
Unraveling the Structural Changes in the DNA-Binding Region of Tumor Protein p53 (TP53) upon Hotspot Mutation p53 Arg248 by Comparative Computational Approach


Abstract
1. Introduction
2. Result
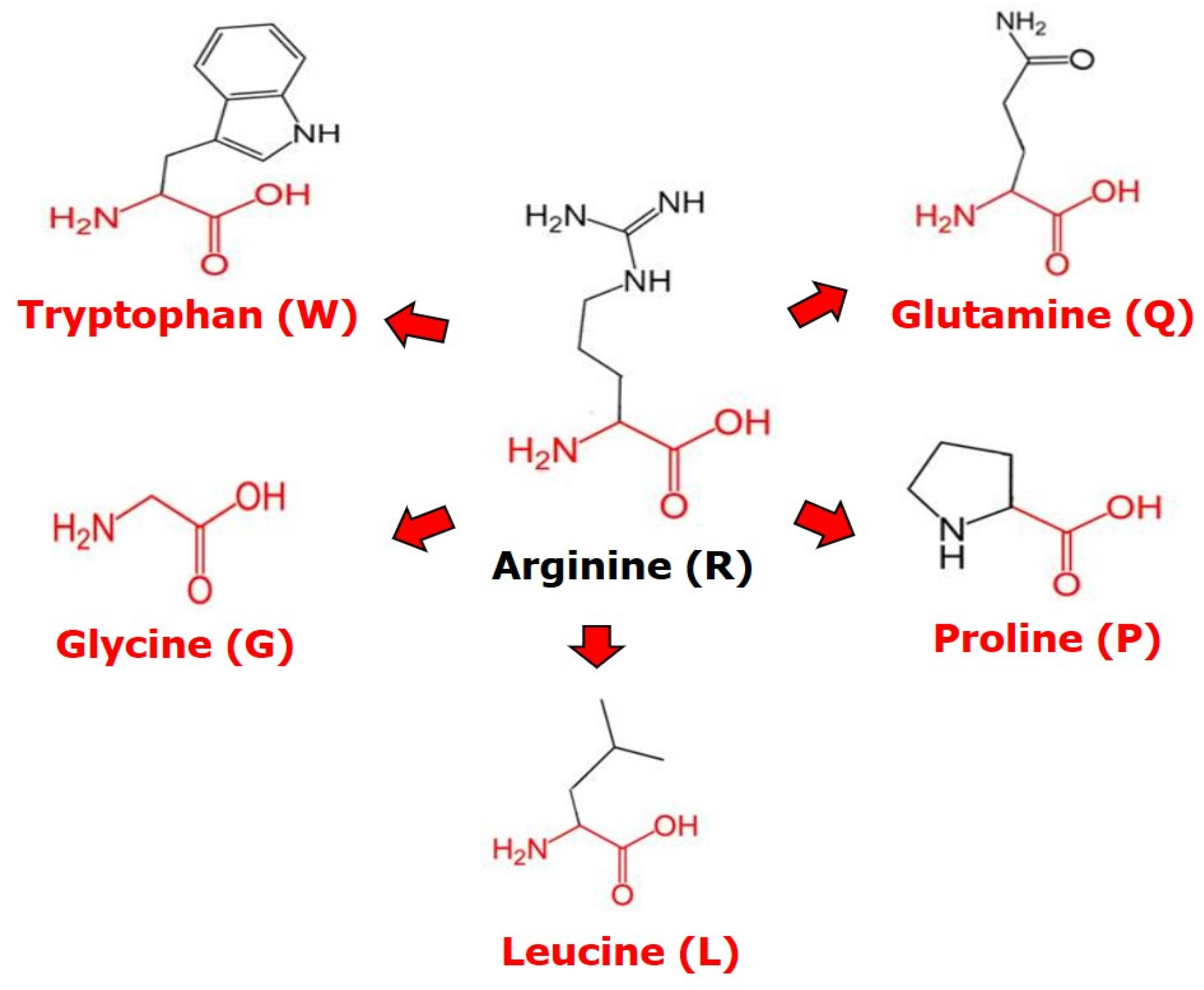
2.1. The p53 Missense Mutations of the Residue R248
2.2. Detection of Deleterious Missense Mutations Using Computational Approaches
2.3. Structural Changes of R248 Missense Mutations in p53 Protein Using HOPE
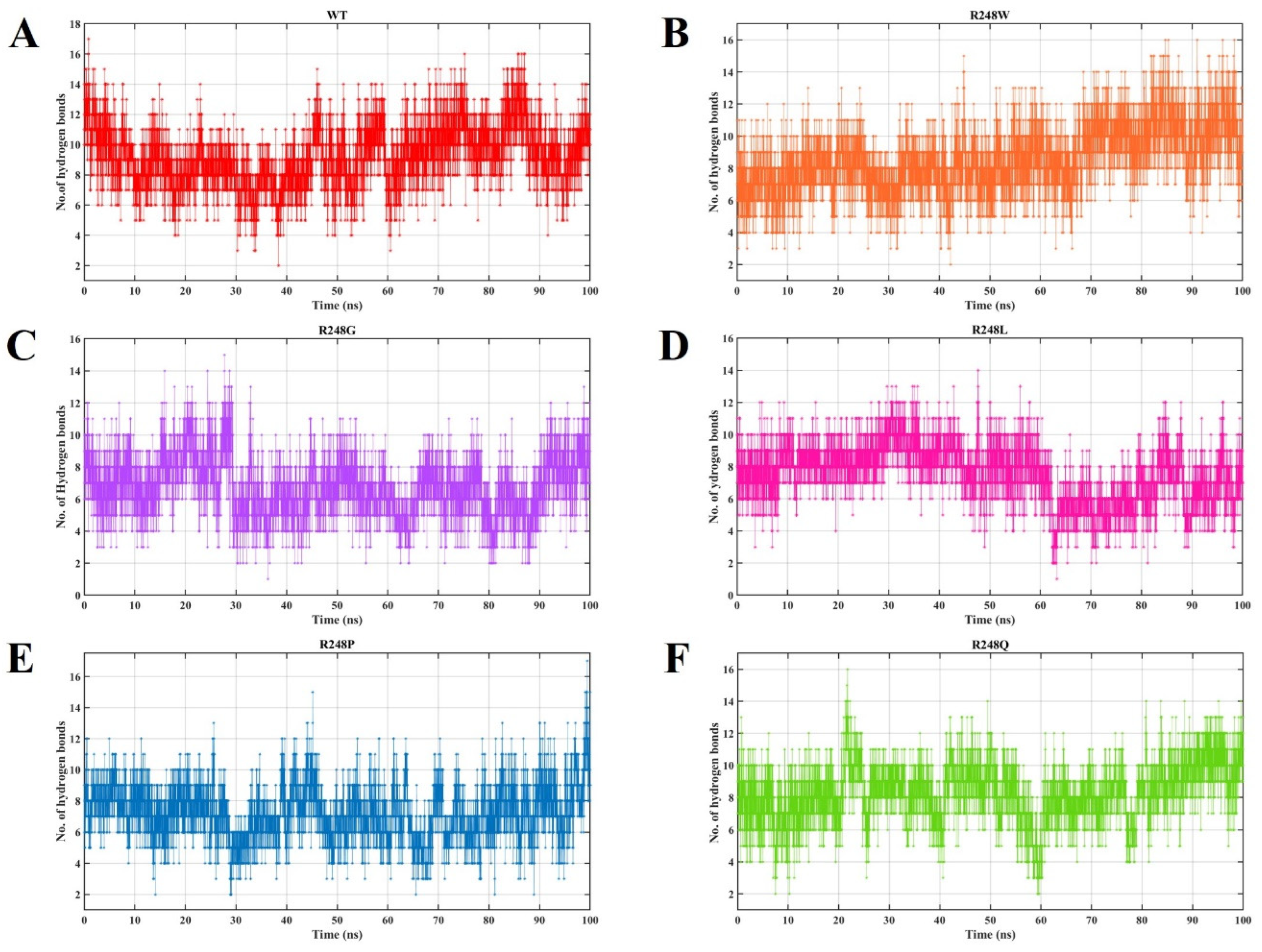
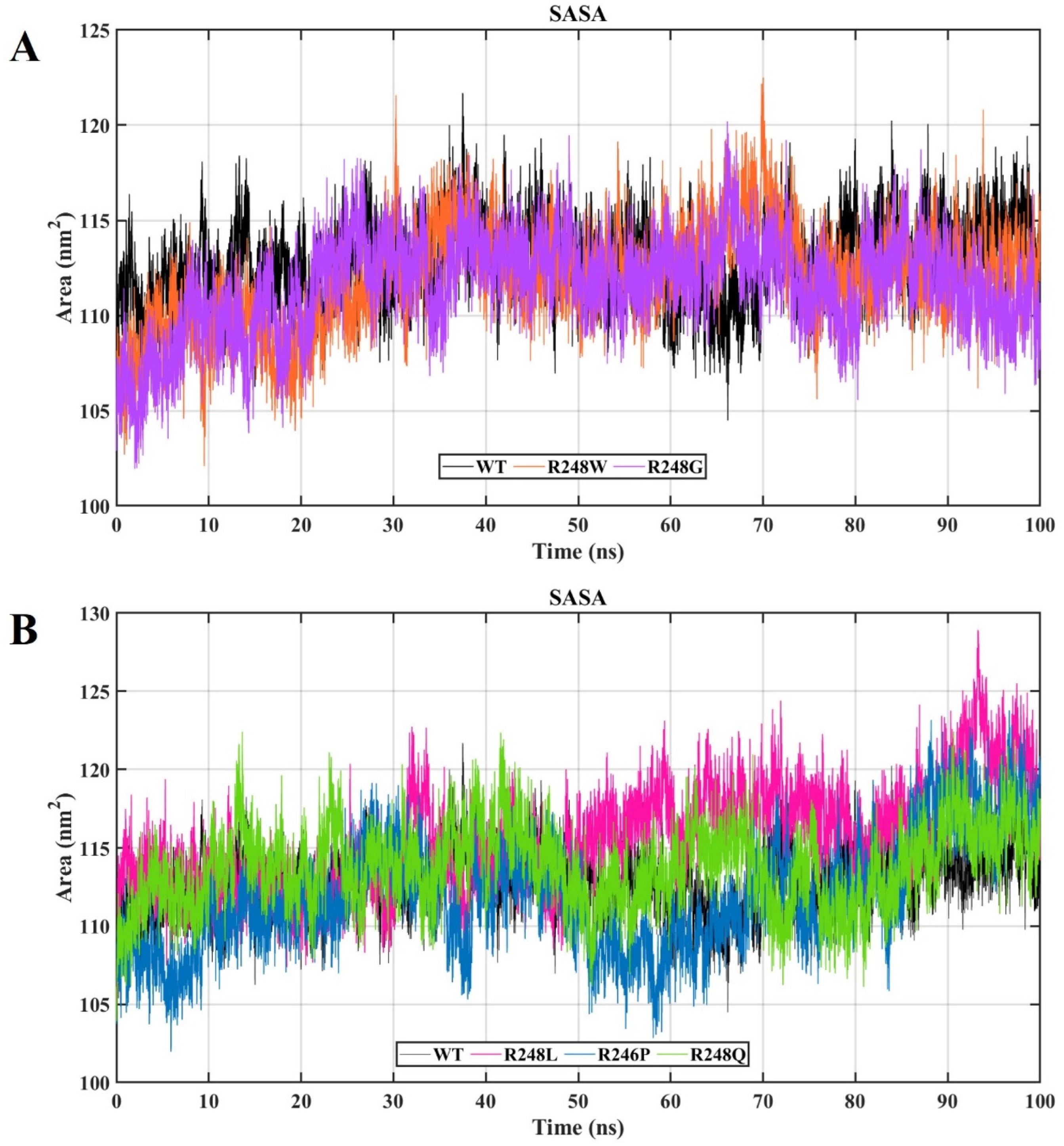
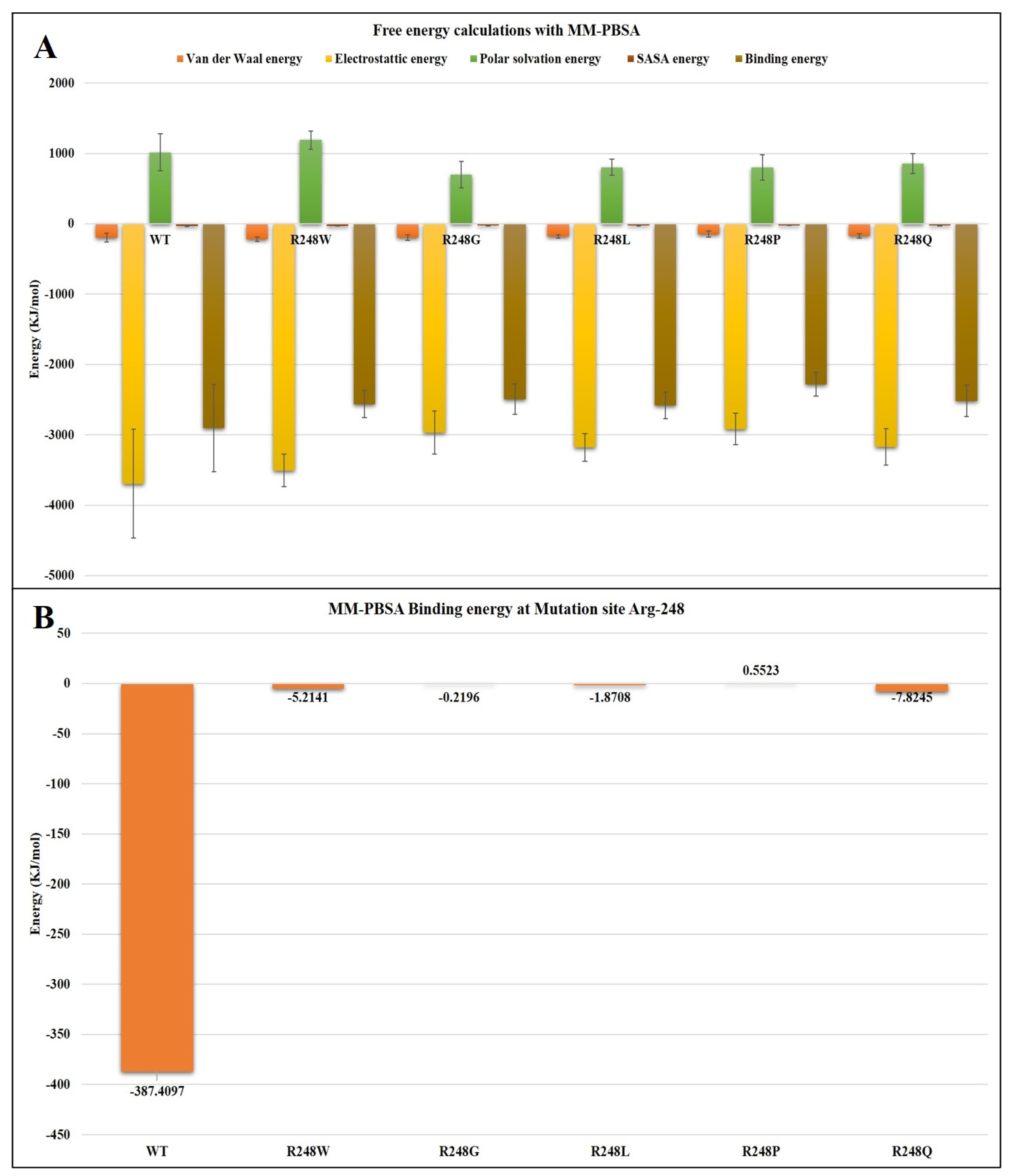
2.4. Molecular Dynamics Simulations
3. Discussion
4. Methodology
4.1. Retrieval of p53 R248 Mutations in the DNA Binding Residues
4.2. Investigation of Functional Consequences of R248 Mutations
4.3. R248 Mutations Impact p53 Protein Stability Using iStable
4.4. Biophysical Characterization by Align GVGD
4.5. Identification of Conserved Regions of p53 Protein Using ConSurf
4.6. Structural Effects of Missense Mutations Using HOPE
4.7. Molecular Dynamics Simulation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sigal, A.; Rotter, V. Oncogenic Mutations of the P53 Tumor Suppressor: The Demons of the Guardian of the Genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar]
- Vousden, K.H.; Lu, X. Live or Let Die: The Cell’s Response to P53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Mogi, A.; Kuwano, H. TP53 Mutations in Nonsmall Cell Lung Cancer. J. Biomed. Biotechnol. 2011, 2011, e583929. [Google Scholar] [CrossRef] [PubMed]
- May, P.; May, E. Twenty Years of P53 Research: Structural and Functional Aspects of the P53 Protein. Oncogene 1999, 18, 7621–7636. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Oren, M. P53 and Apoptosis. Semin. Cancer Biol. 1998, 8, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Gish, K.; Murphy, M.; Yin, Y.; Notterman, D.; Hoffman, W.H.; Tom, E.; Mack, D.H.; Levine, A.J. Analysis of P53-Regulated Gene Expression Patterns Using Oligonucleotide Arrays. Genes Dev. 2000, 14, 981–993. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New Therapeutic Strategies to Treat Human Cancers Expressing Mutant P53 Proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef]
- Hou, H.; Qin, K.; Liang, Y.; Zhang, C.; Liu, D.; Jiang, H.; Liu, K.; Zhu, J.; Lv, H.; Li, T.; et al. Concurrent TP53 Mutations Predict Poor Outcomes of EGFR-TKI Treatments in Chinese Patients with Advanced NSCLC. Cancer Manag. Res. 2019, 11, 5665–5675. [Google Scholar] [CrossRef]
