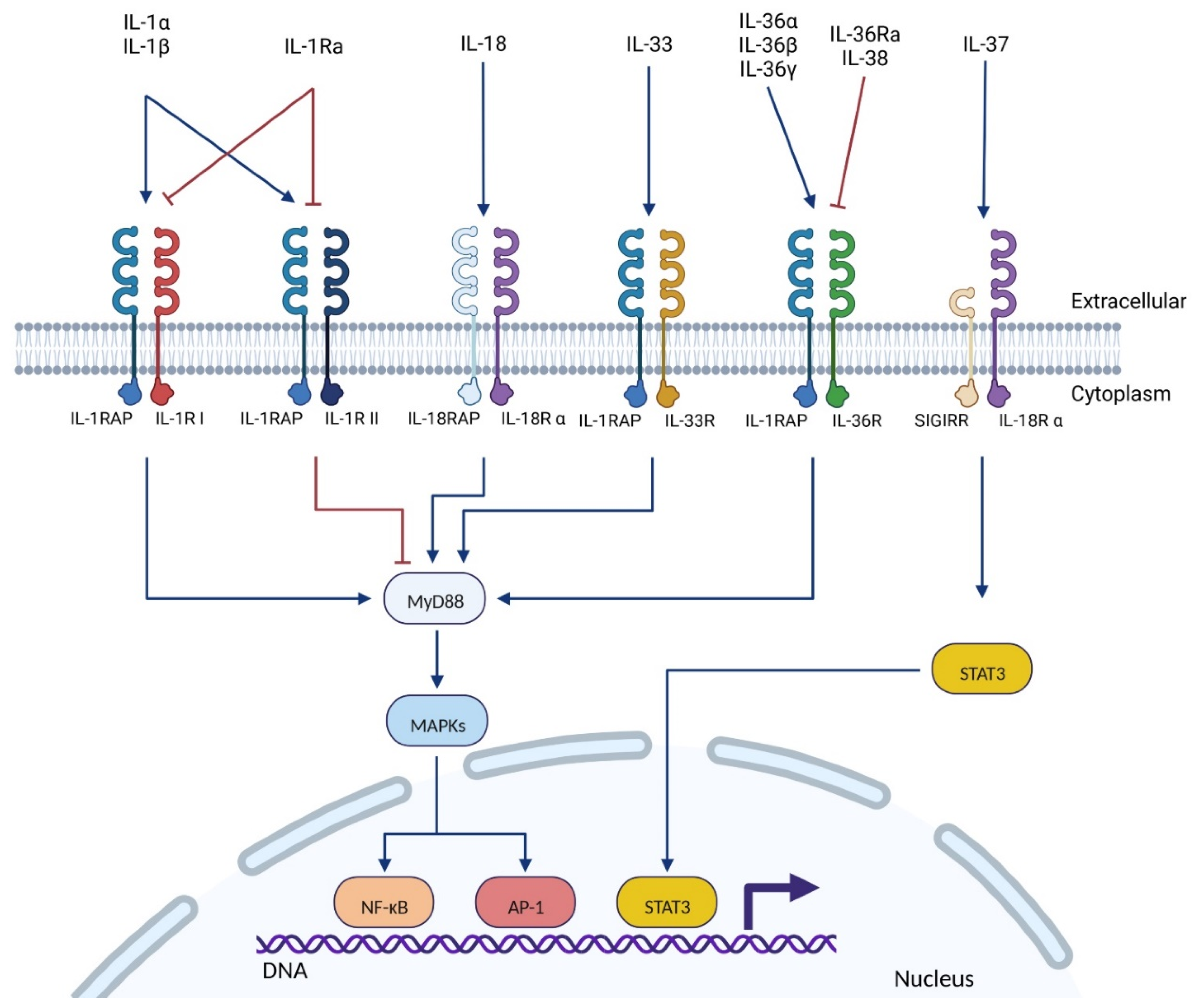
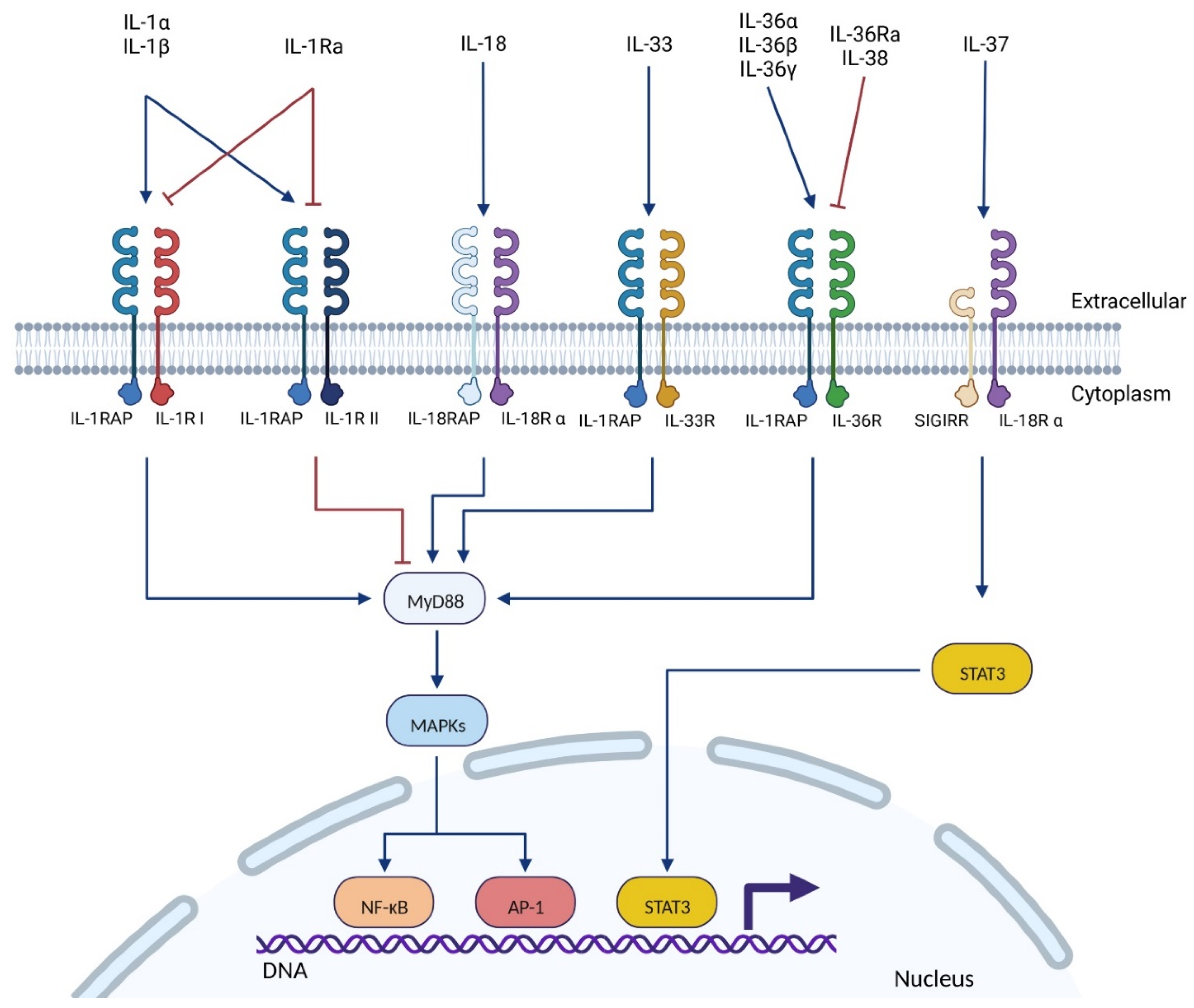
2.1. The IL-1 Superfamily
2.2. IL-1RAP: The Pro-Inflammatory Co-Receptor of the IL-1 Superfamily
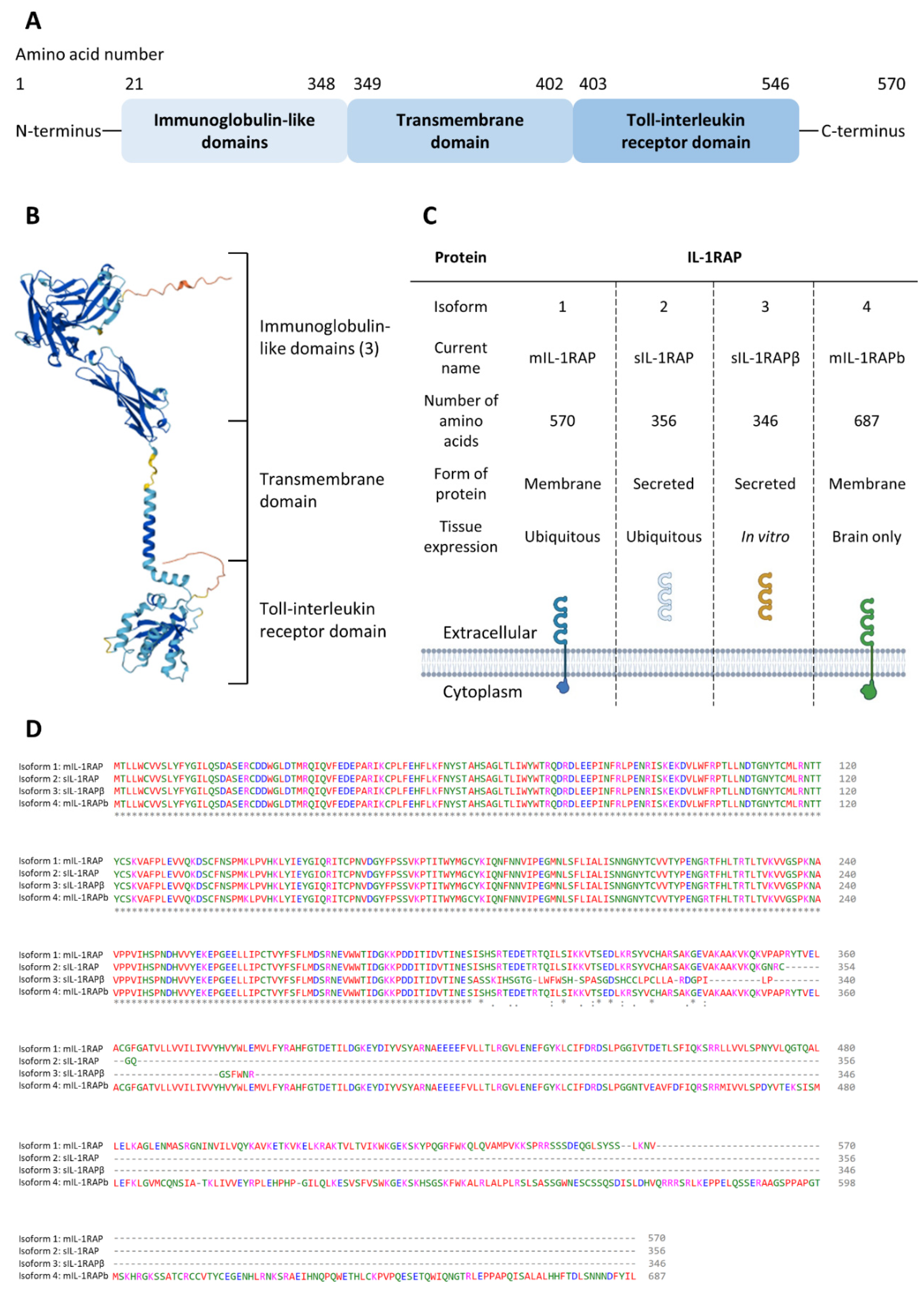
2.2.1. Genetics and Structure of IL-1RAP
- mIL-1RAP: the first form that was described in 1995 by Greenfeder et al. [54]. This mIL-1RAP was first called IL-1-R3, yet IL-1RAP (or IL-1RAcP) is now the usual designation. The mIL-1RAP is involved in the immunity process, and it was recently described in synaptogenesis as being involved in synaptic differentiation [63];
- sIL-1RAP: This isoform is excreted by the cells and is formed by the extracellular domain of mIL-1RAP. The action of sIL-1RAP was explored by Smith et al. in 2003 [64]. They showed that sIL-1RAP could bind the soluble form of IL-1RII and increase its affinity for both IL-1α and IL-1β. However, sIL-1RAP did not increase the affinity of sIL-1R-II for the receptor’s antagonist, IL-1ra. Thus, they suggested that sIL-1RAP plays a role in the negative regulation of IL-1 signaling;
- sIL-1RAP β: in 2003, Jensen and Whitehead discovered a second soluble isoform when cells were treated with staurosporine, which is an apoptosis inducer and an inhibitor of protein kinases. They suggested that under stress conditions, the splicing machinery was shifted to produce sIL-1RAP β instead of mIL-1RAP. Thus, the increase of both the soluble isoforms and the decrease of the membrane form led to the inhibition of IL-1 signaling in order to induce the apoptosis mechanism of the cells. Yet, it appeared that other apoptotic inducers, such as UV light, did not lead to the production of sIL-1RAP β [65];
- IL-1RAPb (also called AcPb): IL-1RAPb structure differs from mIL-1RAP by an extended C-terminal domain (addition of 140 amino acids) and an altered TIR domain;
- This membrane isoform is typically found in the brain and shows different ways of action than classic mIL-1RAP. Indeed, it was first demonstrated in 2009 [66] that IL-1RAPb did not bind the classical IL-1 family signaling pathways protein (MyD88, IRAK-4) and so did not activate the MAPK signaling. Then, in 2011 [67], IL-1RAPb was described as having a role in modulating excitatory neurotransmission by regulating the activation of the kinase Src and NMDA function.
