
Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt–Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene
Abstract
:1. Introduction
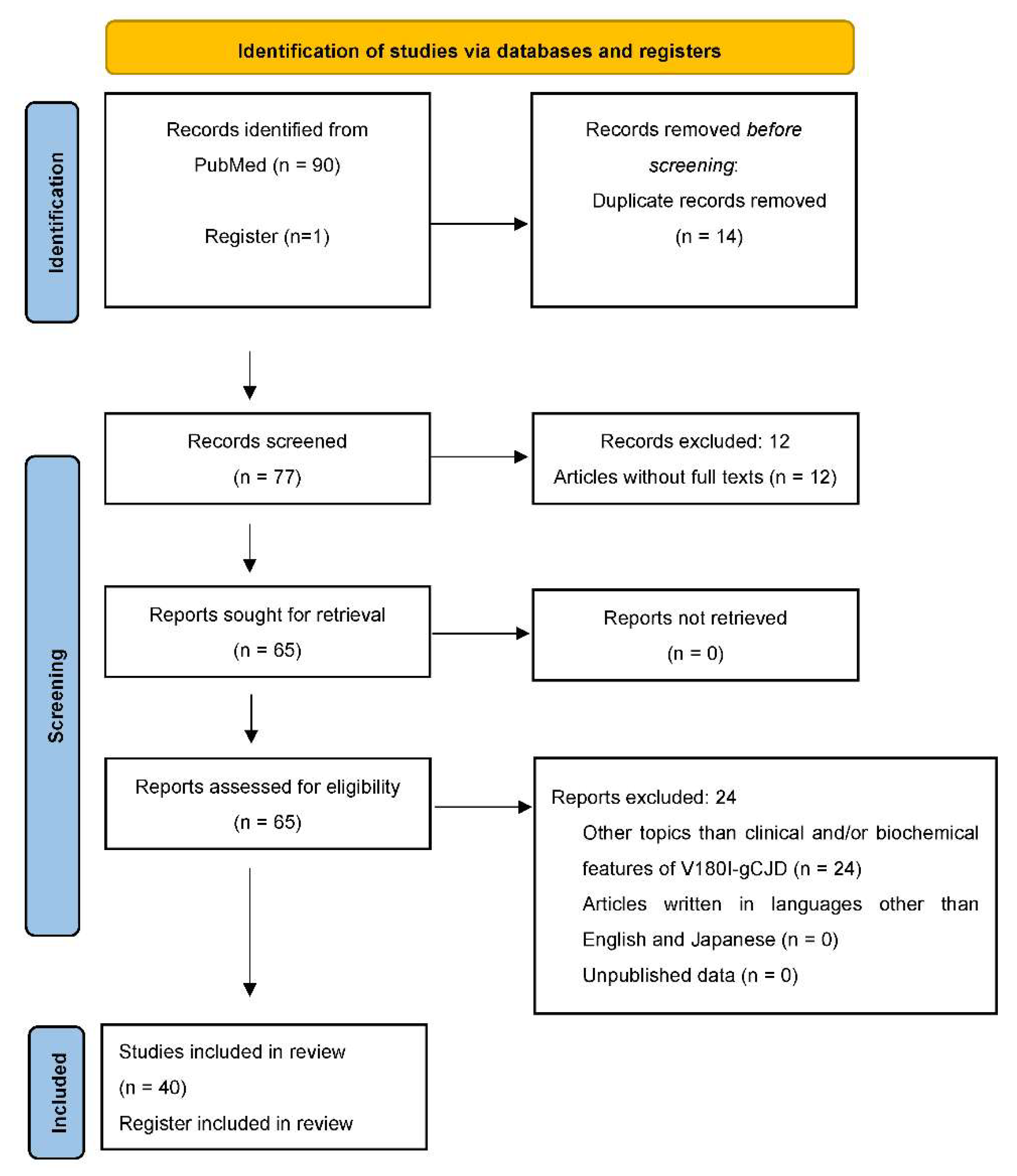
2. Material and Methods
3. Results and Discussion
3.1. Clinical Features of V180I-gCJD
3.1.1. Clinical Features
Epidemiological Factors of V180I-gCJD
Clinical Symptoms and Signs of V180I-gCJD
3.1.2. EEG
3.1.3. Brain Imaging
- bilateral cortical signal hyperintensity on DWI-MRI with or without basal ganglia involvement,
- cortical ribboning edematous swelling on fluid-attenuated inversion recovery and T2-weighted images,
- absence in the occipital lobes, posterior to the parieto-occipital sulcus, brainstem, or cerebellum until the terminal stage.