- Langerød, A.; Zhao, H.; Borgan, Ø.; Nesland, J.M.; Bukholm, I.R.; Ikdahl, T.; Kåresen, R.; Børresen-Dale, A.-L.; Jeffrey, S.S. TP53mutation Status and Gene Expression Profiles Are Powerful Prognostic Markers of Breast Cancer. Cancer Manag. Res. 2007, 9, R30. [Google Scholar] [CrossRef]
- Soussi, T. The P53 Pathway and Human Cancer. Br. J. Surg. 2005, 92, 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Friedler, A.; Veprintsev, D.B.; Freund, S.M.V.; von Glos, K.I.; Fersht, A.R. Modulation of Binding of DNA to the C-Terminal Domain of P53 by Acetylation. Structure 2005, 13, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Bogaerts, A. Structure and Function of P53-DNA Complexes with Inactivation and Rescue Mutations: A Molecular Dynamics Simulation Study. PLoS ONE 2015, 10, e0134638. [Google Scholar] [CrossRef] [PubMed]
- Madapura, H.S.; Salamon, D.; Wiman, K.G.; Lain, S.; Klein, G.; Klein, E.; Nagy, N. P53 Contributes to T Cell Homeostasis through the Induction of Pro-Apoptotic SAP. Cell Cycle 2012, 11, 4563–4569. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When Mutants Gain New Powers: News from the Mutant P53 Field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 Website: An Integrative Resource Centre for the TP53 Mutation Database and TP53 Mutant Analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function P53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Hamada, J.; Tada, M.; Kameyama, T.; Nakagawa, K.; Suzuki, Y.; Ikawa, M.; Hassan, N.M.M.; Kitagawa, Y.; Moriuchi, T. Mutant P53 R248Q but Not R248W Enhances in Vitro Invasiveness of Human Lung Cancer NCI-H1299 Cells. Biomed. Res. 2010, 31, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Gaur, S.; Zhang, K.; Wu, X.; Yuan, Y.-C.; Li, H.; Hu, S.; Weng, Y.; Yen, Y. Mutants TP53 p.R273H and p.R273C but Not p.R273G Enhance Cancer Cell Malignancy. Hum. Mutat. 2014, 35, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The Clinical Value of Somatic TP53 Gene Mutations in 1,794 Patients with Breast Cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Seagle, B.-L.L.; Eng, K.H.; Dandapani, M.; Yeh, J.Y.; Odunsi, K.; Shahabi, S. Survival of Patients with Structurally-Grouped TP53 Mutations in Ovarian and Breast Cancers. Oncotarget 2015, 6, 18641–18652. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Wiman, K.G. Mutant P53 Reactivation by Small Molecules Makes Its Way to the Clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant P53 as a Guardian of the Cancer Cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLOS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive Statistical Study of 452 BRCA1 Missense Substitutions with Classification of Eight Recurrent Substitutions as Neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef]
- Venselaar, H.; te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein Structure Analysis of Mutations Causing Inheritable Diseases. An e-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Chen, C.-W.; Lin, J.; Chu, Y.-W. IStable: Off-the-Shelf Predictor Integration for Predicting Protein Stability Changes. BMC Bioinform. 2013, 14, S5. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.A.; Sidow, A. Physicochemical Constraint Violation by Missense Substitutions Mediates Impairment of Protein Function and Disease Severity. Genome Res. 2005, 15, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human Non-synonymous SNPs: Server and Survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the Insurgence of Human Genetic Diseases Associated to Single Point Protein Mutations with Support Vector Machines and Evolutionary Information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. SNAP: Predict Effect of Non-Synonymous Polymorphisms on Function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhou, M.; Cui, Y. NsSNPAnalyzer: Identifying Disease-Associated Nonsynonymous Single Nucleotide Polymorphisms. Nucleic Acids Res. 2005, 33, W480–W482. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Kejariwal, A. Coding Single-Nucleotide Polymorphisms Associated with Complex vs. Mendelian Disease: Evolutionary Evidence for Differences in Molecular Effects. Proc. Natl. Acad. Sci. USA 2004, 101, 15398–15403. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational Approaches for Predicting the Biological Effect of P53 Missense Mutations: A Comparison of Three Sequence Analysis Based Methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of Protein Stability Changes for Single-Site Mutations Using Support Vector Machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by P53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of P53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. Mutant P53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.W.K.; Lama, D.; Lukman, S.; Lane, D.P.; Verma, C.S.; Sim, A.Y.L. R248Q Mutation—Beyond P53-DNA Binding. Proteins 2015, 83, 2240–2250. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a Consensus Binding Site for P53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Funk, W.D.; Pak, D.T.; Karas, R.H.; Wright, W.E.; Shay, J.W. A Transcriptionally Active DNA-Binding Site for Human P53 Protein Complexes. Mol. Cell. Biol. 1992, 12, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Friedman, P.N.; Chen, X.; Bargonetti, J.; Prives, C. The P53 Protein Is an Unusually Shaped Tetramer That Binds Directly to DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 3319–3323. [Google Scholar] [CrossRef] [PubMed]
- McLure, K.G.; Lee, P.W. How P53 Binds DNA as a Tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef]
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative Binding of Tetrameric P53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef]
- Inga, A.; Storici, F.; Darden, T.A.; Resnick, M.A. Differential Transactivation by the P53 Transcription Factor Is Highly Dependent on P53 Level and Promoter Target Sequence. Mol. Cell. Biol. 2002, 22, 8612–8625. [Google Scholar] [CrossRef]
- Resnick-Silverman, L.; St Clair, S.; Maurer, M.; Zhao, K.; Manfredi, J.J. Identification of a Novel Class of Genomic DNA-Binding Sites Suggests a Mechanism for Selectivity in Target Gene Activation by the Tumor Suppressor Protein P53. Genes Dev. 1998, 12, 2102–2107. [Google Scholar] [CrossRef]
- Andreou, A.M.; Pauws, E.; Jones, M.C.; Singh, M.K.; Bussen, M.; Doudney, K.; Moore, G.E.; Kispert, A.; Brosens, J.J.; Stanier, P. TBX22 Missense Mutations Found in Patients with X-Linked Cleft Palate Affect DNA Binding, Sumoylation, and Transcriptional Repression. Am. J. Hum. Genet. 2007, 81, 700–712. [Google Scholar] [CrossRef]
- Saleem, R.A.; Banerjee-Basu, S.; Berry, F.B.; Baxevanis, A.D.; Walter, M.A. Analyses of the Effects That Disease-Causing Missense Mutations Have on the Structure and Function of the Winged-Helix Protein FOXC1. Am. J. Hum. Genet. 2001, 68, 627–641. [Google Scholar] [CrossRef]
- Thirumal Kumar, D.; Jain, N.; Udhaya Kumar, S.; George Priya Doss, C.; Zayed, H. Identification of Potential Inhibitors against Pathogenic Missense Mutations of PMM2 Using a Structure-Based Virtual Screening Approach. J. Biomol. Struct. Dyn. 2021, 39, 171–187. [Google Scholar] [CrossRef]