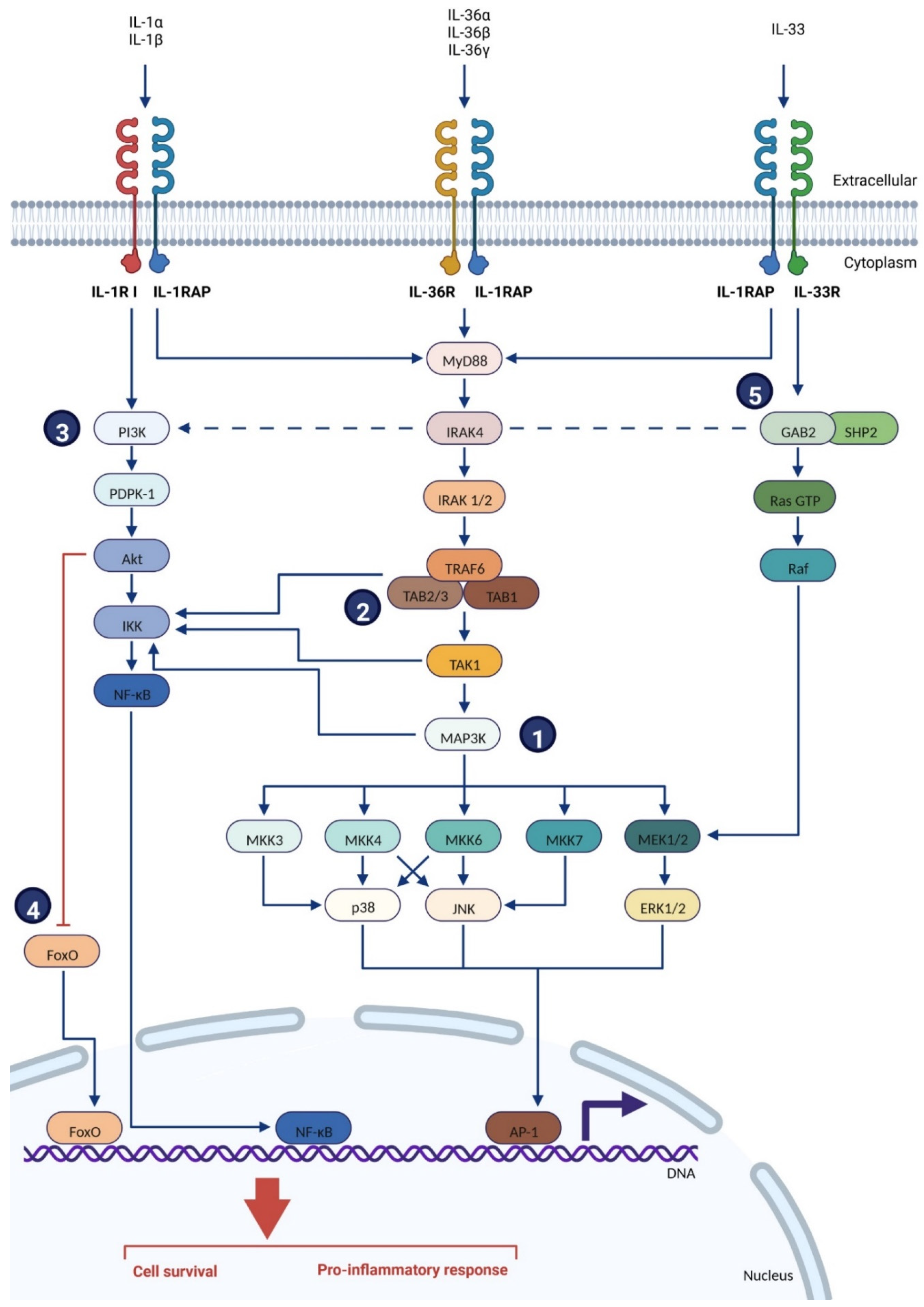
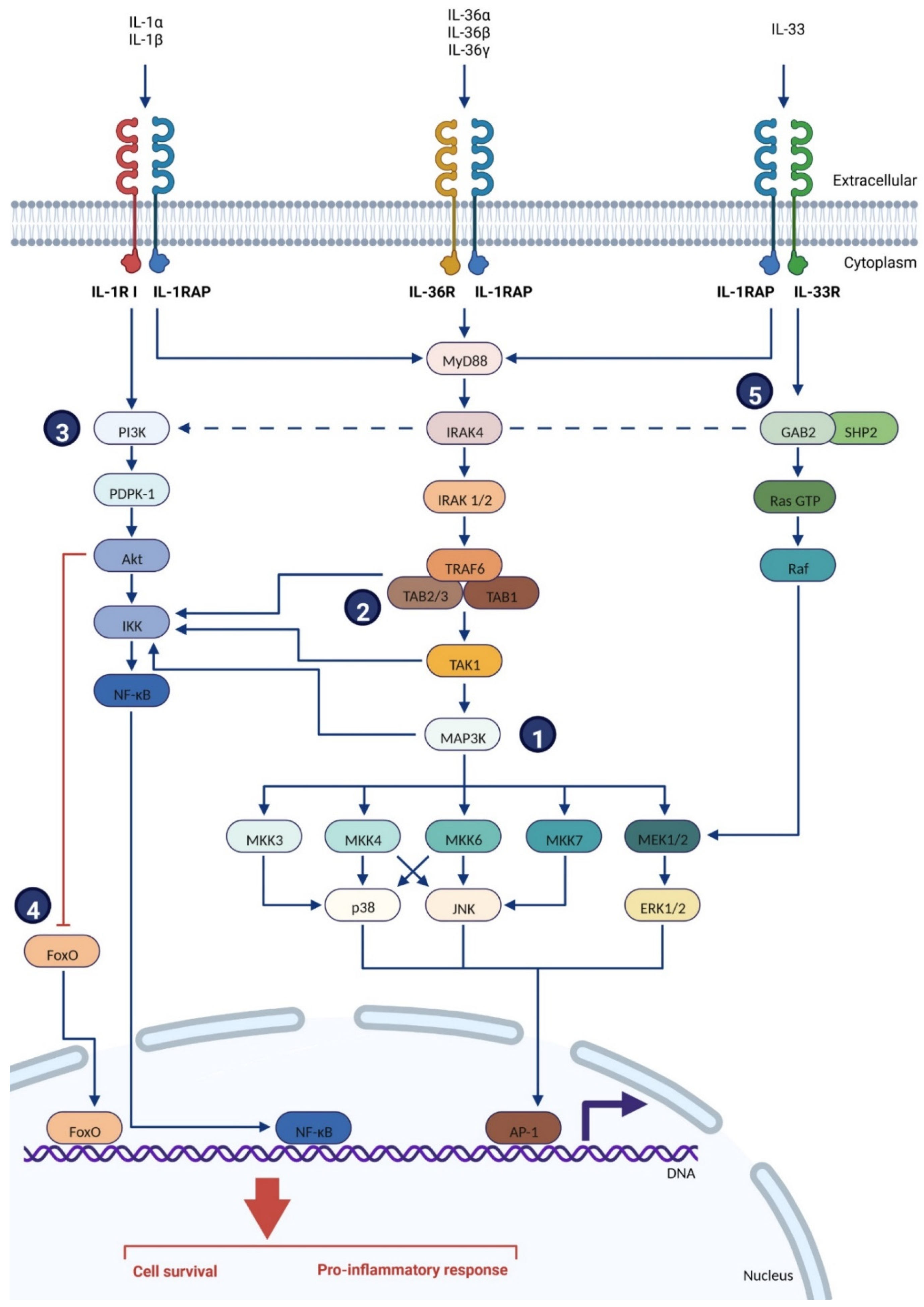
2.2.2. Location & Signaling Pathways
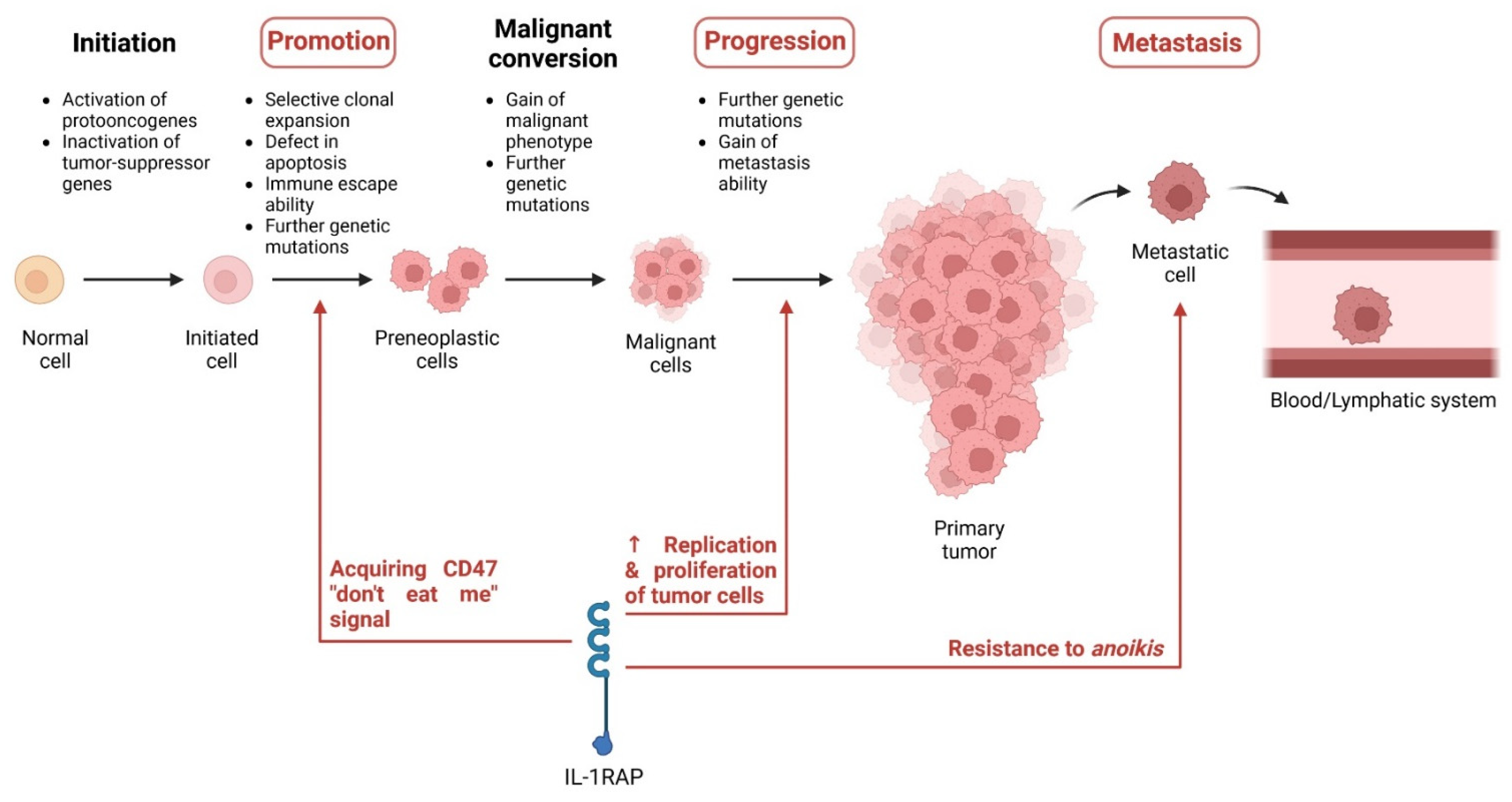
3. Role of IL-1RAP in Tumors
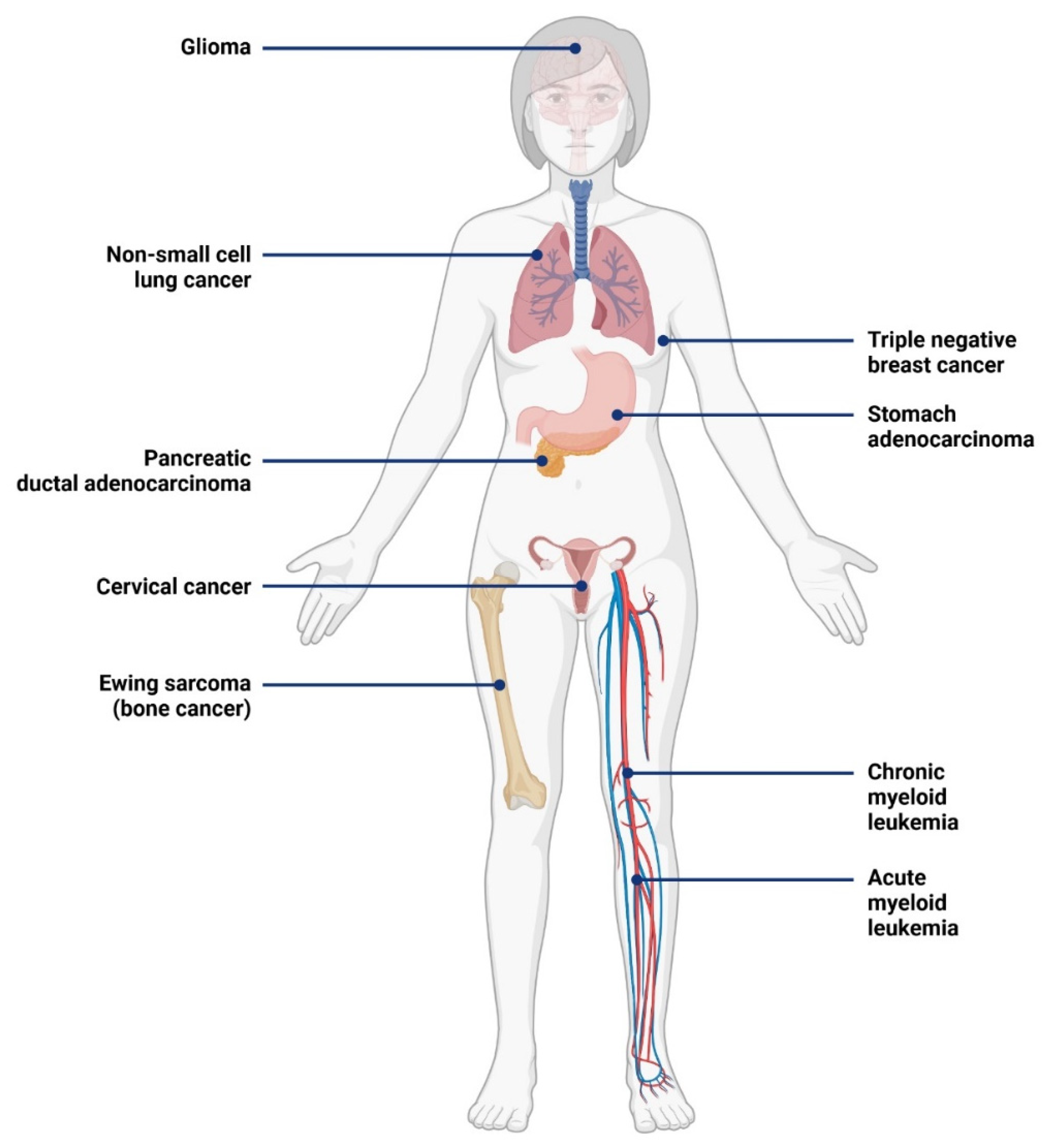
3.1. Expression of IL-1RAP in Cancer
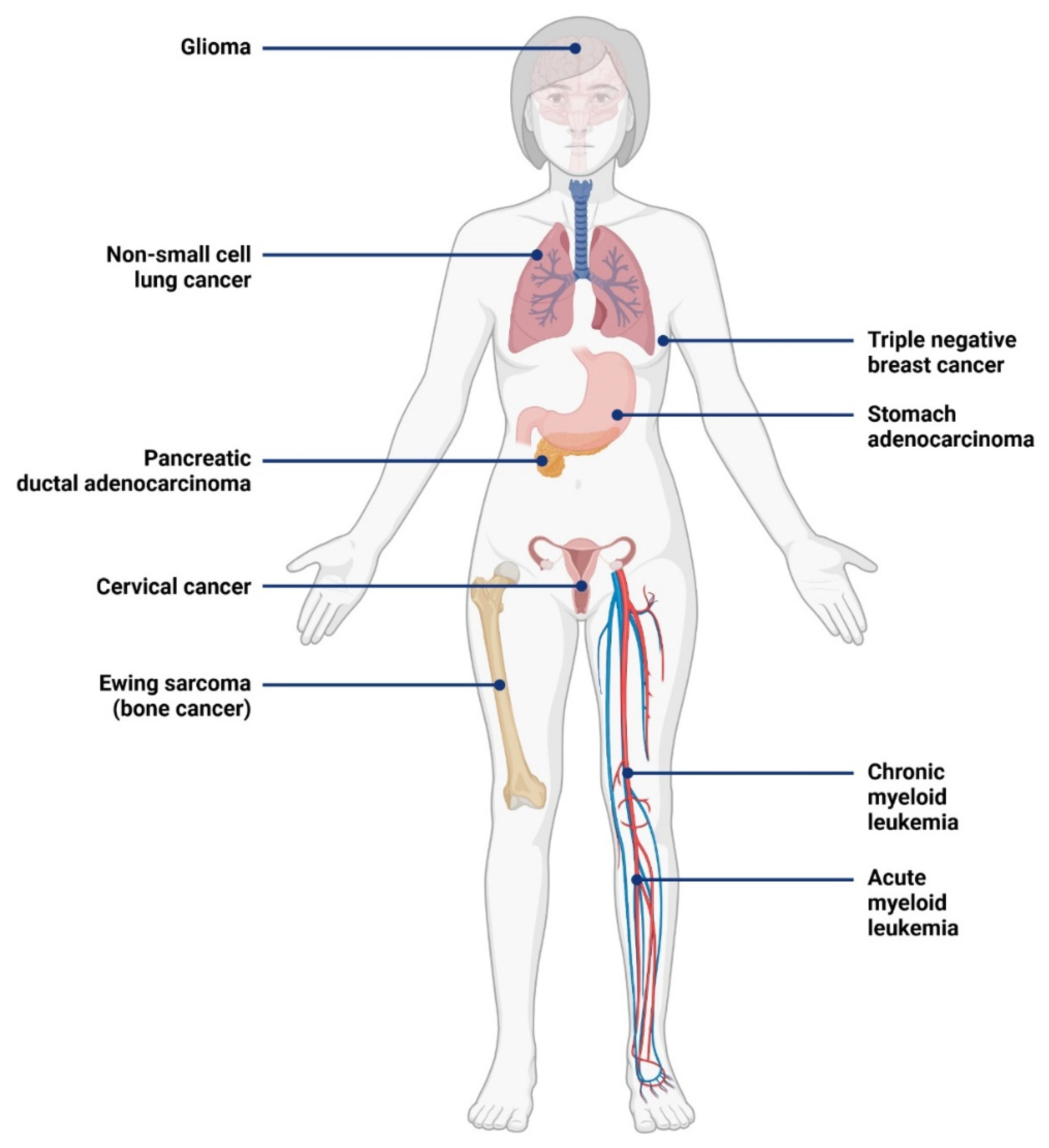
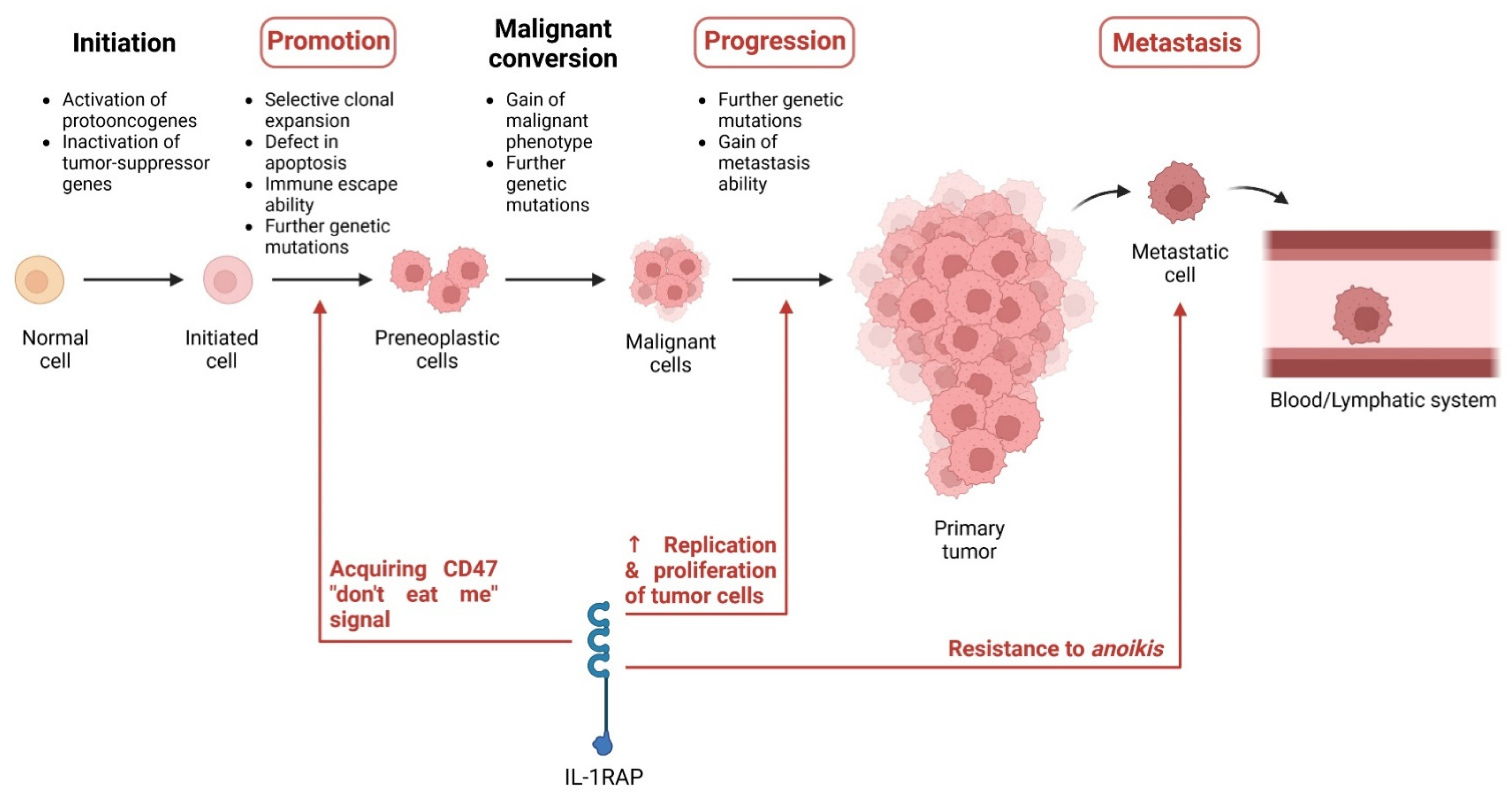
3.2. Role of IL-1RAP in Tumor Progression and Metastases
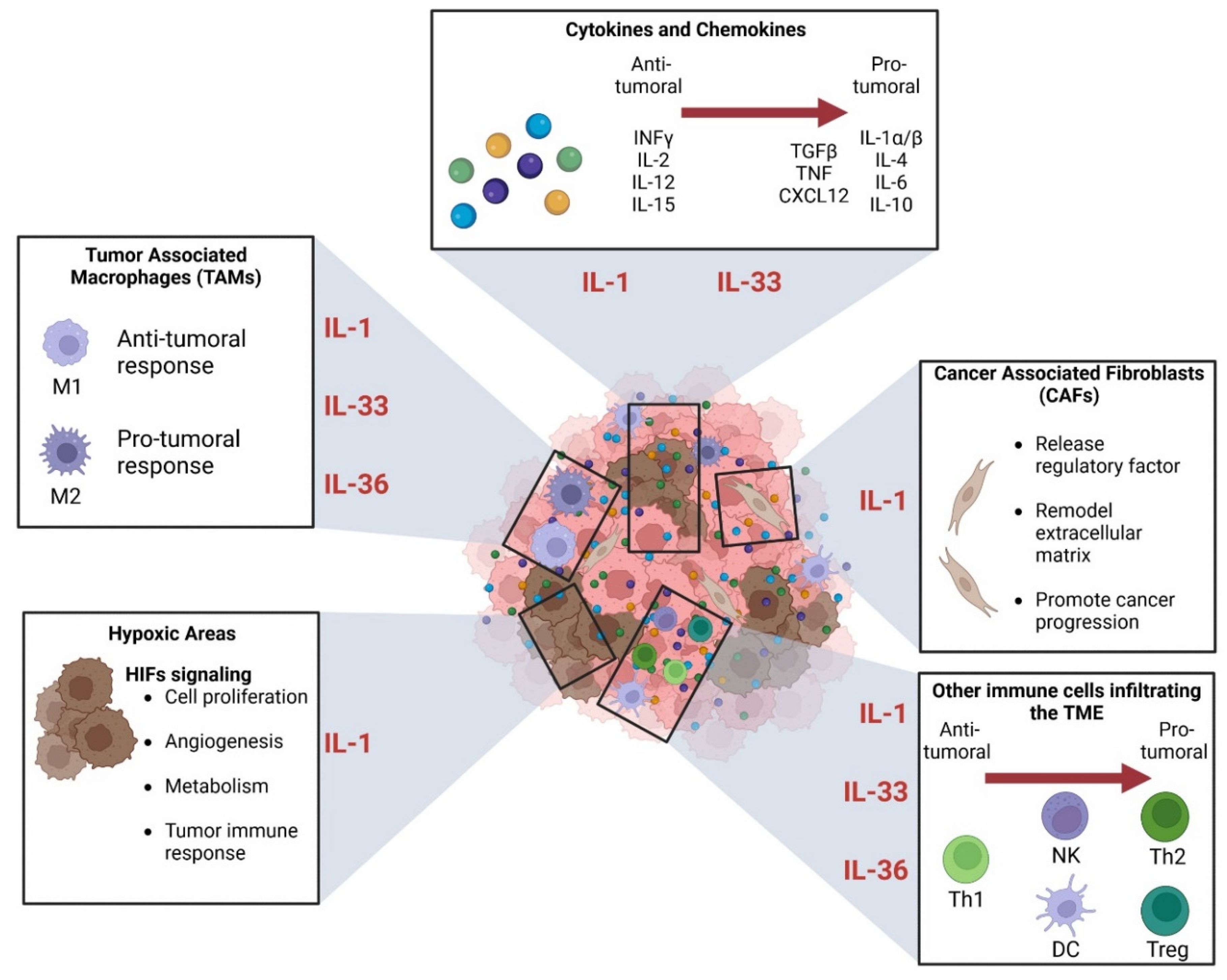
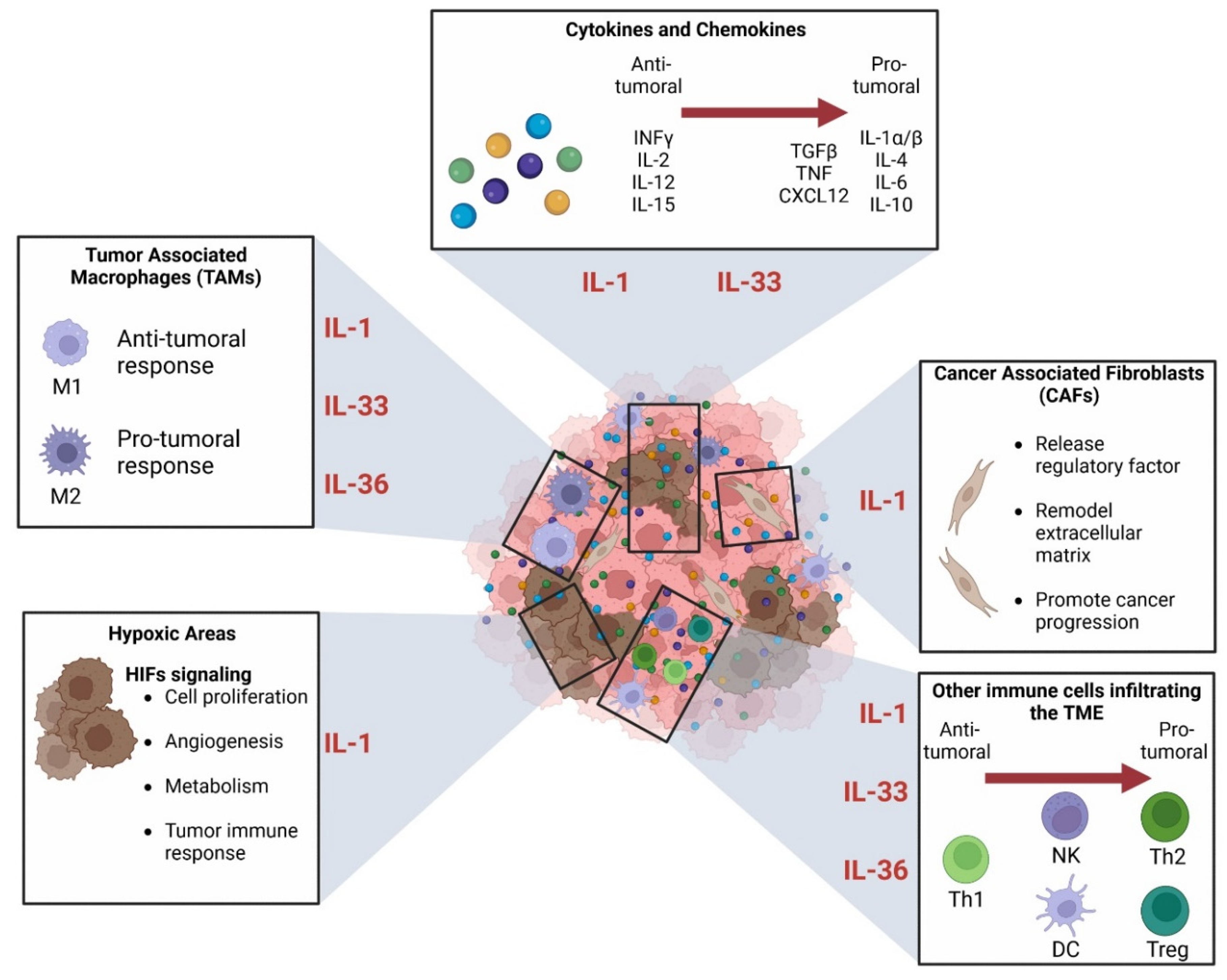
3.3. Role in the Tumor Environment
3.3.1. Connection between IL-1 Superfamily and Immune Cells in TME
3.3.2. Role in Hypoxia
3.3.3. The Relationship between IL-1 Superfamily and the Cancer-Associated Fibroblasts (CAFs)
4. Therapies Targeting IL-1RAP
4.1. Anti-IL-1RAP Antibody in Cancers, Alone and in Combination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-IL-1RAP Treatment | Study | Pathology | Model | Outcomes | References |
|---|---|---|---|---|---|
| Fragment of antibody (scFv 12H7) | In vitro & in vivo (on mice) | TNBC | MDA-MB-231 HCC-70 | Inhibition of the development of TNBC-derived tumors both in vitro and in vivo | [24] |
| Full antibody | In vitro & in vivo (on mice) | CML | KU812 (in vitro) KG-1 (in vitro) CML patients’ cells (in vitro & in vivo) BV-173 (in vivo) | Inhibition of IL-1 signaling and expansion of primitive CML cells | [93,152] |
| Full antibody (mAb81.2) | In vitro | AML | AML patient cells | The antibody can induce ADCC But does not block IL-1 signaling | [96] |
| Full Antibodies (mAb81.2 & mAb3F8) | In vitro & in vivo (on mice) | AML | MA9RasAML patient cells | Antibodies can induce ADCC mAb3F8 can block IL-1 signaling (in vitro) | [153] |
| Full antibody (Nadunolimab) with chemotherapy | In vitro & in vivo (on mice) | NSCLC | LU2503 (PDX) | Nadunolimab works in synergy with chemotherapy by blocking the IL-1α and IL-1β signaling pathway | [28] |
| CAR-T cells (IL1RAP CAR-T) | In vitro & in vivo (on mice) | CML | KU812 cells K562 cells | Cytotoxic activity of autologous CAR-T against CML cells | [27] |
| In vitro & in vivo (on mice) | AML | HL-60 cells Molm-13 cells MonoMac-6 cells PDX | Cytotoxic activity of autologous CAR-T against AML cells | [154] |
| Anti-IL-1RAP Treatment | Treatment in Combination | Clinical Phase | Pathology | CURRENT STATUS | ClinicalTrial ID (Study Name) |
|---|---|---|---|---|---|
| Full antibody (Nadunolimab) | Cisplatin Gemcitabine Nab-paclitaxel | I/IIa | PDAC (phase I/IIa) NSCLC (phase I/IIa) TNBC (phase I) CRC (phase I) | Phase I: Completed Phase IIa: Active, not recruiting (PDAC) Recruiting (NSCLC) | NCT03267316 (CANFOUR) [26,150,151] |
| Full antibody (Nadunolimab) | Pembrolizumab | I | NSCLC UC HNSCC Malignant Melanoma | Active, not recruiting | NCT04452214 (CIRIFOUR) |
| Full antibody (Nadunolimab) | FOLFIRINOX | I | Metastatic PDAC | Active, not recruiting | NCT04990037 (CAPAFOUR) |
| Full antibody (Nadunolimab) | mFOLFOX DTX G/C | I/II | Advanced solid tumors CRC NSCLC BTC | Active, not recruiting | NCT05116891 (CESTAFOUR) |
| Full antibody (Nadunolimab) | Carboplatin Gemcitabine | I/II | TNBC | Recruiting | NCT05181462 (TRIFOUR) |
| CAR-T cells | None | N/A | CML | Completed | NCT02842320 (CAR-LMC) |
| CAR-T cells | None | N/A | AML | Recruiting | NCT04169022 (CAR-LAM) |
- i.
- Preclinical in vitro and in vivo study in CML
- ii.
- Preclinical in vitro and in vivo study in AML
- iii.
- The CAN04 antibody (Nadunolimab) in the clinical trials of Cantargia AB
- Started in August 2017, the CANFOUR clinical trial (ClinicalTrial ID NCT03267316) is a phase I/IIa open study. It is conducted in several European countries (28 locations among 11 countries). The goal is to assess the safety and efficacy of CAN04 in solid tumors. The study is separated into two parts: (i) to assess the safety and tolerability of the product and to evaluate the maximum tolerated dose and recommended phase 2 dose (MTD/RP2D) in PDAC, NSCLC, TNBC, and CRC [26]; and (ii) to evaluate the safety and tolerability at RP2D level in an expanded cohort in PDAC and NSCLC patients. This study aims to enroll 140 participants;
- Starting in September 2020, this second clinical trial on CAN04 (ClinicalTrial ID NCT04452214) is a phase I open-label study. The goal is to assess the safety and to establish a recommended dose of CAN04 in combination with pembrolizumab for the treatment of incurable or metastatic NSCLC, head and neck squamous cell carcinoma, urothelial cancer, or malignant melanoma. This study expects to enroll 15 participants;
- Started in July 2021, the CAPAFOUR study (ClinicalTrial ID NCT04990037) is a phase I open-label study. This clinical trial aims to determine the safety and effectiveness of CAN04 in combination with a modified FOLFIRINOX protocol in metastatic PDAC. The study expects 50 participants;
- The fourth study started in September 2021 (ClinicalTrial ID NCT05116891), and it is a phase I/II open-label study. The phase I goal is to assess the safety, tolerability and MTD/RP2D of CAN04 in combination with standard chemotherapies (mFOLFOX, gemcitabine/cisplatin, docetaxel), while phase II will aim to assess the preliminary efficacy of CAN04 in combination with the same chemotherapies. The two phases will focus on locally advanced or metastatic CRC, NSCLC, or biliary tract cancer. This study aims to enroll 180 participants;
- The last study, TRIFOUR, started in January 2022 (ClinicalTrial ID NCT05181462) and is a phase I/II randomized open-label study. The phase I goal is to assess the safety and highest dose without the serious side effects of CAN04 in combination with gemcitabine and carboplatin. Phase II will aim to assess the efficacy of CAN04 in combination with the same chemotherapy. The two phases will focus on patients with advanced or metastatic TNBC. This study expects to enroll 18 patients in phase I and 98 patients in phase II.
