
Screening of Candidate Genes Associated with Brown Stripe Resistance in Sugarcane via BSR-seq Analysis

 ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Field Resistance Survey of Sugarcane Brown Stripe
2.2. Summary of the BSR-seq and Sequence Alignments
2.3. New Gene Analysis and Differentially Expressed Genes
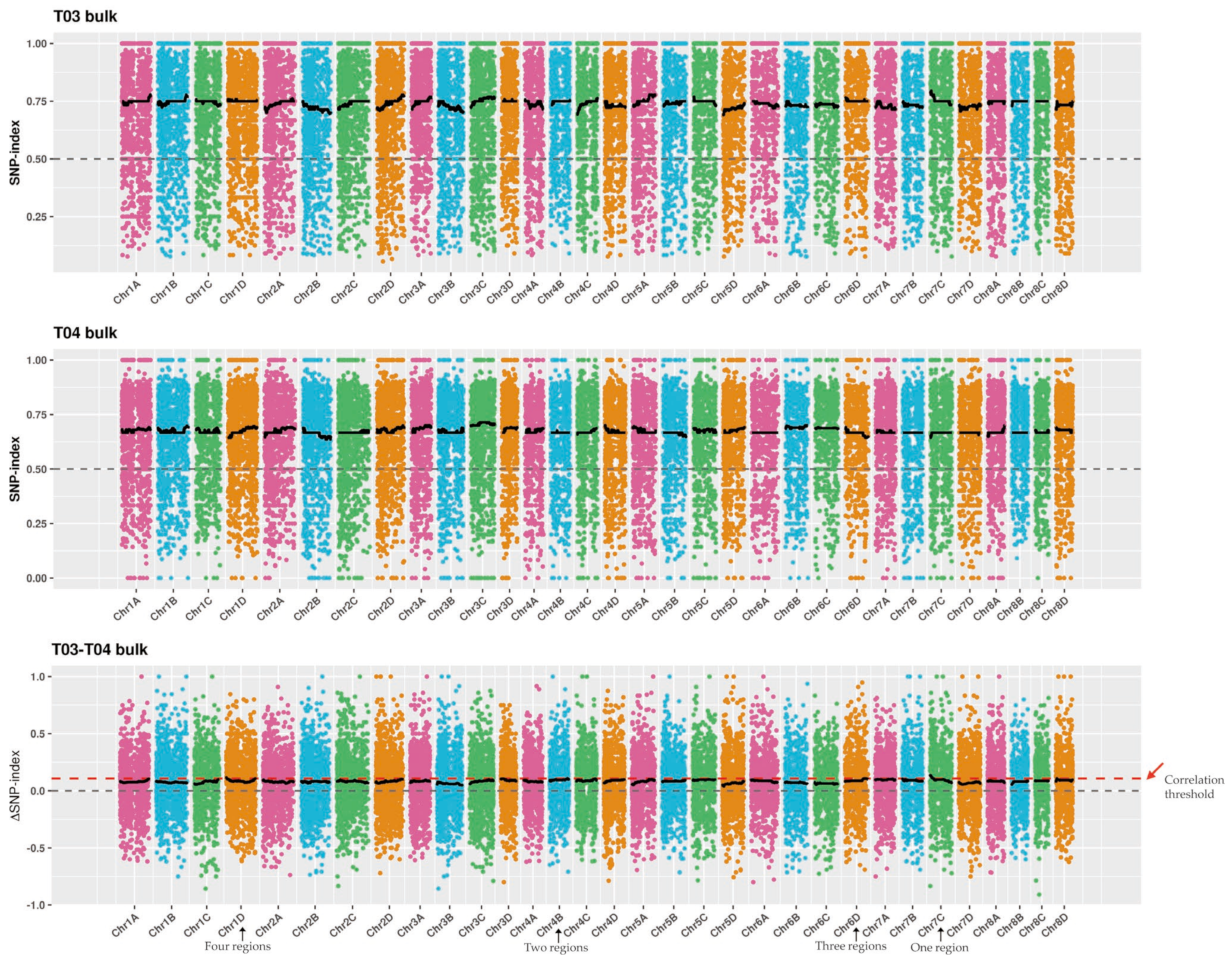
2.4. SNP Calling and Analysis of the Candidate Regions for SBS Resistance
2.5. Functional Annotation, Enrichment Analysis, and DEG Analysis of the Candidate Genes
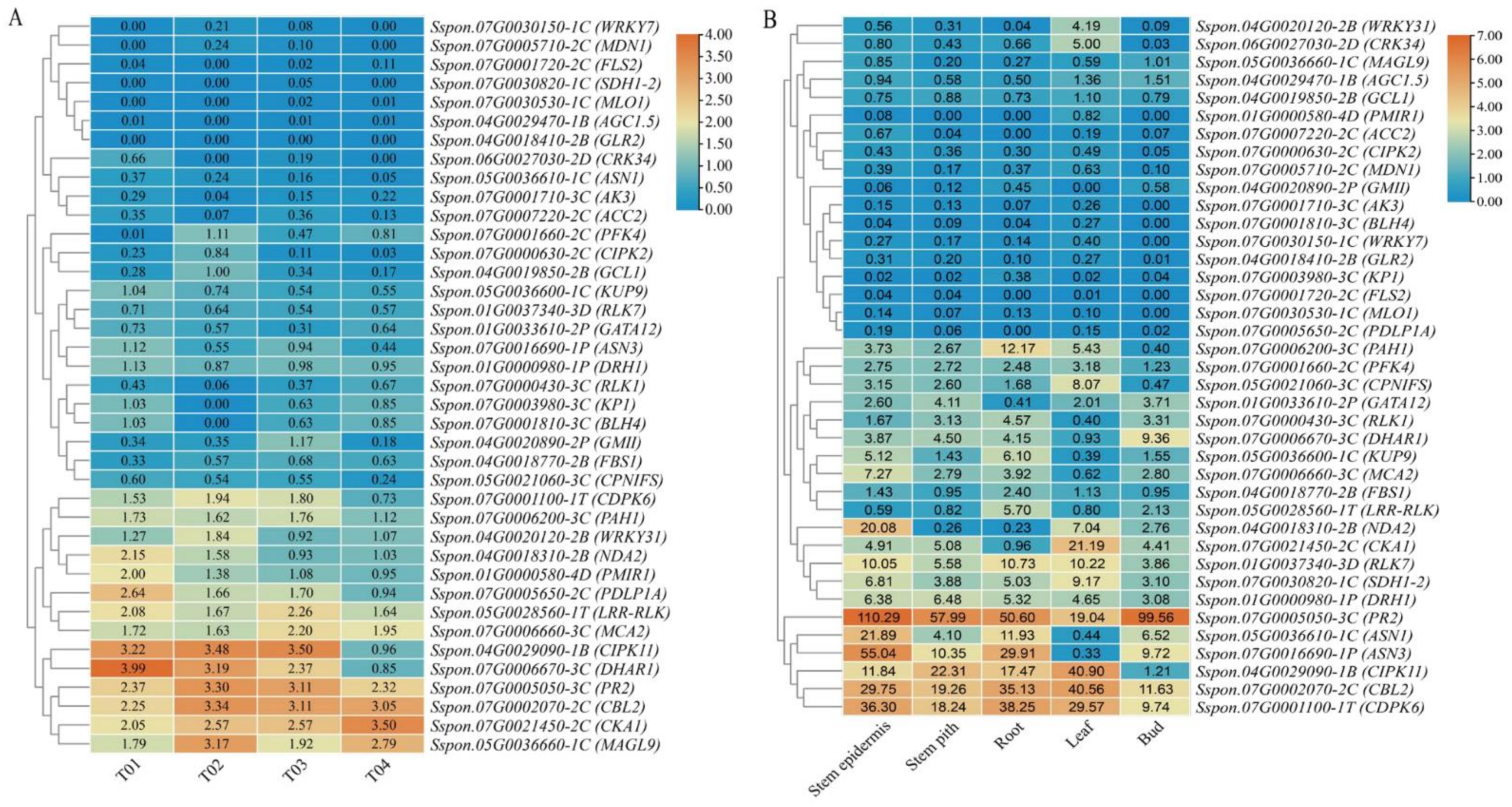
2.6. Expression Profiling Analysis of Candidate Genes
2.7. Validation of Candidate Genes by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, Library Construction, and RNA Sequencing
4.3. New Gene Discovery and Differential Expressed Genes (DEGs) Analysis
4.4. SNP Calling, BSR-seq Association Analysis
4.5. Functional Annotation and Enrichment Analysis
4.6. Expression Profiling of Candidate Genes
4.7. qRT-PCR Verification of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.Y.; Cang, X.Y.; Qin, W.; Shan, H.L.; Zhang, R.Y.; Wang, C.M.; Li, W.F.; Huang, Y.K. Evaluation of field resistance to brown stripe disease in novel and major cultivated sugarcane varieties in China. J. Plant Pathol. 2021, 103, 985–989. [Google Scholar] [CrossRef]
- Ricaud, C.; Egan, B.T.; Gillaspie, A.G.; Hughes, C.G. Diseases of Sugarcane. Major Disease; Elsevier Science Publishers: Amsterdam, The Netherlands, 1988. [Google Scholar]
- You, Q.; Sood, S.; Luo, Z.L.; Liu, H.B.; Sariful Islam, M.; Zhang, M.Q.; Wang, J.P. Identifying genomic regions controlling ratoon stunting disease resistance in sugarcane (Saccharum spp.) clonal F1 population. Crop J. 2021, 9, 1070–1078. [Google Scholar] [CrossRef]
- Le Cunff, L.; Garsmeur, O.; Raboin, L.M.; Pauquet, J.; Telismart, H.; Selvi, A.; Grivet, L.; Philippe, R.; Begum, D.; Deu, M.; et al. Diploid/polyploid syntenic shuttle mapping and haplotype-specific chromosome walking toward a rust resistance gene (Bru1) in highly polyploid sugarcane (2n∼12x∼115). Genetics 2008, 180, 649–660. [Google Scholar] [CrossRef]
- Piperidis, N.; D’Hont, A. Sugarcane genome architecture decrypted with chromosome-specific oligo probes. Plant J. 2020, 103, 2039–2051. [Google Scholar] [CrossRef]
- Piperidis, G.; Piperidis, N.; D’Hont, A. Molecular cytogenetic investigation of chromosome composition and transmission in sugarcane. Mol. Genet. Genomics 2010, 284, 65–73. [Google Scholar] [CrossRef]
- D’Hont, A.; Grivet, L.; Feldmann, P.; Glaszmann, J.C.; Rao, S.; Berding, N. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. Gen. Genet. 1996, 250, 405–413. [Google Scholar] [CrossRef]