- Thirumal Kumar, D.; Udhaya Kumar, S.; Magesh, R.; George Priya Doss, C. Investigating Mutations at the Hotspot Position of the ERBB2 and Screening for the Novel Lead Compound to Treat Breast Cancer—A Computational Approach. Adv. Protein Chem. Struct. Biol. 2021, 123, 49–71. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Baase, W.A.; Kuroki, R.; Matthews, B.W. A Relationship between Protein Stability and Protein Function. Proc. Natl. Acad. Sci. USA 1995, 92, 452–456. [Google Scholar] [CrossRef]
- Sporleder, M.; Zegarra, O.; Hualla, V.; Simon, R.; Kroschel, J. Phthorimaea Operculella Granulovirus: Sequenceanalysis of 5 Genes from 16 Geographical Isolates. In Proceedings of the Brazilian Symposium on Bioinformatics, Angra dos Reis, Brazil, 29–31 August 2007. [Google Scholar]
- Joerger, A.C.; Fersht, A.R. Structure-Function-Rescue: The Diverse Nature of Common P53 Cancer Mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef]
- Rajendran, V.; Purohit, R.; Sethumadhavan, R. In Silico Investigation of Molecular Mechanism of Laminopathy Caused by a Point Mutation (R482W) in Lamin A/C Protein. Amino Acids 2012, 43, 603–615. [Google Scholar] [CrossRef]
- Bartlett, P.A.; Marlowe, C.K. Evaluation of Intrinsic Binding Energy from a Hydrogen Bonding Group in an Enzyme Inhibitor. Science 1987, 235, 569–571. [Google Scholar] [CrossRef]
- Machado, A.C.D.; Saleebyan, S.B.; Holmes, B.T.; Karelina, M.; Tam, J.; Kim, S.Y.; Kim, K.H.; Dror, I.; Hodis, E.; Martz, E.; et al. Proteopedia: 3D Visualization and Annotation of Transcription Factor–DNA Readout Modes. Biochem. Mol. Biol. Educ. 2012, 40, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.U.; Sankar, S.; Kumar, D.T.; Younes, S.; Younes, N.; Siva, R.; Doss, C.G.P.; Zayed, H. Molecular Dynamics, Residue Network Analysis, and Cross-Correlation Matrix to Characterize the Deleterious Missense Mutations in GALE Causing Galactosemia III. Cell Biochem. Biophys. 2021, 79, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Chen, Z.; Li, Q.; Liu, R.; Song, W.; Kong, R.; Chang, S. Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3. Int. J. Mol. Sci. 2019, 20, 224. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.B.; DeDecker, B.S.; Freund, S.M.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-Spot Mutants of P53 Core Domain Evince Characteristic Local Structural Changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442. [Google Scholar] [CrossRef]