3.1.4. CSF Biomarkers
RT-QuIC
14-3-3 Protein
Tau Protein
3.2. Biochemical Features of V180I-gCJD
3.2.1. Neuropathological Examination
Neuropathological Findings of sCJD and gCJD
Neuropathological Findings of V180I-gCJD
3.2.2. Western Blot Analysis and Pathogenicity of V180I Mutation
4. Conclusions
- an extremely low frequency of family history;
- a late onset age and slow progression;
- major initial symptoms of cerebral cortical dysfunctions and a low frequency of myoclonus, visual disturbance, and cerebellar signs;
- a low positive rate of PSWCs on EEGs;
- a predominant involvement of the cerebral cortex in DWI-MRI and SPECT imaging, and the absence of abnormal findings in occipital lobes, posterior to the parieto-occipital sulcus, brainstem, or the cerebellum;
- a relatively low positive rate of PrPSc by CSF RT-QuIC, and a high positive rate of 14-3-3 protein and t-tau protein in CSF examination.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. The prion diseases. Brain Pathol. 1998, 8, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Harris, J.O.; Gajdusek, D.C.; Gibbs, C.J.; Bernoulli, C.; Asher, D.M. Creutzfeldt-Jakob disease: Patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann. Neurol. 1979, 5, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Ladogana, A.; Puopolo, M.; Croes, E.A.; Budka, H.; Jarius, C.; Collins, S.; Klug, G.M.; Sutcliffe, T.; Giulivi, A.; Alperovitch, A.; et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005, 64, 1586–1591. [Google Scholar] [CrossRef]
- Kretzschmar, H.A.; Neumann, M.; Stavrou, D. Codon 178 mutation of the human prion protein gene in a German family (Backer family): Sequencing data from 72-year-old celloidin-embedded brain tissue. Acta Neuropathol. 1995, 89, 96–98. [Google Scholar] [CrossRef]
- Mead, S.; Lloyd, S.; Collinge, J. Genetic factors in mammalian prion diseases. Annu. Rev. Genet. 2019, 53, 117–147. [Google Scholar] [CrossRef]
- Kim, M.O.; Takada, L.T.; Wong, K.; Forner, S.A.; Geschwind, M.D. Genetic PrP prion diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033134. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.; Mead, S.; Collinge, J. Genetics of prion disease. Top. Curr. Chem. 2011, 305, 1–22. [Google Scholar] [CrossRef]
- Kovács, G.G.; Puopolo, M.; Ladogana, A.; Pocchiari, M.; Budka, H.; van Duijn, C.; Collins, S.J.; Boyd, A.; Giulivi, A.; Coulthart, M.; et al. Genetic prion disease: The EUROCJD experience. Hum. Genet. 2005, 118, 166–174. [Google Scholar] [CrossRef]
- Nozaki, I.; Hamaguchi, T.; Sanjo, N.; Noguchi-Shinohara, M.; Sakai, K.; Nakamura, Y.; Sato, T.; Kitamoto, T.; Mizusawa, H.; Moriwaka, F.; et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010, 133, 3043–3057. [Google Scholar] [CrossRef]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Dong, X.P. Epidemiological characteristics of human prion diseases. Infect. Dis. Poverty 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G.; Seguin, J.; Quadrio, I.; Höftberger, R.; Kapás, I.; Streichenberger, N.; Biacabe, A.G.; Meyronet, D.; Sciot, R.; Vandenberghe, R.; et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: Characterization of a complex proteinopathy. Acta Neuropathol. 