- Chen, N.; Juric, I.; Cosgrove, E.J.; Bowman, R.; Fitzpatrick, J.W.; Schoech, S.J.; Clark, A.G.; Coop, G. Allele frequency dynamics in a pedigreed natural population. Proc. Natl. Acad. Sci. USA 2019, 116, 2158–2164. [Google Scholar] [CrossRef]
- Li, R.; Hou, Z.L.; Gao, L.W.; Xiao, D.; Hou, X.L.; Zhang, C.W.; Yan, J.Y.; Song, L.X. Conjunctive analyses of BSA-Seq and BSR-Seq to reveal the molecular pathway of leafy head formation in Chinese cabbage. Plants 2019, 8, 603. [Google Scholar] [CrossRef]
- Ma, P.T.; Wu, L.R.; Xu, Y.F.; Xu, H.X.; Zhang, X.; Wang, W.R.; Liu, C.; Wang, B. Bulked segregant RNA-Seq provides distinctive expression profile against powdery mildew in the wheat genotype YD588. Front. Plant Sci. 2021, 22, 72. [Google Scholar] [CrossRef]
- Shi, L.J.; Jiang, C.C.; He, Q.; Habekuß, A.; Ordon, F.; Luan, H.Y.; Shen, H.Q.; Liu, J.; Feng, Z.Y.; Zhang, J.; et al. Bulked segregant RNA-sequencing (BSR-Seq) identified a novel rare allele of eIF4E effective against multiple isolates of BaYMV/BaMMV. Theor. Appl. Genet. 2019, 132, 1777–1788. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef]
- Liu, S.Z. Gene mapping via bulked segregant RNA-seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef]
- Du, H.W.; Zhu, J.X.; Su, H.; Huang, M.; Wang, H.W.; Ding, S.C.; Zhang, B.L.; Luo, A.; Wei, S.D.; Tian, X.H.; et al. Bulked segregant RNA-Seq reveals differential expression and SNPs of candidate genes associated with waterlogging tolerance in maize. Front. Plant Sci. 2017, 8, 1022. [Google Scholar] [CrossRef]
- Zhang, M.D.; Huang, S.N.; Gao, Y.; Fu, W.; Qu, G.Y.; Zhao, Y.H.; Shi, F.Y.; Liu, Z.Y.; Feng, H. Fine mapping of a leaf flattening gene Bralcm through BSR-Seq in Chinese cabbage (Brassica rapa L. ssp. Pekinensis). Sci. Rep. 2020, 10, 13924. [Google Scholar] [CrossRef]
- Pu, X.; Tang, Y.Y.; Zhang, M.H.; Li, T.; Qiu, X.B.; Zhang, J.Y.; Wang, J.H.; Li, L.L.; Yang, Z.; Su, Y.; et al. Identification and candidate gene mining of HvSS1, a novel qualitative locus on chromosome 6H, regulating the uppermost internode elongation in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2021, 134, 2481–2494. [Google Scholar] [CrossRef]
- Gao, J.; Dai, G.X.; Zhou, W.Y.; Liang, H.F.; Huang, J.; Qing, D.J.; Chen, W.W.; Wu, H.; Yang, X.H.; Li, D.T.; et al. Mapping and identifying a candidate gene Plr4, a recessive gene regulating purple leaf in rice, by using bulked segregant and transcriptome analysis with next-generation sequencing. Int. J. Mol. Sci. 2019, 20, 4335. [Google Scholar] [CrossRef]