4.2. Anti-IL-1RAP Chimeric Antigen Receptor T (CAR-T) Cells in Cancers
4.2.1. Preclinical Studies
4.2.2. Clinical Trials
- The CAR-LMC clinical trial (ClinicalTrials ID NCT02842320): started in October 2015, this trial enrolled 53 patients and monitored them for 24 months in an open-label single-armed study. The first step was to collect T-cells from the patient and engineer them with a CAR. Then, they infused them back into the patient and started the monitoring with blood samples. These samples were analyzed by flow cytometry to detect IL-1RAP expression on the surface of cells. At the time of writing this review, the study is ending, and no results have been published yet;
- The CAR-LAM clinical trial (ClinicalTrials ID NCT04169022): started in July 2019, this trial is still recruiting, and the team aims for the enrollment of 50 patients. It is an open-label, non-randomized two-armed study. The first arm is comprised of AML patients at diagnosis (except AML3), while the second arm is made up of AML patients at relapse after chemotherapy, targeted therapy, or allografts. The protocol will be similar to the CAR-LMC study: a two-year follow-up with flow cytometry analysis to explore the IL-1RAP expression of cells in blood samples. At the time of writing this review, the study is starting, and no results have been published yet.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dagenais, G.R.; Leong, D.P.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Gupta, R.; Diaz, R.; Avezum, A.; Oliveira, G.B.F.; Wielgosz, A.; et al. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): A prospective cohort study. Lancet 2020, 395, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Spath, S.S.; Marjani, S.L.; Zhang, W.; Pan, X. Characterization of cancer genomic heterogeneity by next-generation sequencing advances precision medicine in cancer treatment. Precis. Clin. Med. 2018, 1, 29–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Salas-Benito, D.; Perez-Gracia, J.L.; Ponz-Sarvise, M.; Rodriguez-Ruiz, M.E.; Martinez-Forero, I.; Castanon, E.; Lopez-Picazo, J.M.; Sanmamed, M.F.; Melero, I. Paradigms on Immunotherapy Combinations with Chemotherapy. Cancer Discov. 2021, 11, 1353–1367. [Google Scholar] [CrossRef] [PubMed]
- Peckham, M.J.; Barrett, A.; Liew, K.H.; Horwich, A.; Robinson, B.; Dobbs, H.J.; McElwain, T.J.; Hendry, W.F. The treatment of metastatic germ-cell testicular tumours with bleomycin, etoposide and cis-platin (BEP). Br. J. Cancer 1983, 47, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Spiers, A.S.; Roberts, P.D.; Marsh, G.W.; Parekh, S.J.; Franklin, A.J.; Galton, D.A.; Szur, Z.L.; Paul, E.A.; Husband, P.; Wiltshaw, E. Acute lymphoblastic leukaemia: Cyclical chemotherapy with three combinations of four drugs (COAP-POMP-CART regimen). Br. Med. J. 1975, 4, 614–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Afify, S.M.; Hassan, G.; Seno, A.; Seno, M. Cancer-inducing niche: The force of chronic inflammation. Br. J. Cancer 2022, 127, 193–201. [Google Scholar] [CrossRef]
- Danforth, D.N. The Role of Chronic Inflammation in the Development of Breast Cancer. Cancers 2021, 13, 3918. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Piotrowski, I.; Kulcenty, K.; Suchorska, W. Interplay between inflammation and cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 422–427. [Google Scholar] [CrossRef]
- Borish, L.C.; Steinke, J.W. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111 (Suppl. S2), S460––S475. [Google Scholar] [CrossRef]
- Atsumi, T.; Singh, R.; Sabharwal, L.; Bando, H.; Meng, J.; Arima, Y.; Yamada, M.; Harada, M.; Jiang, J.J.; Kamimura, D.; et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 2014, 74, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Barreyro, L.; Will, B.; Bartholdy, B.; Zhou, L.; Todorova, T.I.; Stanley, R.F.; Ben-Neriah, S.; Montagna, C.; Parekh, S.; Pellagatti, A.; et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 2012, 120, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family--Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 76, 25–37. [Google Scholar] [CrossRef]
- Fields, J.K.; Gunther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [Green Version]
- Eldesouki, R.E.; Wu, C.; Saleh, F.M.; Mohammed, E.A.; Younes, S.; Hassan, N.E.; Brown, T.C.; Alt, E.U.; Robinson, J.E.; Badr, F.M.; et al. Identification and Targeting of Thomsen-Friedenreich and IL1RAP Antigens on Chronic Myeloid Leukemia Stem Cells Using Bi-Specific Antibodies. Onco. Targets Ther. 2021, 14, 609–621. [Google Scholar] [CrossRef]
- Blatt, K.; Menzl, I.; Eisenwort, G.; Cerny-Reiterer, S.; Herrmann, H.; Herndlhofer, S.; Stefanzl, G.; Sadovnik, I.; Berger, D.; Keller, A.; et al. Phenotyping and Target Expression Profiling of CD34(+)/CD38(-) and CD34(+)/CD38(+) Stem- and Progenitor cells in Acute Lymphoblastic Leukemia. Neoplasia 2018, 20, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zhang, Y.; Zhang, B.; Wang, Y.; Wang, Y.; Yang, L. Synthetic human monoclonal antibody targets hIL1 receptor accessory protein chain with therapeutic potential in triple-negative breast cancer. Biomed. Pharmacother. 2018, 107, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Wang, H.; Gordon-Mitchell, S.; Sahu, S.; Bhagat, T.D.; Choudhary, G.; Aluri, S.; Pradhan, K.; Sahu, P.; et al. Innate immune mediator, Interleukin-1 receptor accessory protein (IL1RAP), is expressed and pro-tumorigenic in pancreatic cancer. J. Hematol. Oncol. 2022, 15, 70. [Google Scholar] [CrossRef] [PubMed]