2011, 121, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, T.; Ohta, M.; Dohura, K.; Hitoshi, S.; Terao, Y.; Tateishi, J. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem. Biophys. Res. Commun. 1993, 191, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Chasseigneaux, S.; Haïk, S.; Laffont-Proust, I.; De Marco, O.; Lenne, M.; Brandel, J.P.; Hauw, J.J.; Laplanche, J.L.; Peoc’h, K. V180I mutation of the prion protein gene associated with atypical PrPSc glycosylation. Neurosci. Lett. 2006, 408, 165–169. [Google Scholar] [CrossRef]
- Yang, T.I.; Jung, D.S.; Ahn, B.Y.; Jeong, B.H.; Cho, H.J.; Kim, Y.S.; Na, D.L.; Geschwind, M.D.; Kim, E.J. Familial Creutzfeldt-Jakob disease with V180I mutation. J. Korean Med. Sci. 2010, 25, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- De Souza, R.K.M.; Josviak, N.D.; Batistela, M.S.; Santos, P.S.F.; Landemberger, M.C.; Ramina, R. First case of V180I rare mutation in a Brazilian patient with Creutzfeldt-Jakob disease. Prion 2017, 11, 465–468. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Shen, X.J.; Zhou, W.; Xiao, K.; Zhang, X.M.; Zhang, B.Y.; Dong, X.P. Rare V180I mutation in PRNP gene of a Chinese patient with Creutzfeldt-Jakob disease. Prion 2014, 8, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, N.; Yi, S.; An, S.S.A.; Park, Y.H.; Kim, S. A Creutzfeldt–Jakob disease patient with V180I mutation survived for 16.5 years after diagnosis. Acta Neurol. Belg. 2022, 122, 249–250. [Google Scholar] [CrossRef]
- Jin, K.; Shiga, Y.; Shibuya, S.; Chida, K.; Sato, Y.; Konno, H.; Dohura, K.; Kitamoto, T.; Itoyama, Y. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology 2004, 62, 502–505. [Google Scholar] [CrossRef]
- Qina, T.; Sanjo, N.; Hizume, M.; Higuma, M.; Tomita, M.; Atarashi, R.; Satoh, K.; Nozaki, I.; Hamaguchi, T.; Nakamura, Y.; et al. Clinical features of genetic Creutzfeldt-Jakob disease with V180I mutation in the prion protein gene. BMJ Open 2014, 4, e004968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe Lee, S.; Ran Ju, Y.; Choi, B.Y.; Wook Hyeon, J.; Sun Park, J.; Kyeong Kim, C.; Yeon Kim, S. Genotype patterns and characteristics of PRNP in the Korean population. Prion 2012, 6, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Sanjo, N.; Hizume, M.; Kobayashi, A.; Ohgami, T.; Satoh, K.; Hamaguchi, T.; Yamada, M.; Kitamoto, T.; Mizusawa, H.; et al. Biochemical features of genetic Creutzfeldt-Jakob disease with valine-to-isoleucine substitution at codon 180 on the prion protein gene. Biochem. Biophys. Res. Commun. 2018, 496, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Mutsukura, K.; Satoh, K.; Shirabe, S.; Tomita, I.; Fukutome, T.; Morikawa, M.; Iseki, M.; Sasaki, K.; Shiaga, Y.; Kitamoto, T.; et al. Familial Creutzfeldt-Jakob disease with a V180I mutation: Comparative analysis with pathological findings and diffusion-weighted images. Dement. Geriatr. Cogn. Disord. 2009, 28, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuma, M.; Sanjo, N.; Satoh, K.; Shiga, Y.; Sakai, K.; Nozaki, I.; Hamaguchi, T.; Nakamura, Y.; Kitamoto, T.; Shirabe, S.; et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS ONE 2013, 8, e60003. [Google Scholar] [CrossRef] [Green Version]