- Wang, Z.T.; Ren, H.; Pang, C.; Lu, G.L.; Xu, F.; Cheng, W.; Que, Y.X.; Xu, L.P. An autopolyploid-suitable polyBSA-seq strategy for screening candidate genetic markers linked to leaf blight resistance in sugarcane. Theor. Appl. Genet. 2022, 135, 623–636. [Google Scholar] [CrossRef]
- Ramirez-Gonzalez, R.H.; Segovia, V.; Bird, N.; Fenwick, P.; Holdgate, S.; Berry, S.; Jack, P.; Caccamo, M.; Uauy, C. RNA-seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. J. 2015, 13, 613–624. [Google Scholar] [CrossRef]
- Cao, Y.Y.; Diao, Q.N.; Chen, Y.Y.; Jin, H.J.; Zhang, Y.P.; Zhang, H.M. Development of KASP markers and identification of a QTL underlying powdery mildew resistance in melon (Cucumis melo L.) by bulked segregant analysis and RNA-seq. Front. Plant Sci. 2021, 11, 593207. [Google Scholar] [CrossRef]
- Edae, E.A.; Rouse, M.N. Bulked segregant analysis RNA-seq (BSR-Seq) validated a stem resistance locus in Aegilops umbellulata, a wild relative of wheat. PLoS ONE 2019, 14, e0215492. [Google Scholar] [CrossRef]
- Hou, X.G.; Guo, Q.; Wei, W.Q.; Guo, L.L.; Guo, D.L.; Zhang, L. Screening of genes related to early and late flowering in tree peony based on bulked segregant RNA sequencing and verification by quantitative real-time PCR. Molecules 2018, 23, 689. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.L.; Pan, Y.B.; Wang, Z.T.; Xu, F.; Cheng, W.; Huang, X.G.; Ren, H.; Pang, C.; Que, Y.X.; Xu, L.P. Utilization of a sugarcane100k single nucleotide polymorphisms microarray-derived high-density genetic map in quantitative trait loci mapping and function role prediction of genes related to chlorophyll content in sugarcane. Front. Plant Sci. 2021, 12, 817875. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.R.; Kuang, L.; Gao, Y.Q.; Wang, Y.L.; Salt, D.E.; Chao, D.Y. AtHMA4 drives natural variation in leaf Zn concentration of Arabidopsis thaliana. Front. Plant Sci. 2018, 9, 270. [Google Scholar] [CrossRef]
- Guo, J.J.; Fan, J.; Hauser, B.A.; Rhee, S.Y. Target enrichment improves mapping of complex traits by deep sequencing. G3 Genes|Genome|Genet. 2016, 6, 67–77. [Google Scholar] [CrossRef]
- Li, F.; Numa, H.; Hara, N.; Sentoku, N.; Ishii, T.; Fukuta, Y.; Nishimura, N.; Kato, H. Identification of a locus for seed shattering in rice (Oryza sativa L.) by combining bulked segregant analysis with whole-genome sequencing. Mol. Breed. 2019, 39, 36. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wu, Q.B.; Pan, Y.B.; Su, Y.C.; Zou, W.H.; Xu, F.; Sun, T.T.; Grisham, M.P.; Yang, S.L.; Xu, L.P.; Que, Y.X. WGCNA identifies a comprehensive and dynamic gene co-expression network that associates with smut resistance in sugarcane. Int. J. Mol. Sci. 2022, 23, 10770. [Google Scholar] [CrossRef]