- Robbrecht, D.; Jungels, C.; Sorensen, M.M.; Spanggaard, I.; Eskens, F.; Fretland, S.O.; Guren, T.K.; Aftimos, P.; Liberg, D.; Svedman, C.; et al. First-in-human phase 1 dose-escalation study of CAN04, a first-in-class interleukin-1 receptor accessory protein (IL1RAP) antibody in patients with solid tumours. Br. J. Cancer 2022, 126, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Warda, W.; Larosa, F.; Neto Da Rocha, M.; Trad, R.; Deconinck, E.; Fajloun, Z.; Faure, C.; Caillot, D.; Moldovan, M.; Valmary-Degano, S.; et al. CML Hematopoietic Stem Cells Expressing IL1RAP Can Be Targeted by Chimeric Antigen Receptor-Engineered T Cells. Cancer Res. 2019, 79, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Rydberg Millrud, C.; Deronic, A.; Gronberg, C.; Jaensson Gyllenback, E.; von Wachenfeldt, K.; Forsberg, G.; Liberg, D. Blockade of IL-1alpha and IL-1beta signaling by the anti-IL1RAP antibody nadunolimab (CAN04) mediates synergistic anti-tumor efficacy with chemotherapy. Cancer Immunol. Immunother. 2022. [Google Scholar] [CrossRef]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Chang, H.K.; Mohan, S.K.; Chin, Y. 1H, 13C and 15N backbone and side chain resonance assignments of human interleukin 1alpha. Biomol. NMR Assign. 2010, 4, 59–60. [Google Scholar] [CrossRef]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Nussinov, R.; Keskin, O. The structural pathway of interleukin 1 (IL-1) initiated signaling reveals mechanisms of oncogenic mutations and SNPs in inflammation and cancer. PLoS Comput. Biol. 2014, 10, e1003470. [Google Scholar] [CrossRef] [Green Version]
- Kuo, W.C.; Lee, C.C.; Chang, Y.W.; Pang, W.; Chen, H.S.; Hou, S.C.; Lo, S.Y.; Yang, A.S.; Wang, A.H. Structure-based Development of Human Interleukin-1beta-Specific Antibody That Simultaneously Inhibits Binding to Both IL-1RI and IL-1RAcP. J. Mol. Biol. 2021, 433, 166766. [Google Scholar] [CrossRef]
- Stockman, B.J.; Scahill, T.A.; Strakalaitis, N.A.; Brunner, D.P.; Yem, A.W.; Deibel, M.R., Jr. Solution structure of human interleukin-1 receptor antagonist protein. FEBS Lett. 1994, 349, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, N.; Kimura, T.; Arita, K.; Ariyoshi, M.; Ohnishi, H.; Yamamoto, T.; Zuo, X.; Maenaka, K.; Park, E.Y.; Kondo, N.; et al. The structural basis for receptor recognition of human interleukin-18. Nat. Commun. 2014, 5, 5340. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Liu, X.; Hammel, M.; He, Y.; Tainer, J.A.; Jeng, U.S.; Zhang, L.; Wang, S.; Wang, X. Structural insights into the interaction of IL-33 with its receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 14918–14923. [Google Scholar] [CrossRef] [Green Version]
- Elias, M.; Zhao, S.; Le, H.T.; Wang, J.; Neurath, M.F.; Neufert, C.; Fiocchi, C.; Rieder, F. IL-36 in chronic inflammation and fibrosis—bridging the gap? J. Clin. Investig. 2021, 131, e144336. [Google Scholar] [CrossRef]
- Goradia, N.; Wissbrock, A.; Wiedemann, C.; Bordusa, F.; Ramachandran, R.; Imhof, D.; Ohlenschlager, O. (1)H, (13)C, and (15)N resonance assignments for the pro-inflammatory cytokine interleukin-36alpha. Biomol. NMR Assign. 2016, 10, 329–333. [Google Scholar] [CrossRef]
- Todorovic, V.; Su, Z.; Putman, C.B.; Kakavas, S.J.; Salte, K.M.; McDonald, H.A.; Wetter, J.B.; Paulsboe, S.E.; Sun, Q.; Gerstein, C.E.; et al. Small Molecule IL-36gamma Antagonist as a Novel Therapeutic Approach for Plaque Psoriasis. Sci. Rep. 2019, 9, 9089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunther, S.; Sundberg, E.J. Molecular determinants of agonist and antagonist signaling through the IL-36 receptor. J. Immunol. 2014, 193, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front. Immunol. 2021, 12, 696605. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, A.M.; Nold-Petry, C.A.; D’Andrea, L.; Cho, S.X.; Lao, J.C.; Rudloff, I.; Ngo, D.; Lo, C.Y.; Soares da Costa, T.P.; Perugini, M.A.; et al. Homodimerization attenuates the anti-inflammatory activity of interleukin-37. Sci. Immunol. 2017, 2, eaaj1548. [Google Scholar] [CrossRef]
- Xie, L.; Huang, Z.; Li, H.; Liu, X.; Zheng, S.; Su, W. IL-38: A New Player in Inflammatory Autoimmune Disorders. Biomolecules 2019, 9, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Veerdonk, F.L.; de Graaf, D.M.; Joosten, L.A.; Dinarello, C.A. Biology of IL-38 and its role in disease. Immunol. Rev. 2018, 281, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Guenther, S.S.; Sundberg, E.J. Crystal Structure of IL-38. Available online: https://www.rcsb.org/structure/5bow (accessed on 30 August 2022).
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef] [Green Version]
- Pavlowsky, A.; Gianfelice, A.; Pallotto, M.; Zanchi, A.; Vara, H.; Khelfaoui, M.; Valnegri, P.; Rezai, X.; Bassani, S.; Brambilla, D.; et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr. Biol. 2010, 20, 103–115. [Google Scholar] [CrossRef]
- Pavlowsky, A.; Zanchi, A.; Pallotto, M.; Giustetto, M.; Chelly, J.; Sala, C.; Billuart, P. Neuronal JNK pathway activation by IL-1 is mediated through IL1RAPL1, a protein required for development of cognitive functions. Commun. Integr. Biol 2010, 3, 245–247. [Google Scholar] [CrossRef]
- Nimma, S.; Gu, W.; Manik, M.K.; Ve, T.; Nanson, J.D.; Kobe, B. Crystal structure of the Toll/interleukin-1 receptor (TIR) domain of IL-1R10 provides structural insights into TIR domain signalling. FEBS Lett. 2022, 596, 886–897. [Google Scholar] [CrossRef]
- Greenfeder, S.A.; Nunes, P.; Kwee, L.; Labow, M.; Chizzonite, R.A.; Ju, G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J. Biol. Chem. 1995, 270, 13757–13765. [Google Scholar] [CrossRef] [Green Version]