- Akagi, A.; Iwasaki, Y.; Mimuro, M.; Kitamoto, T.; Yamada, M.; Yoshida, M. Pathological progression of genetic Creutzfeldt-Jakob disease with a PrP V180I mutation. Prion 2018, 12, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Iwasaki, Y.; Waza, M.; Kato, S.; Akagi, A.; Kimura, A.; Inuzuka, T.; Satoh, K.; Kitamoto, T.; Yoshida, M.; et al. Clinicopathological findings of a long-term survivor of V180I genetic Creutzfeldt-Jakob disease. Prion 2020, 14, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Nomura, T.; Iwata, I.; Naganuma, R.; Matsushima, M.; Satoh, K.; Kitamoto, T.; Yabe, I. A patient with spastic paralysis finally diagnosed as V180I genetic Creutzfeldt-Jakob disease 9 years after onset. Prion 2020, 14, 226–231. [Google Scholar] [CrossRef]
- Kunieda, K.; Hayashi, Y.; Yamada, M.; Waza, M.; Yaguchi, T.; Fujishima, I.; Shimohata, T. Serial evaluation of swallowing function in a long-term survivor of V180I genetic Creutzfeldt-Jakob disease. Prion 2020, 14, 180–184. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Mori, K.; Ito, M.; Nagaoka, M.; Ieda, T.; Kitamoto, T.; Yoshida, M.; Hashizume, Y. An autopsied case of V180I Creutzfeldt-Jakob disease presenting with panencephalopathic-type pathology and a characteristic prion protein type. Neuropathology 2011, 31, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Mori, K.; Ito, M.; Akagi, A.; Mimuro, M.; Kitamoto, T.; Yoshida, M. An autopsy case of Creutzfeldt-Jakob disease with a prion protein gene codon 180 mutation presenting with pathological laughing and an exaggerated startle reaction. Neuropathology 2017, 37, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Mori, K.; Ito, M.; Kawai, Y. A case of V180I genetic Creutzfeldt-Jakob disease presenting with conspicuous facial mimicry. Prion 2019, 13, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Matsumura, N.; Suzuki, T.; Nakano, H.; Nagayama, H.; Yokoo, H.; Tamura, K.; Katayama, Y.; Sugisaki, Y.; Oba, K. A case of Creutzfeldt-Jakob disease with codon 129 polymorphism and codon 180 point mutation. Nihon Ronen Igakkai Zasshi 2008, 45, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y. Three cases of Creutzfeldt-Jakob disease with prion protein gene codon180 mutation presenting with pathological laughing and crying. J. Neurol. Sci. 2012, 319, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Nagata, T.; Shinagawa, S.; Kobayashi, N.; Kondo, K.; Shigeta, M. A case of V180I genetic mutation Creutzfeldt Jakob disease (CJD) with delusional misidentification as an initial symptom. Prion 2022, 16, 7–13. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yoshikura, N.; Takekoshi, A.; Yamada, M.; Asano, T.; Kimura, A.; Satoh, K.; Kitamoto, T.; Inuzuka, T. Preserved regional cerebral blood flow in the occipital cortices, brainstem, and cerebellum of patients with V180I-129M genetic Creutzfeldt-Jakob disease in serial SPECT studies. J. Neurol. Sci. 2016, 370, 145–151. [Google Scholar] [CrossRef]