- Wang, Z.T.; Lu, G.L.; Wu, Q.B.; Li, A.T.; Que, Y.X.; Xu, L.P. Isolating QTL controlling sugarcane leaf blight resistance using a two-way pseudo-testcross strategy. Crop J. 2021, 10, 1131–1140. [Google Scholar] [CrossRef]
- Liu, X.M.; Bi, B.; Xu, X.; Li, B.H.; Tian, S.M.; Wang, J.P.; Zhang, H.; Wang, G.Q.; Han, Y.J.; McElroy, J.S. Rapid identification of a candidate nicosulfuron sensitivity gene (Nss) in maize (Zea mays L.) via combining bulked segregant analysis and RNA-seq. Theor. Appl. Genet. 2019, 132, 1351–1361. [Google Scholar] [CrossRef]
- Hao, Z.M.; Geng, M.M.; Hao, Y.R.; Zhang, Y.; Zhang, L.J.; Wen, S.M.; Wang, R.H.; Liu, G.R. Screening for differential expression of genes for resistance to Sitodiplosis mosellana in bread wheat via BSR-Seq analysis. Theor. Appl. Genet. 2019, 132, 3201–3221. [Google Scholar] [CrossRef]
- Gao, Y.J.; Zhou, S.; Huang, Y.X.; Zhang, B.Q.; Xu, Y.H.; Zhang, G.M.; Lakshmanan, P.; Yang, R.Z.; Zhou, H.; Huang, D.L.; et al. Quantitative trait loci mapping and development of KASP marker smut screening assay using high-density genetic map and bulked segregant RNA sequencing in sugarcane (Saccharum spp.). Front. Plant Sci. 2022, 12, 796189. [Google Scholar] [CrossRef]
- Wu, Q.B.; Su, Y.C.; Pan, Y.B.; Xu, F.; Zou, W.H.; Que, B.B.; Lin, P.X.; Sun, T.T.; Grisham, M.P.; Xu, L.P. Genetic identification of SNP markers and candidate genes associated with sugarcane smut resistance using BSR-Seq. Front. Plant Sci. 2022, 13, 1035266. [Google Scholar] [CrossRef]
- Zhu, T.; Wu, L.R.; He, H.G.; Song, J.C.; Jia, M.S.; Liu, L.C.; Wang, X.L.; Han, R.; Niu, L.P.; Du, W.X.; et al. Bulked segregant RNA-seq reveals distinct expression profiling in Chinese wheat cultivar Jimai 23 responding to powdery mildew. Front. Genet. 2020, 11, 474. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, J.D.; Wang, S.L.; Chen, L.; Ma, C.L.; Yao, M.Z. Repressed gene expression of photosynthetic antenna proteins associated with yellow leaf variation as revealed by bulked segregant RNA-seq in tea plant Camellia sinensis. J. Agric. Food Chem. 2020, 68, 8068–8079. [Google Scholar] [CrossRef]
- Yang, X.P.; Islam, M.S.; Sushma, S.; Stephanie, M.; Hanson, E.A.; Jack, C.; Wang, J. Identifying quantitative trait loci (QTLs) and developing diagnostic markers linked to orange rust resistance in sugarcane (Saccharum spp.). Front. Plant Sci. 2018, 9, 350. [Google Scholar] [CrossRef]
- Zhang, S.P.; Liu, S.L.; Miao, H.; Shi, Y.X.; Wang, M.; Wang, Y.; Li, B.J.; Gu, X.F. Inheritance and QTL mapping of resistance to gummy stem blight in cucumber stem. Mol. Breed. 2017, 37, 49. [Google Scholar] [CrossRef]
- Arora, R.; Kumar, A.; Singh, I.K.; Singh, A. Pathogenesis related proteins: A defensin for plants but an allergen for humans. Int. J. Biol. Macromol. 2020, 157, 659–672. [Google Scholar] [CrossRef]