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debets, R.; Timans, J.C.; Churakowa, T.; Zurawski, S.; de Waal Malefyt, R.; Moore, K.W.; Abrams, J.S.; O’Garra, A.; Bazan, J.F.; Kastelein, R.A. IL-18 receptors, their role in ligand binding and function: Anti-IL-1RAcPL antibody, a potent antagonist of IL-18. J. Immunol. 2000, 165, 4950–4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, M.; Hammond, D.W.; Cox, A.; Nicklin, M.J. The human gene encoding the interleukin-1 receptor accessory protein (IL1RAP) maps to chromosome 3q28 by fluorescence in situ hybridization and radiation hybrid mapping. Genomics 1998, 47, 325–326. [Google Scholar] [CrossRef]
- Radons, J.; Dove, S.; Neumann, D.; Altmann, R.; Botzki, A.; Martin, M.U.; Falk, W. The interleukin 1 (IL-1) receptor accessory protein Toll/IL-1 receptor domain: Analysis of putative interaction sites in vitro mutagenesis and molecular modeling. J. Biol. Chem. 2003, 278, 49145–49153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingel, A.; Weiss, T.M.; Niebuhr, M.; Pan, B.; Appleton, B.A.; Wiesmann, C.; Bazan, J.F.; Fairbrother, W.J. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors--insight into heterotrimeric IL-1 signaling complexes. Structure 2009, 17, 1398–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadio, R.; Frigimelica, E.; Bossu, P.; Neumann, D.; Martin, M.U.; Tagliabue, A.; Boraschi, D. Model of interaction of the IL-1 receptor accessory protein IL-1RAcP with the IL-1beta/IL-1R(I) complex. FEBS Lett. 2001, 499, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.; Ybe, J.A.; Saha, S.S.; Caviness, G.; Raymond, E.; Ganesan, R.; Mbow, M.L.; Kao, C.C. Structural and Functional Attributes of the Interleukin-36 Receptor. J. Biol. Chem. 2016, 291, 16597–16609. [Google Scholar] [CrossRef]
- Stroud, R.M.; Wells, J.A. Mechanistic diversity of cytokine receptor signaling across cell membranes. Sci. STKE 2004, 2004, re7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagata, A.; Yoshida, T.; Sato, Y.; Goto-Ito, S.; Uemura, T.; Maeda, A.; Shiroshima, T.; Iwasawa-Okamoto, S.; Mori, H.; Mishina, M.; et al. Mechanisms of splicing-dependent trans-synaptic adhesion by PTPdelta-IL1RAPL1/IL-1RAcP for synaptic differentiation. Nat. Commun. 2015, 6, 6926. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.E.; Hanna, R.; Della, F.; Moore, H.; Chen, H.; Farese, A.M.; MacVittie, T.J.; Virca, G.D.; Sims, J.E. The soluble form of IL-1 receptor accessory protein enhances the ability of soluble type II IL-1 receptor to inhibit IL-1 action. Immunity 2003, 18, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, L.E.; Whitehead, A.S. Expression of alternatively spliced interleukin-1 receptor accessory protein mRNAs is differentially regulated during inflammation and apoptosis. Cell Signal. 2003, 15, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Lipsky, B.P.; Russell, C.; Ketchem, R.R.; Kirchner, J.; Hensley, K.; Huang, Y.; Friedman, W.J.; Boissonneault, V.; Plante, M.M.; et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 2009, 30, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Smith, D.E.; Ibanez-Sandoval, O.; Sims, J.E.; Friedman, W.J. Neuron-specific effects of interleukin-1beta are mediated by a novel isoform of the IL-1 receptor accessory protein. J. Neurosci. 2011, 31, 18048–18059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Pascual-Figal, D.A.; Januzzi, J.L. The biology of ST2: The International ST2 Consensus Panel. Am. J. Cardiol. 2015, 115 (Suppl. S7), 3B–7B. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penha, R.; Higgins, J.; Mutamba, S.; Barrow, P.; Mahida, Y.; Foster, N. IL-36 receptor is expressed by human blood and intestinal T lymphocytes and is dose-dependently activated via IL-36beta and induces CD4+ lymphocyte proliferation. Cytokine 2016, 85, 18–25. [Google Scholar] [CrossRef]
- Kopp, E.; Medzhitov, R.; Carothers, J.; Xiao, C.; Douglas, I.; Janeway, C.A.; Ghosh, S. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 1999, 13, 2059–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karandikar, M.; Xu, S.; Cobb, M.H. MEKK1 binds raf-1 and the ERK2 cascade components. J. Biol. Chem. 2000, 275, 40120–40127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Tuckerman, J.; Nechushtan, H.; Schutz, G.; Razin, E.; Angel, P. c-Fos as a regulator of degranulation and cytokine production in FcepsilonRI-activated mast cells. J. Immunol. 2004, 173, 2571–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; He, H.; Jonsson, P.; Sinha, I.; Zhao, C.; Dahlman-Wright, K. AP-1 Is a Key Regulator of Proinflammatory Cytokine TNFalpha-mediated Triple-negative Breast Cancer Progression. J. Biol. Chem. 2016, 291, 5068–5079. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar]
- Mun, S.H.; Ko, N.Y.; Kim, H.S.; Kim, J.W.; Kim, D.K.; Kim, A.R.; Lee, S.H.; Kim, Y.G.; Lee, C.K.; Lee, S.H.; et al. Interleukin-33 stimulates formation of functional osteoclasts from human CD14(+) monocytes. Cell Mol. Life Sci. 2010, 67, 3883–3892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.A.; Lin, Y.F.; Huang, H.J.; Samanta, A.K.; Liao, W.S. The IL-1 receptor accessory protein is essential for PI 3-kinase recruitment and activation. Biochem. Biophys. Res. Commun. 2004, 316, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inatomi, O.; Fujimoto, T.; Imaeda, H.; Tani, M.; Andoh, A. Interleukin-36alpha Induces Inflammatory Mediators From Human Pancreatic Myofibroblasts Via a MyD88 Dependent Pathway. Pancreas 2017, 46, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; Neufert, C. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2017, 66, 823–838. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, Y.; Feng, S.; Ishida, Y.; Chiu, T.P.; Saito, T.; Wang, S.; Ann, D.K.; Ou, J.J. Macrophages activated by hepatitis B virus have distinct metabolic profiles and suppress the virus via IL-1beta to downregulate PPARalpha and FOXO3. Cell Rep. 2022, 38, 110284. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. FOXO transcription factors in ageing and cancer. Acta Physiol. 2008, 192, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Qiao, Q.; Ferrao, R.; Shen, C.; Hatcher, J.M.; Buhrlage, S.J.; Gray, N.S.; Wu, H. Crystal structure of human IRAK1. Proc. Natl. Acad. Sci. USA 2017, 114, 13507–13512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Kanakaraj, P.; Schafer, P.H.; Cavender, D.E.; Wu, Y.; Ngo, K.; Grealish, P.F.; Wadsworth, S.A.; Peterson, P.A.; Siekierka, J.J.; Harris, C.A.; et al. Interleukin (IL)-1 receptor-associated kinase (IRAK) requirement for optimal induction of multiple IL-1 signaling pathways and IL-6 production. J. Exp. Med. 1998, 187, 2073–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Keshava Prasad, T.S.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal. 2018, 12, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.B.; Yu, W.N.; Feng, J.H.; Luo, J.M. Structure and function of Gab2 and its role in cancer (Review). Mol. Med. Rep. 2015, 12, 4007–4014. [Google Scholar] [CrossRef] [Green Version]
- Melo, J.V. The molecular biology of chronic myeloid leukaemia. Leukemia 1996, 10, 751–756. [Google Scholar] [PubMed]
- Heisterkamp, N.; Stam, K.; Groffen, J.; de Klein, A.; Grosveld, G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature 1985, 315, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Jaras, M.; Johnels, P.; Hansen, N.; Agerstam, H.; Tsapogas, P.; Rissler, M.; Lassen, C.; Olofsson, T.; Bjerrum, O.W.; Richter, J.; et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl. Acad. Sci. USA 2010, 107, 16280–16285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosberg, S.; Greif, P.A. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.; Barreyro, L.; Todorova, T.I.; Taylor, S.J.; Antony-Debre, I.; Narayanagari, S.R.; Carvajal, L.A.; Leite, J.; Piperdi, Z.; Pendurti, G.; et al. IL1RAP potentiates multiple oncogenic signaling pathways in AML. J. Exp. Med. 2018, 215, 1709–1727. [Google Scholar] [CrossRef] [PubMed]
- Askmyr, M.; Agerstam, H.; Hansen, N.; Gordon, S.; Arvanitakis, A.; Rissler, M.; Juliusson, G.; Richter, J.; Jaras, M.; Fioretos, T. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 2013, 121, 3709–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Rulicke, T.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of target expression profiles in CD34+/CD38- and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020, 4, 5118–5132. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef]
- Zhang, H.F.; Hughes, C.S.; Li, W.; He, J.Z.; Surdez, D.; El-Naggar, A.M.; Cheng, H.; Prudova, A.; Delaidelli, A.; Negri, G.L.; et al. Proteomic Screens for Suppressors of Anoikis Identify IL1RAP as a Promising Surface Target in Ewing Sarcoma. Cancer Discov. 2021, 11, 2884–2903. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Liberg, D.S.W.; Riva, M.; Massoumi, R.; Forsberg, G.; Von Wachenfeldt, K. The CAN04 Antibody Targets IL1RAP and Inhibits Tumor Growth in a PDX Model for NSCLC. Available online: https://cantargia.com/assets/uploads/Poster-PEGS-Europe-final.pdf (accessed on 24 October 2022).
- Rasmussen, B.K.; Hansen, S.; Laursen, R.J.; Kosteljanetz, M.; Schultz, H.; Norgard, B.M.; Guldberg, R.; Gradel, K.O. Epidemiology of glioma: Clinical characteristics, symptoms, and predictors of glioma patients grade I-IV in the the Danish Neuro-Oncology Registry. J. Neurooncol. 2017, 135, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro. Oncol. 2018, 20 (Suppl. S4), iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Zhang, W.; Wang, M.; Jia, P. IL1RAP regulated by PRPRD promotes gliomas progression via inducing neuronal synapse development and neuron differentiation in vitro. Pathol Res. Pract. 2020, 216, 153141. [Google Scholar] [CrossRef]
- Yu, K.; Rohr, J.; Liu, Y.; Li, M.; Xu, J.; Wang, K.; Chai, J.; Zhao, D.; Liu, Y.; Ma, J.; et al. Progress in triple negative breast carcinoma pathophysiology: Potential therapeutic targets. Pathol. Res. Pract. 2020, 216, 152874. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rivandi, M.; Abedini, S.; Pasdar, A.; Sahebkar, A. Regulators and mechanisms of anoikis in triple-negative breast cancer (TNBC): A review. Crit. Rev. Oncol. Hematol. 2019, 140, 17–27. [Google Scholar] [CrossRef]
- Waldum, H.L.; Fossmark, R. Types of Gastric Carcinomas. Int. J. Mol. Sci. 2018, 19, 4109. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Xia, Q.; Li, A.; Wang, Z. The Potential Role of IL1RAP on Tumor Microenvironment-Related Inflammatory Factors in Stomach Adenocarcinoma. Technol. Cancer Res. Treat. 2021, 20, 5282. [Google Scholar] [CrossRef]
- Johnson, C.A.; James, D.; Marzan, A.; Armaos, M. Cervical Cancer: An Overview of Pathophysiology and Management. Semin. Oncol. Nurs. 2019, 35, 166–174. [Google Scholar] [CrossRef]
- Liu, F.; Dai, M.; Xu, Q.; Zhu, X.; Zhou, Y.; Jiang, S.; Wang, Y.; Ai, Z.; Ma, L.; Zhang, Y.; et al. SRSF10-mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP-NF-kappaB-CD47 axis. Oncogene 2018, 37, 2394–2409. [Google Scholar] [CrossRef] [Green Version]