- Fujita, H.; Ogaki, K.; Shiina, T.; Onuma, H.; Sakuramoto, H.; Satoh, K.; Suzuki, K. V180I genetic Creutzfeldt-Jakob disease with cardiac sympathetic nerve denervation masquerading as Parkinson’s disease: A case report. Medicine 2021, 100, e24294. [Google Scholar] [CrossRef]
- Tomizawa, Y.; Taniguchi, D.; Furukawa, Y. Genetic Creutzfeldt-Jakob disease mimicking dementia with Lewy bodies: Clinical and radiological findings. J. Neurol. Sci. 2020, 409, 116604. [Google Scholar] [CrossRef]
- Koizumi, R.; Ueda, N.; Mugita, A.; Kimura, K.; Kishida, H.; Tanaka, F. Case report: Extremely early detection of preclinical magnetic resonance imaging abnormality in Creutzfeldt–Jakob disease with the V180I mutation. Front. Neurol. 2021, 12, 751750. [Google Scholar] [CrossRef]
- Deguchi, K.; Takamiya, M.; Deguchi, S.; Morimoto, N.; Kurata, T.; Ikeda, Y.; Abe, K. Spreading brain lesions in a familial Creutzfeldt-Jakob disease with V180I mutation over 4 years. BMC Neurol. 2012, 12, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, Y.; Mori, K.; Ito, M.; Kawai, Y.; Hoshino, K.I.; Kawabata, Y.; Mimuro, M.; Yoshida, M. Gastrostomy in patients with prion disease. Prion 2017, 11, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, S.; Manabe, Y.; Fujii, D.; Sakai, Y.; Narai, H.; Omori, N.; Kitamoto, T.; Abe, K. Serial diffusion-weighted MRI and SPECT findings in a Creutzfeldt-Jakob disease patient with V180I mutation. J. Neurol. Sci. 2011, 301, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, Y.; Fujita, K.; Izumi, Y.; Kaji, R. Early detection of familial Creutzfeldt-Jakob disease on diffusion-weighted imaging before symptom onset. J. Neurol. Sci. 2012, 319, 130–132. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sugiyama, A.; Muto, M.; Satoh, K.; Kitamoto, T.; Kuwabara, S. Early diagnosis of V180I genetic Creutzfeldt-Jakob disease at the preserved cognitive function stage. Cureus 2022, 14, e23374. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Kato, H.; Ando, T.; Akagi, A.; Mimuro, M.; Miyahara, H.; Kitamoto, T.; Yoshida, M. Autopsy case of V180I genetic Creutzfeldt-Jakob disease presenting with early disease pathology. Neuropathology 2018, 38, 638–645. [Google Scholar] [CrossRef]
- Yoshida, H.; Terada, S.; Ishizu, H.; Ikeda, K.; Hayabara, T.; Ikeda, K.; Deguchi, K.; Touge, T.; Kitamoto, T.; Kuroda, S. An autopsy case of Creutzfeldt-Jakob disease with a V180I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology 2010, 30, 159–164. [Google Scholar] [CrossRef]
- Yeo, M.J.; Lee, S.H.; Lee, S.Y.; Jeon, Y.C.; Park, S.J.; Cho, H.J.; Choi, K.C.; Kim, Y.S.; Kim, S.H. Familial Creutzfeldt-Jakob disease with a mutation at codon 180 presenting with an atypical phenotype. J. Clin. Neurosci. 2013, 20, 180–182. [Google Scholar] [CrossRef]
- Xiao, X.; Yuan, J.; Haïk, S.; Cali, I.; Zhan, Y.; Moudjou, M.; Li, B.; Laplanche, J.L.; Laude, H.; Langeveld, J.; et al. Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS ONE 2013, 8, e58786. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, J.; Shen, P.; Abskharon, R.; Lang, Y.; Dang, J.; Adornato, A.; Xu, L.; Chen, J.; Feng, J.; et al. In vitro seeding activity of glycoform-deficient prions from variably protease-sensitive prionopathy and familial CJD associated with PrPV180I mutation. Mol. Neurobiol. 2019, 56, 5456–5469. [Google Scholar] [CrossRef]