- Sels, J.; Mathys, J.; De Coninck, B.M.A.; Cammue, B.P.A.; De Bolle, M.F.C. Plant pathogenesis-related (PR) proteins: A focus on PR peptides. Plant Physiol. Biochem. 2008, 46, 941–950. [Google Scholar] [CrossRef]
- Hou, M.M.; Xu, W.J.; Bai, H.; Liu, Y.M.; Li, L.; Liu, L.Y.; Liu, L.J.; Liu, B.; Liu, G.Z. Characteristic expression of rice pathogenesis-related proteins in rice leaves during interactions with Xanthomonas oryzae pv. Oryzae. Plant Cell Rep. 2012, 31, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.; Zhou, J.R.; Rott, P.C.; Li, J.; Fu, H.Y.; Huang, M.T.; Zhang, H.L.; Gao, S.J. SCPR1 plays a positive role in the regulation of resistance to diverse stresses in sugarcane (Saccharum spp.) and Arabidopsis thaliana. Ind. Crops Prod. 2022, 180, 114736. [Google Scholar] [CrossRef]
- Peng, Q.; Su, Y.C.; Ling, H.; Ahmad, W.; Gao, S.W.; Guo, J.L.; Que, Y.X.; Xu, L.P. A sugarcane pathogenesis-related protein, ScPR10, plays a positive role in defense responses under Sporisorium scitamineum, SrMV, SA, and MeJA stresses. Plant Cell Rep. 2017, 36, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.L.; Du, L.; Huang, Y.; Gao, S.M.; Yu, M. Origin and diversification of leucine-rich repeat receptor-like protein kinase (LRR-RLK) genes in plants. BMC Evol. Biol. 2017, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Pi, L.Y.; Chen, X.H.; Chakrabarty, P.K.; Jiang, J.; De Leon, A.L.; Liu, G.Z.; Li, L.; Benny, U.; Oard, J.; et al. Rice Xa21 binding protein 3 is a ubiquitin ligase required for full Xa21-mediated disease resistance. Plant Cell 2006, 18, 3635–3646. [Google Scholar] [CrossRef]
- Chinchilla, D.; Zipfel, C.; Robatzek, S.; Kemmerling, B.; Nürnberger, T.; Jones, J.D.G.; Felix, G.; Boller, T. A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 2007, 448, 497–500. [Google Scholar] [CrossRef]
- Kudla, J.; Becker, D.; Grill, E.; Hedrich, R.; Hippler, M.; Kummer, U.; Parniske, M.; Romeis, T.; Schumacher, K. Advances and current challenges in calcium signaling. New Phytol. 2018, 218, 414–431. [Google Scholar] [CrossRef]
- Heyer, M.; Scholz, S.S.; Reichelt, M.; Kunert, G.; Oelmüller, R.; Mithöfer, A. The Ca2+ sensor proteins CMl37 and CMl42 antagonistically regulate plant stress responses by altering phytohormone signals. Plant Mol. Biol. 2022, 109, 611–625. [Google Scholar] [CrossRef]
- Boudsocq, M.; Sheen, J. CDPKs in immune and stress signaling. Trends Plant Sci. 2013, 18, 30–40. [Google Scholar] [CrossRef]
- Ma, X.; Li, Q.H.; Yu, Y.N.; Qiao, Y.M.; Haq, S.U.; Gong, Z.H. The CBL–CIPK pathway in plant response to stress signals. Int. J. Mol. Sci. 2020, 21, 5668. [Google Scholar] [CrossRef]