- Hojen, J.F.; Kristensen, M.L.V.; McKee, A.S.; Wade, M.T.; Azam, T.; Lunding, L.P.; de Graaf, D.M.; Swartzwelter, B.J.; Wegmann, M.; Tolstrup, M.; et al. IL-1R3 blockade broadly attenuates the functions of six members of the IL-1 family, revealing their contribution to models of disease. Nat. Immunol. 2019, 20, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Attard, C.; Kulkarni, H.; Cummings, N.; Diego, V.P.; Carless, M.A.; Shields, K.A.; Johnson, M.P.; Kowlessur, S.; Dyer, T.D.; et al. Plasma levels of soluble interleukin 1 receptor accessory protein are reduced in obesity. J. Clin. Endocrinol. Metab. 2014, 99, 3435–3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.F.; Chi, W.; Lin, D.; Dai, M.L.; Wang, Y.L.; Yang, Y.M.; Jin, Z.B.; Wang, Y. Association of IL33 and IL1RAP Polymorphisms With Acute Anterior Uveitis. Curr Mol. Med. 2018, 17, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Sanna, V.; Ahmad, N.; Sechi, M.; Mukhtar, H. Resveratrol nanoformulation for cancer prevention and therapy. Ann. N. Y. Acad. Sci. 2015, 1348, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W.; Holland, J.F.; Frei, E. Hamilton, ON, Canada, 2003. In Cancer Medicine 6; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Dou, X.; Tong, P.; Huang, H.; Zellmer, L.; He, Y.; Jia, Q.; Zhang, D.; Peng, J.; Wang, C.; Xu, N.; et al. Evidence for immortality and autonomy in animal cancer models is often not provided, which causes confusion on key issues of cancer biology. J. Cancer 2020, 11, 2887–2920. [Google Scholar] [CrossRef]
- Pitot, H.C.; Hikita, H.; Dragan, Y.; Sargent, L.; Haas, M. Review article: The stages of gastrointestinal carcinogenesis--application of rodent models to human disease. Aliment. Pharmacol. Ther. 2000, 14 (Suppl. S1), 153–160. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. Steps in metastasis: An updated review. Med. Oncol. 2021, 38, 3. [Google Scholar] [CrossRef]
- Riera-Domingo, C.; Audige, A.; Granja, S.; Cheng, W.C.; Ho, P.C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, Hypoxia, and Metabolism-the Menage a Trois of Cancer: Implications for Immunotherapy. Physiol. Rev. 2020, 100, 1–102. [Google Scholar] [CrossRef]
- Karin, N.; Wildbaum, G. The Role of Chemokines in Shaping the Balance Between CD4(+) T Cell Subsets and Its Therapeutic Implications in Autoimmune and Cancer Diseases. Front. Immunol. 2015, 6, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, L.; Li, X.F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 6304. [Google Scholar] [CrossRef] [PubMed]
- Yuen, V.W.; Wong, C.C. Hypoxia-inducible factors and innate immunity in liver cancer. J. Clin. Invest. 2020, 130, 5052–5062. [Google Scholar] [CrossRef]
- Multhoff, G.; Vaupel, P. Hypoxia Compromises Anti-Cancer Immune Responses. Adv. Exp. Med. Biol. 2020, 1232, 131–143. [Google Scholar] [PubMed]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef] [Green Version]
- Laye, S.; Liege, S.; Li, K.S.; Moze, E.; Neveu, P.J. Physiological significance of the interleukin 1 receptor accessory protein. Neuroimmunomodulation 2001, 9, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Huber, M.; Kollewe, C.; Bischoff, S.C.; Falk, W.; Martin, M.U. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18660–18665. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wang, D.; Pei, Y.; Sun, L. Systematic identification of key genes and pathways in the development of invasive cervical cancer. Gene 2017, 618, 28–41. [Google Scholar] [CrossRef]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Yu, X.Q.; Wang, P.H. Inflammasome activation and Th17 responses. Mol. Immunol. 2019, 107, 142–164. [Google Scholar] [CrossRef] [PubMed]
- Al-Sahaf, S.; Hendawi, N.B.; Ollington, B.; Bolt, R.; Ottewell, P.D.; Hunter, K.D.; Murdoch, C. Increased Abundance of Tumour-Associated Neutrophils in HPV-Negative Compared to HPV-Positive Oropharyngeal Squamous Cell Carcinoma Is Mediated by IL-1R Signalling. Front. Oral Health 2021, 2, 604565. [Google Scholar] [CrossRef] [PubMed]
- Fournie, J.J.; Poupot, M. The Pro-tumorigenic IL-33 Involved in Antitumor Immunity: A Yin and Yang Cytokine. Front. Immunol. 2018, 9, 2506. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, D.; Martin, P.; Flacher, V.; Sun, Y.; Jarrossay, D.; Brembilla, N.; Mueller, C.; Arnett, H.A.; Palmer, G.; Towne, J.; et al. Interleukin-36 potently stimulates human M2 macrophages, Langerhans cells and keratinocytes to produce pro-inflammatory cytokines. Cytokine 2016, 84, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Hu, R.; Hambly, B.D. IL-34, IL-36 and IL-38 in colorectal cancer-key immunoregulators of carcinogenesis. Biophys. Rev. 2020, 12, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. IL-36 in chronic inflammation and cancer. Cytokine Growth Factor Rev. 2020, 55, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.M.; Giraldo, N.A.; Petitprez, F.; Julie, C.; Lacroix, L.; Peschaud, F.; Emile, J.F.; Marisa, L.; Fridman, W.H.; Storkus, W.J.; et al. Association of IL-36gamma with tertiary lymphoid structures and inflammatory immune infiltrates in human colorectal cancer. Cancer Immunol. Immunother. 2019, 68, 109–120. [Google Scholar] [CrossRef]
- Diaz-Barreiro, A.; Huard, A.; Palmer, G. Multifaceted roles of IL-38 in inflammation and cancer. Cytokine 2022, 151, 155808. [Google Scholar] [CrossRef]
- Lappano, R.; Talia, M.; Cirillo, F.; Rigiracciolo, D.C.; Scordamaglia, D.; Guzzi, R.; Miglietta, A.M.; De Francesco, E.M.; Belfiore, A.; Sims, A.H.; et al. The IL1beta-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2020, 39, 153. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjomsland, V.; Spangeus, A.; Valila, J.; Sandstrom, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1alpha sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Landberg, N.; Hansen, N.; Askmyr, M.; Agerstam, H.; Lassen, C.; Rissler, M.; Hjorth-Hansen, H.; Mustjoki, S.; Jaras, M.; Richter, J.; et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia 2016, 30, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Guo, W.; Ren, T.; Huang, Y.; Sun, K.; Zhang, H.; Yu, Y.; Wang, W.; Niu, J. Macrophages reduce the sensitivity of osteosarcoma to neoadjuvant chemotherapy drugs by secreting Interleukin-1 beta. Cancer Lett. 2020, 480, 4–14. [Google Scholar] [CrossRef]