- Kosami, K.; Ae, R.; Hamaguchi, T.; Sanjo, N.; Tsukamoto, T.; Kitamoto, T.; Yamada, M.; Mizusawa, H.; Nakamura, Y. Methionine homozygosity for PRNP polymorphism and susceptibility to human prion diseases. J. Neurol. Neurosurg. Psychiatry 2022, 93, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.H.; Lee, K.H.; Kim, N.H.; Jin, J.K.; Kim, J.I.; Carp, R.I.; Kim, Y.S. Association of sporadic Creutzfeldt-Jakob disease with homozygous genotypes at PRNP codons 129 and 219 in the Korean population. Neurogenetics 2005, 6, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, S.; Higuchi, J.; Shin, R.W.; Tateishi, J.; Kitamoto, T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 1998, 43, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.G.; Trabattoni, G.; Hainfellner, J.A.; Ironside, J.W.; Knight, R.S.; Budka, H. Mutations of the prion protein gene phenotypic spectrum. J. Neurol. 2002, 249, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Mao, Z.H. Progression of motor symptoms in Parkinson’s disease. Neurosci. Bull. 2012, 28, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s disease: Current status and future developments. Curr. Neuropharmacol. 2018, 16, 1239–1252. [Google Scholar] [CrossRef]
- Arnts, H.; van Erp, W.S.; Lavrijsen, J.C.M.; van Gaal, S.; Groenewegen, H.J.; van den Munckhof, P. On the pathophysiology and treatment of akinetic mutism. Neurosci. Biobehav. Rev. 2020, 112, 270–278. [Google Scholar] [CrossRef]
- Steinhoff, B.J.; Räcker, S.; Herrendorf, G.; Poser, S.; Grosche, S.; Zerr, I.; Kretzschmar, H.; Weber, T. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch. Neurol. 1996, 53, 162–166. [Google Scholar] [CrossRef]
- Le Bihan, D.; Breton, E.; Lallemand, D.; Grenier, P.; Cabanis, E.; Laval-Jeantet, M. MR imaging of intravoxel incoherent motions: Application to diffusion and perfusion in neurologic disorders. Radiology 1986, 161, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Harada, M.; Sasaki, M.; Yuasa, T.; Sakai, K.; Hamaguchi, T.; Sanjo, N.; Shiga, Y.; Satoh, K.; Atarashi, R.; et al. Multicentre multiobserver study of diffusion-weighted and fluid-attenuated inversion recovery MRI for the diagnosis of sporadic Creutzfeldt-Jakob disease: A reliability and agreement study. BMJ Open 2012, 2, e000649. [Google Scholar] [CrossRef]
- Vitali, P.; Maccagnano, E.; Caverzasi, E.; Henry, R.G.; Haman, A.; Torres-Chae, C.; Johnson, D.Y.; Miller, B.L.; Geschwind, M.D. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011, 76, 1711–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzi, A.; Pascuzzo, R.; Blevins, J.; Grisoli, M.; Lodi, R.; Moscatelli, M.E.M.; Castelli, G.; Cohen, M.L.; Schonberger, L.B.; Foutz, A.; et al. Evaluation of a new criterion for detecting prion disease with diffusion magnetic resonance imaging. JAMA Neurol. 