- Nakazawa, S.; Sakata, K.-I.; Liang, S.; Yoshikawa, K.; Iizasa, H.; Tada, M.; Hamada, J.-I.; Kashiwazaki, H.; Kitagawa, Y.; Yamazaki, Y. Dominant-Negative P53 Mutant R248Q Increases the Motile and Invasive Activities of Oral Squamous Cell Carcinoma Cells. Biomed. Res. 2019, 40, 37–49. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant P53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.-C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant P53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.W.; Jeon, H.-Y.; Jin, X.; Kim, E.-J.; Kim, J.-K.; Shin, Y.J.; Lee, Y.; Kim, S.H.; Lee, S.Y.; Seo, S.; et al. TP53 Gain-of-Function Mutation Promotes Inflammation in Glioblastoma. Cell Death Differ. 2019, 26, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Costa, C.; Orozco, M.; de la Cruz, X. Characterization of Disease-Associated Single Amino Acid Polymorphisms in Terms of Sequence and Structure Properties. J. Mol. Biol. 2002, 315, 771–786. [Google Scholar] [CrossRef]
- Livesey, B.J.; Marsh, J.A. The Properties of Human Disease Mutations at Protein Interfaces. PLOS Comput. Biol. 2022, 18, e1009858. [Google Scholar] [CrossRef]
- Matthews, B.W. Structural and Genetic Analysis of Protein Stability. Annu. Rev. Biochem. 1993, 62, 139–160. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Sternberg, M.J.E. The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef]
- Li, E.; You, M.; Hristova, K. FGFR3 Dimer Stabilization Due to a Single Amino Acid Pathogenic Mutation. J. Mol. Biol. 2006, 356, 600–612. [Google Scholar] [CrossRef]
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread Macromolecular Interaction Perturbations in Human Genetic Disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef]
- Kamaraj, B.; Al-Subaie, A.M.; Ahmad, F.; Surapaneni, K.M.; Alsamman, K. Effect of Novel Leukemia Mutations (K75E & E222K) on Interferon Regulatory Factor 1 and Its Interaction with DNA: Insights from Molecular Dynamics Simulations and Docking Studies. J. Biomol. Struct. Dyn. 2021, 39, 5235–5247. [Google Scholar] [CrossRef]
- Sneha, P.; Thirumal Kumar, D.; George Priya Doss, C.; Siva, R.; Zayed, H. Determining the Role of Missense Mutations in the POU Domain of HNF1A That Reduce the DNA-Binding Affinity: A Computational Approach. PLoS ONE 2017, 12, e0174953. [Google Scholar] [CrossRef]
- Salsbury, F.R. Molecular Dynamics Simulations of Protein Dynamics and Their Relevance to Drug Discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef]