- Paulus, A.; Cicenas, S.; Zvirbule, Z.; Paz-Ares, L.G.; Awada, A.; Garcia-Ribas, I.; Losic, N.; Zemaitis, M. Phase 1/2a trial of nadunolimab, a first-in-class fully humanized monoclonal antibody against IL1RAP, in combination with cisplatin and gemcitabine (CG) in patients with non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 9020. [Google Scholar] [CrossRef]
- Cutsem, E.V.; Eefsen, R.L.; Ochsenreither, S.; Zvirbule, Z.; Ivanauskas, A.; Arnold, D.; Baltruskeviciene, E.; Pfeiffer, P.; Yachnin, J.; Garcia-Carbonero, R.; et al. Phase 1/2a trial of nadunolimab, a first-in-class fully humanized monoclonal antibody against IL1RAP, in combination with gemcitabine and nab-paclitaxel (GN) in patients with pancreatic adenocarcinoma (PDAC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 4141. [Google Scholar] [CrossRef]
- Agerstam, H.; Hansen, N.; von Palffy, S.; Sanden, C.; Reckzeh, K.; Karlsson, C.; Lilljebjorn, H.; Landberg, N.; Askmyr, M.; Hogberg, C.; et al. IL1RAP antibodies block IL-1-induced expansion of candidate CML stem cells and mediate cell killing in xenograft models. Blood 2016, 128, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Agerstam, H.; Karlsson, C.; Hansen, N.; Sanden, C.; Askmyr, M.; von Palffy, S.; Hogberg, C.; Rissler, M.; Wunderlich, M.; Juliusson, G.; et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2015, 112, 10786–10791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trad, R.; Warda, W.; Alcazer, V.; Neto da Rocha, M.; Berceanu, A.; Nicod, C.; Haderbache, R.; Roussel, X.; Desbrosses, Y.; Daguindau, E.; et al. Chimeric antigen receptor T-cells targeting IL-1RAP: A promising new cellular immunotherapy to treat acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e00422. [Google Scholar] [CrossRef]
- Fields, J.K.; Kihn, K.; Birkedal, G.S.; Klontz, E.H.; Sjostrom, K.; Gunther, S.; Beadenkopf, R.; Forsberg, G.; Liberg, D.; Snyder, G.A.; et al. Molecular Basis of Selective Cytokine Signaling Inhibition by Antibodies Targeting a Shared Receptor. Front. Immunol. 2021, 12, 779100. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Mason, N.J.; Posey, A.D. Keeping the Engine Running: The Relevance and Predictive Value of Preclinical Models for CAR-T Cell Development. ILAR J. 2018, 59, 276–285. [Google Scholar] [CrossRef]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Stromberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Frenay, F.B.P.S.; Moreau, M.; Rossi, C.; Casasnovas, O.; Ringenbach, L.; Ferrand, C.; Collin, B. Theranostic Approach of CD38 and IL-1RAP Positive Haematological Malignancies Based on Radiolabelled Antibodies. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, S417–S418. [Google Scholar]
| Cytokine | FAMILY NAME | Known Structure (Uniprot ID *) | Biological Effect | Reference |
|---|---|---|---|---|
| IL-1α | IL-1F1 | Yes (P01583) | Proinflammatory | [21,29,30,31] |
| IL-1β | IL-1F2 | Yes (P01584) | Proinflammatory | [21,29,31,32] |
| IL-1Ra (Anakinra) | IL-1F3 | Yes (P18510) | Anti-inflammatory | [21,29,33] |
| IL-18 | IL-1F4 | Yes (Q14116) | Proinflammatory | [21,34,35,36] |
| IL-33 (NF-HEV *) | IL-1F11 | Yes (O95760) | Proinflammatory | [21,34,37,38] |
| IL-36α | IL-1F6 | Yes (Q9UHA7) | Proinflammatory | [21,39,40] |
| IL-36β (IL1-H2) | IL-1F8 | No (Q9NZH7) | Proinflammatory | [21,39] |
| IL-36γ | IL-1F9 | Yes (Q9NZH8) | Proinflammatory | [21,39,41] |
| IL-36Ra | IL-1F5 | Yes (Q9UBH0) | Anti-inflammatory | [21,39,42] |
| IL-37 | IL-1F7 | Yes (Q9NZH6) | Anti-inflammatory | [21,43,44] |
| IL-38 | IL-1F10 | Yes (Q8WWZ1) | Anti-inflammatory | [21,45,46,47] |
| Receptor (RNA Tissues Expression) * | Co-Receptor (RNA Tissues Expression) * | Signaling Pathways | Ligand | Reference |
|---|---|---|---|---|
| IL-1RI (ubiquitous) | IL-1RAP ** (ubiquitous) | MyD88 **: MAPK ** (MAP3K **) NF-κB **, AP-1 ** | IL-1α IL-1β IL-1Ra | [20,48,49] |
| IL-1RII (appendix, bone marrow, colon, esophagus, placenta, skin, stomach, spleen) | IL-1RAP (ubiquitous) | MyD88: MAPK NF-κB, AP-1 | IL-1α IL-1β IL-1Ra | [20,48,49] |
| IL-18Rα (ubiquitous) | IL-18RAP (bone marrow, appendix, spleen) | MyD88: MAPK (JNK1 **) NF-κB, AP-1 | IL-18 | [20,35,48,49] |
| SIGIRR ** (ubiquitous, spleen, kidney) | STAT3**: MAPK, NF-κB, AP-1 (inhibition) | IL-37 | [20,35,48,49] | |
| IL-33R (placenta, kidney, lung, adrenal, gall bladder) | IL-1RAP (ubiquitous) | MyD88: MAPK, NF-κB, AP-1 | IL-33 | [20,48,49] |
| IL-36R (ubiquitous, skin, thyroid, esophagus, kidney) | IL-1RAP (ubiquitous) | MyD88: MAPK, NF-κB, AP-1 | IL-36α IL-36β IL-36γ IL-36Ra IL-38 | [20,48,49,50] |
| Unknown | IL-1RAPL1 ** (brain) | PSD-95 **, JNK ** | Unknown | [20,48,49,51,52] |
| Unknown | IL-1RAPL2 (adrenal, urinary bladder, testis, brain) | Unknown | Unknown | [20,48,49,53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frenay, J.; Bellaye, P.-S.; Oudot, A.; Helbling, A.; Petitot, C.; Ferrand, C.; Collin, B.; Dias, A.M.M. IL-1RAP, a Key Therapeutic Target in Cancer. Int. J. Mol. Sci. 2022, 23, 14918. https://doi.org/10.3390/ijms232314918
Frenay J, Bellaye P-S, Oudot A, Helbling A, Petitot C, Ferrand C, Collin B, Dias AMM. IL-1RAP, a Key Therapeutic Target in Cancer. International Journal of Molecular Sciences. 2022; 23(23):14918. https://doi.org/10.3390/ijms232314918
Chicago/Turabian StyleFrenay, Jame, Pierre-Simon Bellaye, Alexandra Oudot, Alex Helbling, Camille Petitot, Christophe Ferrand, Bertrand Collin, and Alexandre M. M. Dias. 2022. "IL-1RAP, a Key Therapeutic Target in Cancer" International Journal of Molecular Sciences 23, no. 23: 14918. https://doi.org/10.3390/ijms232314918