2020, 77, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Podreka, I.; Baumgartner, C.; Suess, E.; Müller, C.; Brücke, T.; Lang, W.; Holzner, F.; Steiner, M.; Deecke, L. Quantification of regional cerebral blood flow with IMP-SPECT. Reproducibility and clinical relevance of flow values. Stroke 1989, 20, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.; Appleby, B.; Brandel, J.P.; Caughey, B.; Collins, S.; Geschwind, M.D.; Green, A.; Haïk, S.; Kovacs, G.G.; Ladogana, A.; et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021, 20, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.; Laux, M.; Glatzel, M.; Matschke, J.; Knipper, T.; Goebel, S.; Treig, J.; Schulz-Schaeffer, W.; Cramm, M.; Schmitz, M.; et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology 2018, 91, e331–e338. [Google Scholar] [CrossRef]
- Rhoads, D.D.; Wrona, A.; Foutz, A.; Blevins, J.; Glisic, K.; Person, M.; Maddox, R.A.; Belay, E.D.; Schonberger, L.B.; Tatsuoka, C.; et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology 2020, 95, e1017–e1026. [Google Scholar] [CrossRef]
- Fiorini, M.; Iselle, G.; Perra, D.; Bongianni, M.; Capaldi, S.; Sacchetto, L.; Ferrari, S.; Mombello, A.; Vascellari, S.; Testi, S.; et al. High diagnostic accuracy of RT-QuIC assay in a prospective study of patients with suspected sCJD. Int. J. Mol. Sci. 2020, 21, 880. [Google Scholar] [CrossRef] [Green Version]
- Mammana, A.; Baiardi, S.; Rossi, M.; Franceschini, A.; Donadio, V.; Capellari, S.; Caughey, B.; Parchi, P. Detection of prions in skin punch biopsies of Creutzfeldt-Jakob disease patients. Ann. Clin. Transl. Neurol. 2020, 7, 559–564. [Google Scholar] [CrossRef]
- Baiardi, S.; Rossi, M.; Mammana, A.; Appleby, B.S.; Barria, M.A.; Calì, I.; Gambetti, P.; Gelpi, E.; Giese, A.; Ghetti, B.; et al. Phenotypic diversity of genetic Creutzfeldt-Jakob disease: A histo-molecular-based classification. Acta Neuropathol. 2021, 142, 707–728. [Google Scholar] [CrossRef]
- Schelzke, G.; Kretzschmar, H.A.; Zerr, I. Clinical aspects of common genetic Creutzfeldt-Jakob disease. Eur. J. Epidemiol. 2012, 27, 147–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, L.G.; Petersen, R.B.; Tabaton, M.; Brown, P.; LeBlanc, A.C.; Montagna, P.; Cortelli, P.; Julien, J.; Vital, C.; Pendelbury, W.W. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Disease phenotype determined by a DNA polymorphism. Science 1992, 258, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.B.; Parchi, P.; Richardson, S.L.; Urig, C.B.; Gambetti, P. Effect of the D178N mutation and the codon 129 polymorphism on the metabolism of the prion protein. J. Biol. Chem. 1996, 271, 12661–12668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, D.; Holzmann, C.; Riess, O. 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 2003, 4, 752–762. [Google Scholar] [CrossRef]
- Hsich, G.; Kenney, K.; Gibbs, C.J.; Lee, K.H.; Harrington, M.G. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N. Engl. J. Med. 1996, 335, 924–930. [Google Scholar] [CrossRef]
- World Health Organization. WHO Manual for Strengthening Diagnosis and Surveillance of Creutzfeldt-Jakob Disease; Zeidler, M., Gibbs, C.J., Meslin, F., Eds.; World Health Organization: Geneva, Switzerland, 1998; Available online: https://apps.who.int/iris/handle/10665/66394 (accessed on 24 August 2022).