- Ma, X.; Gai, W.X.; Qiao, Y.M.; Ali, M.; Wei, A.M.; Luo, D.X.; Li, Q.H.; Gong, Z.H. Identification of CBL and CIPK gene families and functional characterization of CaCIPK1 under phytophthora capsici in pepper (Capsicum annuum L.). BMC Genomics 2019, 20, 775. [Google Scholar] [CrossRef]
- Liu, P.; Guo, J.; Zhang, R.M.; Zhao, J.X.; Liu, C.; Qi, T.; Duan, Y.H.; Kang, Z.S.; Guo, J. TaCIPK10 interacts with and phosphorylates TaNH2 to activate wheat defense responses to stripe rust. Plant Biotechnol. J. 2019, 17, 956–968. [Google Scholar] [CrossRef]
- Liu, P.; Duan, Y.H.; Liu, C.; Xue, Q.H.; Guo, J.; Qi, T.; Kang, Z.S.; Guo, J. The calcium sensor TaCBL4 and its interacting protein TaCIPK5 are required for wheat resistance to stripe rust fungus. J. Exp. Bot. 2018, 69, 4443–4457. [Google Scholar] [CrossRef]
- Zhang, H.F.; Yang, B.; Liu, W.Z.; Li, H.W.; Wang, L.; Wang, B.Y.; Deng, M.; Liang, W.W.; Deyholos, M.K.; Jiang, Y.Q. Identification and characterization of CBL and CIPK gene families in canola (Brassica napus L.). BMC Plant Biol. 2014, 14, 8. [Google Scholar] [CrossRef]
- Zhu, X.L.; Wang, B.Q.; Wang, X.; Wei, X.H. Identification of the CIPK-CBL family gene and functional characterization of CqCIPK14 gene under drought stress in quinoa. BMC Genomics 2022, 23, 447. [Google Scholar]
- Zhang, Y.; Zhou, X.N.; Liu, S.Y.; Yu, A.Z.; Yang, C.M.; Chen, X.L.; Liu, J.Y.; Wang, A.X. Identification and functional analysis of tomato CIPK gene family. Int. J. Mol. Sci. 2019, 21, 110. [Google Scholar] [CrossRef]
- Poddar, N.; Deepika, D.; Chitkara, P.; Singh, A.; Kumar, S. Molecular and expression analysis indicate the role of CBL interacting protein kinases (CIPKs) in abiotic stress signaling and development in chickpea. Sci. Rep. 2022, 12, 16862. [Google Scholar] [CrossRef]
- Su, W.H.; Ren, Y.J.; Wang, D.J.; Huang, L.; Fu, X.Q.; Ling, H.; Su, Y.C.; Huang, N.; Tang, H.C.; Xu, L.P. New insights into the evolution and functional divergence of the CIPK gene family in Saccharum. BMC Genomics 2020, 21, 868. [Google Scholar] [CrossRef]
- Zhang, J.Z. Overexpression analysis of plant transcription factors. Curr. Opin. Plant Biol. 2003, 6, 430–440. [Google Scholar] [CrossRef]
- Fountain, J.C.; Khera, P.; Yang, L.M.; Nayak, S.N.; Scully, B.T.; Lee, R.D.; Chen, Z.Y.; Kemerait, R.C.; Varshney, R.K.; Guo, B.Z. Resistance to Aspergillus flavus in maize and peanut: Molecular biology, breeding, environmental stress, and future perspectives. Crop J. 2015, 3, 229–237. [Google Scholar] [CrossRef]