- Benz, R.W.; Castro-Román, F.; Tobias, D.J.; White, S.H. Experimental Validation of Molecular Dynamics Simulations of Lipid Bilayers: A New Approach. Biophys. J. 2005, 88, 805–817. [Google Scholar] [CrossRef]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted Rescue of a Destabilized Mutant of P53 by an in Silico Screened Drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small Molecule Induced Reactivation of Mutant P53 in Cancer Cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Reorganizing the Protein Space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar] [CrossRef] [PubMed]
- Berezin, C.; Glaser, F.; Rosenberg, J.; Paz, I.; Pupko, T.; Fariselli, P.; Casadio, R.; Ben-Tal, N. ConSeq: The Identification of Functionally and Structurally Important Residues in Protein Sequences. Bioinformatics 2004, 20, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The Projection of Evolutionary Conservation Scores of Residues on Protein Structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Jaidhan, B.J.; Rao, P.S.; Apparao, A. Energy Minimization and Conformation Analysis of Molecules Using Steepest Descent Method. Int. J. Comput. Sci. Inf. Technol. 2014, 5, 4. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Desdouits, N.; Nilges, M.; Blondel, A. Principal Component Analysis Reveals Correlation of Cavities Evolution and Functional Motions in Proteins. J. Mol. Graph. Model. 2015, 55, 13–24. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | R248W | R248G | R248Q | R248P | R248L |
|---|---|---|---|---|---|
| PredictSNP prediction | D | D | D | D | D |
| MAPP prediction | D | D | D | D | D |
| PhD-SNP prediction | D | D | D | D | D |
| PolyPhen-1 prediction | D | D | D | D | D |
| SIFT prediction | D | D | D | D | D |
| SNAP prediction | D | D | D | D | D |
| Mutation | R248W | R248G | R248Q | R248P | R248L | |
|---|---|---|---|---|---|---|
| Align GVGD | Class C65 | Class C65 | Class C35 | Class C65 | Class C65 | |
| ConSurf | 9 | 9 | 9 | 9 | 9 | |
| iStable | I-Mutant2.0 SEQ | D | D | D | D | D |
| DDG | −0.83 | −1.74 | −1.74 | −1.07 | −0.83 | |
| MUpro | D | D | D | D | I | |
| Conf. Score | −0.65126505 | −0.89185965 | −0.89185965 | −0.57518851 | 0.67336898 | |
| iStable | D | D | D | D | I | |
| Conf. Score | 0.831653 | 0.859332 | 0.859332 | 0.835782 | 0.506999 | |
| Wild Type | R248W | R248G | R248L | R248P | R24Q | |