- Hamlin, C.; Puoti, G.; Berri, S.; Sting, E.; Harris, C.; Cohen, M.; Spear, C.; Bizzi, A.; Debanne, S.M.; Rowland, D.Y. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology 2012, 79, 547–552. [Google Scholar] [CrossRef]
- Stoeck, K.; Sanchez-Juan, P.; Gawinecka, J.; Green, A.; Ladogana, A.; Pocchiari, M.; Sanchez-Valle, R.; Mitrova, E.; Sklaviadis, T.; Kulczycki, J.; et al. Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: A longitudinal multicentre study over 10 years. Brain 2012, 135, 3051–3061. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Lattanzio, F.; Abu-Rumeileh, S.; Franceschini, A.; Kai, H.; Amore, G.; Poggiolini, I.; Rossi, M.; Baiardi, S.; McGuire, L.; Ladogana, A.; et al. Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: Diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels. Acta Neuropathol. 2017, 133, 559–578. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Mastrianni, J.A. The prion diseases. J. Geriatr. Psychiatry Neurol. 2010, 23, 277–298. [Google Scholar] [CrossRef]
- Kübler, E.; Oesch, B.; Raeber, A.J. Diagnosis of prion diseases. Br. Med. Bull. 2003, 66, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.; Trojanowski, J.Q.; et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 1996, 39, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y. Creutzfeldt-Jakob disease. Neuropathology 2017, 37, 174–188. [Google Scholar] [CrossRef] [Green Version]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial CJD: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Tatsumi, S.; Mimuro, M.; Kitamoto, T.; Hashizume, Y.; Yoshida, M. Relation between clinical findings and progression of cerebral cortical pathology in MM1-type sporadic Creutzfeldt-Jakob disease: Proposed staging of cerebral cortical pathology. J. Neurol. Sci. 2014, 341, 97–104. [Google Scholar] [CrossRef]
- Satoh, K.; Muramoto, T.; Tanaka, T.; Kitamoto, N.; Ironside, J.W.; Nagashima, K.; Yamada, M.; Sato, T.; Mohri, S.; Kitamoto, T. Association of an 11-12 kDa protease-resistant prion protein fragment with subtypes of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J. Gen. Virol. 2003, 84, 2885–2893. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Article * | Dataset Size ** |
|---|---|
| Nozaki et al. 2010 [10] | 89 |
| Yang et al. 2010 [16] | 1 |
| Shi et al. 2014 [18] | 1 |
| Ryoo et al. 2022 [19] | 1 |
| Jin et al. 2004 [20] | 9 |
| Qina et al. 2014 [21] | 186 |
| Ito et al. 2018 [24] | 7 |
| Kutsukura et al. 2009 [25] | 3 |
| Higuma et al. 2013 [26] | 151 |
| Akagi et al. 2018 [27] | 6 |
| Hayashi et al. 2020 [28] | 1 |
| Nomura et al. 2020 [29] | 1 |
| Kunieda et al. 2020 [30] | 1 |
| Iwasaki et al. 2011 [31] | 1 |
| Iwasaki et al. 2017 [32] | 1 |
| Iwasaki et al. 2019 [33] | 1 |
| Suzuki et al. 2008 [34] | 1 |
| Iwasaki et al. 2012 [35] | 3 |
| Nagata et al. 2022 [36] | 1 |
| Hayashi et al. 2016 [37] | 3 |
| Fujita et al. 2021 [38] | 1 |
| Tomizawa et al. 2020 [39] | 1 |
| Koizumi et al. 2021 [40] | 1 |
| Deguchi et al. 2012 [41] | 1 |
| Iwasaki et al. 2017 [42] | 3 |
| Kono et al. 2011 [43] | 1 |
| Terasawa et al. 2012 [44] | 1 |
| Suzuki et al. 2022 [45] | 1 |
| Iwasaki et al. 2018 [46] | 1 |
| Yoshida et al. 2010 [47] | 1 |
| Yeo et al. 2013 [48] | 1 |
| Xiao et al. 2013 [49] | 6 |
| Wang et al. 2019 [50] | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsubayashi, T.; Sanjo, N. Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt–Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene. Int. J. Mol. Sci. 2022, 23, 15172. https://doi.org/10.3390/ijms232315172
Matsubayashi T, Sanjo N. Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt–Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene. International Journal of Molecular Sciences. 2022; 23(23):15172. https://doi.org/10.3390/ijms232315172
Chicago/Turabian StyleMatsubayashi, Taiki, and Nobuo Sanjo. 2022. "Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt–Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene" International Journal of Molecular Sciences 23, no. 23: 15172. https://doi.org/10.3390/ijms232315172
APA StyleMatsubayashi, T., & Sanjo, N. (2022). Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt–Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene. International Journal of Molecular Sciences, 23(23), 15172. https://doi.org/10.3390/ijms232315172