- Fountain, J.C.; Raruang, Y.; Luo, M.; Brown, R.L.; Guo, B.Z.; Chen, Z.Y. Potential roles of WRKY transcription factors in regulating host defense responses during Aspergillus flavus infection of immature maize kernels. Physiol. Mol. Plant Pathol. 2015, 89, 31–40. [Google Scholar] [CrossRef]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Liu, H.B.; Deng, Y.; Xiao, J.H.; Li, X.H.; Wang, S.P. The WRKY45-2 WRKY13 WRKY42 transcriptional regulatory cascade is required for rice resistance to fungal pathogen. Plant Physiol. 2015, 167, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- YYokotani, N.; Sato, Y.; Tanabe, S.; Chujo, T.; Shimizu, T.; Okada, K.; Yamane, H.; Shimono, M.; Sugano, S.; Takatsuji, H. WRKY76 is a rice transcriptional repressor playing opposite roles in blast disease resistance and cold stress tolerance. J. Exp. Bot. 2013, 64, 5085–5097. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.J.; Wang, L.; Su, W.H.; Ren, Y.J.; You, C.H.; Zhang, C.; Que, Y.X.; Su, Y.C. A class III WRKY transcription factor in sugarcane was involved in biotic and abiotic stress responses. Sci. Rep. 2020, 10, 20964. [Google Scholar] [CrossRef]
- Wen, J.; Shen, Y.; Xing, Y.; Wang, Z.; Han, S.; Li, S.; Yang, C.; Hao, D.; Zhang, Y. QTL mapping of Fusarium ear rot resistance in maize. Plant Dis. 2020, 105, 558–565. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Albaradei, S.; Thafar, M.; Alsaedi, A.; Van Neste, C.; Gojobori, T.; Essack, M.; Gao, X. Machine learning and deep learning methods that use omics data for metastasis prediction. Comput. Struct. Biotec. 2021, 19, 5008–5018. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Reumers, J.; De Rijk, P.; Zhao, H.; Liekens, A.; Smeets, D.; Cleary, J.; Van Loo, P.; Van Den Bossche, M.; Catthoor, K.; Sabbe, B.; et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 2012, 30, 61–68. [Google Scholar] [CrossRef]
- Takagi, H.; Uemura, A.; Yaegashi, H.; Tamiru, M.; Abe, A.; Mitsuoka, C.; Utsushi, H.; Natsume, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap-Gap: Whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 2013, 200, 276–283. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.Y.; He, J.; Lanczycki, C.J.; Lu, S.N.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, 200–203. [Google Scholar] [CrossRef]
- Chen, C.G.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Ling, H.; Wu, Q.B.; Guo, J.L.; Xu, L.P.; Que, Y.X. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. PLoS ONE 2014, 9, e97469. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | SBS-Resistant Parent YT93-159 (T01) | SBS-Susceptible Parent ROC22 (T02) | SBS-Susceptible Bulk (T03) | SBS-Resistant Bulk (T04) |
|---|---|---|---|---|
| Clean bases a | 17,433,975,290 | 20,136,050,680 | 71,212,242,046 | 61,215,473,752 |