|---|---|---|---|---|---|---|
| Van der Waals energy (±SD) | −196.928 +/−63.403 | −217.153 +/−31.264 | −195.339 +/−37.430 | −179.873 +/−22.772 | −145.227 +/−44.175 | −172.756 +/−32.282 |
| Electrostatic energy (±SD) | −3694.303 +/−774.611 | −3507.850 +/−230.814 | −2969.158 +/−304.942 | −3180.017 +/−199.262 | −2917.121 +/−223.878 | −3172.238 +/−257.610 |
| Polar solvation energy (±SD) | 1013.332 +/−262.915 | 1191.171 +/−129.089 | 696.856 +/−189.826 | 802.547 +/−114.081 | 801.287 +/−179.410 | 853.801 +/−141.240 |
| SASA energy (±SD) | −28.858 +/−7.108 | −31.040 +/−4.048 | −24.867 +/−4.462 | −26.327 +/−3.180 | −22.537 +/−4.612 | −25.422 +/−3.371 |
| Total Binding energy (±SD) | −2906.756 +/−621.185 | −2564.871 +/−194.196 | −2492.508 +/−215.762 | −2583.670 +/−186.701 | −2283.598 +/−169.333 | −2516.614 +/−227.002 |
| No. of H-Binding Site at Arg248 | Conventional H-Binding of Arg248 with DNA | |
|---|---|---|
| WT | 3 | A:Arg248:NH2-E:Gua12:O2P |
| A:Arg248:NH2-F:Ade6:O3′ | ||
| A:Arg248:NE-F:Thy7:O1P | ||
| R248W | 1 | A:TRP248:HD1-E:THY7:O1P |
| R248G | 0 | Nil |
| R248L | 0 | Nil |
| R248P | 0 | Nil |
| R24Q | 1 | A: GLN248:HE21-E:THY7:O1P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balasundaram, A.; Doss, C.G.P. Unraveling the Structural Changes in the DNA-Binding Region of Tumor Protein p53 (TP53) upon Hotspot Mutation p53 Arg248 by Comparative Computational Approach. Int. J. Mol. Sci. 2022, 23, 15499. https://doi.org/10.3390/ijms232415499
Balasundaram A, Doss CGP. Unraveling the Structural Changes in the DNA-Binding Region of Tumor Protein p53 (TP53) upon Hotspot Mutation p53 Arg248 by Comparative Computational Approach. International Journal of Molecular Sciences. 2022; 23(24):15499. https://doi.org/10.3390/ijms232415499
Chicago/Turabian StyleBalasundaram, Ambritha, and C. George Priya Doss. 2022. "Unraveling the Structural Changes in the DNA-Binding Region of Tumor Protein p53 (TP53) upon Hotspot Mutation p53 Arg248 by Comparative Computational Approach" International Journal of Molecular Sciences 23, no. 24: 15499. https://doi.org/10.3390/ijms232415499
APA StyleBalasundaram, A., & Doss, C. G. P. (2022). Unraveling the Structural Changes in the DNA-Binding Region of Tumor Protein p53 (TP53) upon Hotspot Mutation p53 Arg248 by Comparative Computational Approach. International Journal of Molecular Sciences, 23(24), 15499. https://doi.org/10.3390/ijms232415499