| Clean reads b | 58,329,199 | 67,443,343 | 239,023,714 | 205,084,682 |
| Q30 (%) c | 93.33 | 93.06 | 94.05 | 94.25 |
| GC (%) d | 52.27 | 53.00 | 52.84 | 52.55 |
| Total reads e | 116,658,398 | 134,886,686 | 478,047,428 | 410,169,364 |
| Mapped reads f | 81,605,146 (69.95%) g | 92,357,559 (68.47%) | 323,730,767 (67.72%) | 286,908,836 (69.95%) |
| Unique mapped reads h | 39,889,476 (34.19%) i | 44,895,323 (33.28%) | 166,031,678 (34.73%) | 150,882,351 (36.79%) |
| Tarit | Grade | Symptom Description |
|---|---|---|
| SBS-resistance | 1 | No disease symptom on the surface of sugarcane leaves |
| 2 | Disease symptoms appear sporadically on the surface of sugarcane leaves | |
| 3 | Mild occurrence. The details are as follows: no more than 1/3 of the sugarcane leaves with disease symptoms, and the upper young leaves (+1, +2, +3 leaves) have no disease symptoms or sporadic disease symptoms | |
| 4 | Moderate occurrence. The details are as follows: more than 1/3 of the sugarcane leaves with disease symptoms, and some disease symptoms appear in the upper young leaves (+1, +2, +3 leaves) | |
| 5 | Severe occurrence. The details are as follows: more than 1/3 of the sugarcane leaves with disease symptoms, and the upper young leaves (+1, +2, +3 leaves) have more disease symptoms or more than 2/3 of the sugarcane leaves with disease symptoms, and the upper young leaves (+1, +2, +3 leaves) have some disease symptoms |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.; Wang, Z.; Xu, F.; Lu, G.; Su, Y.; Wu, Q.; Wang, T.; Que, Y.; Xu, L. Screening of Candidate Genes Associated with Brown Stripe Resistance in Sugarcane via BSR-seq Analysis. Int. J. Mol. Sci. 2022, 23, 15500. https://doi.org/10.3390/ijms232415500
Cheng W, Wang Z, Xu F, Lu G, Su Y, Wu Q, Wang T, Que Y, Xu L. Screening of Candidate Genes Associated with Brown Stripe Resistance in Sugarcane via BSR-seq Analysis. International Journal of Molecular Sciences. 2022; 23(24):15500. https://doi.org/10.3390/ijms232415500
Chicago/Turabian StyleCheng, Wei, Zhoutao Wang, Fu Xu, Guilong Lu, Yachun Su, Qibin Wu, Ting Wang, Youxiong Que, and Liping Xu. 2022. "Screening of Candidate Genes Associated with Brown Stripe Resistance in Sugarcane via BSR-seq Analysis" International Journal of Molecular Sciences 23, no. 24: 15500. https://doi.org/10.3390/ijms232415500
APA StyleCheng, W., Wang, Z., Xu, F., Lu, G., Su, Y., Wu, Q., Wang, T., Que, Y., & Xu, L. (2022). Screening of Candidate Genes Associated with Brown Stripe Resistance in Sugarcane via BSR-seq Analysis. International Journal of Molecular Sciences, 23(24), 15500. https://doi.org/10.3390/ijms232415500