
Acetylation State of Lysine 14 of Histone H3.3 Affects Mutant Huntingtin Induced Pathogenesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
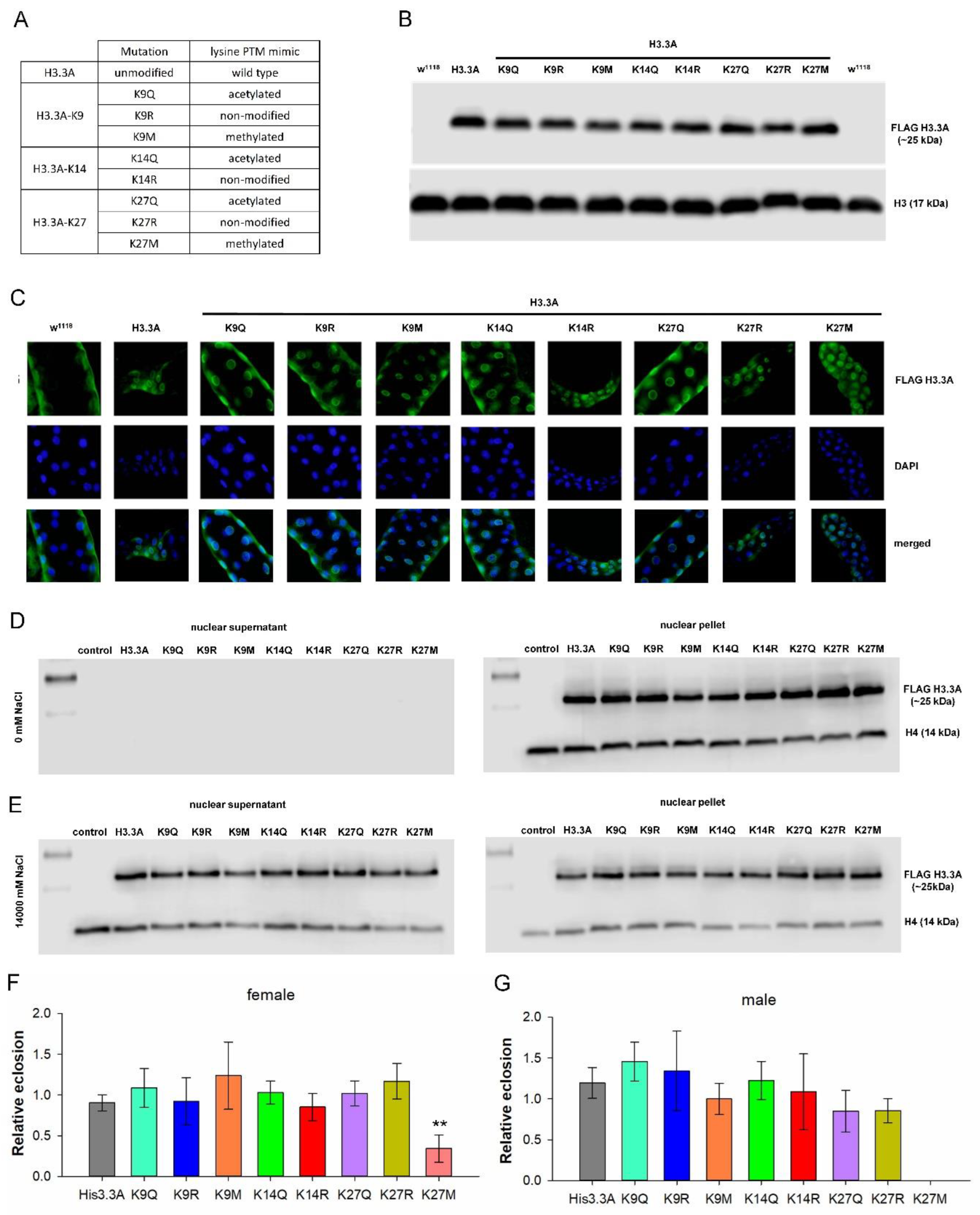
2.1. H3.3 PTM Mimetic Mutants Localize to Chromatin
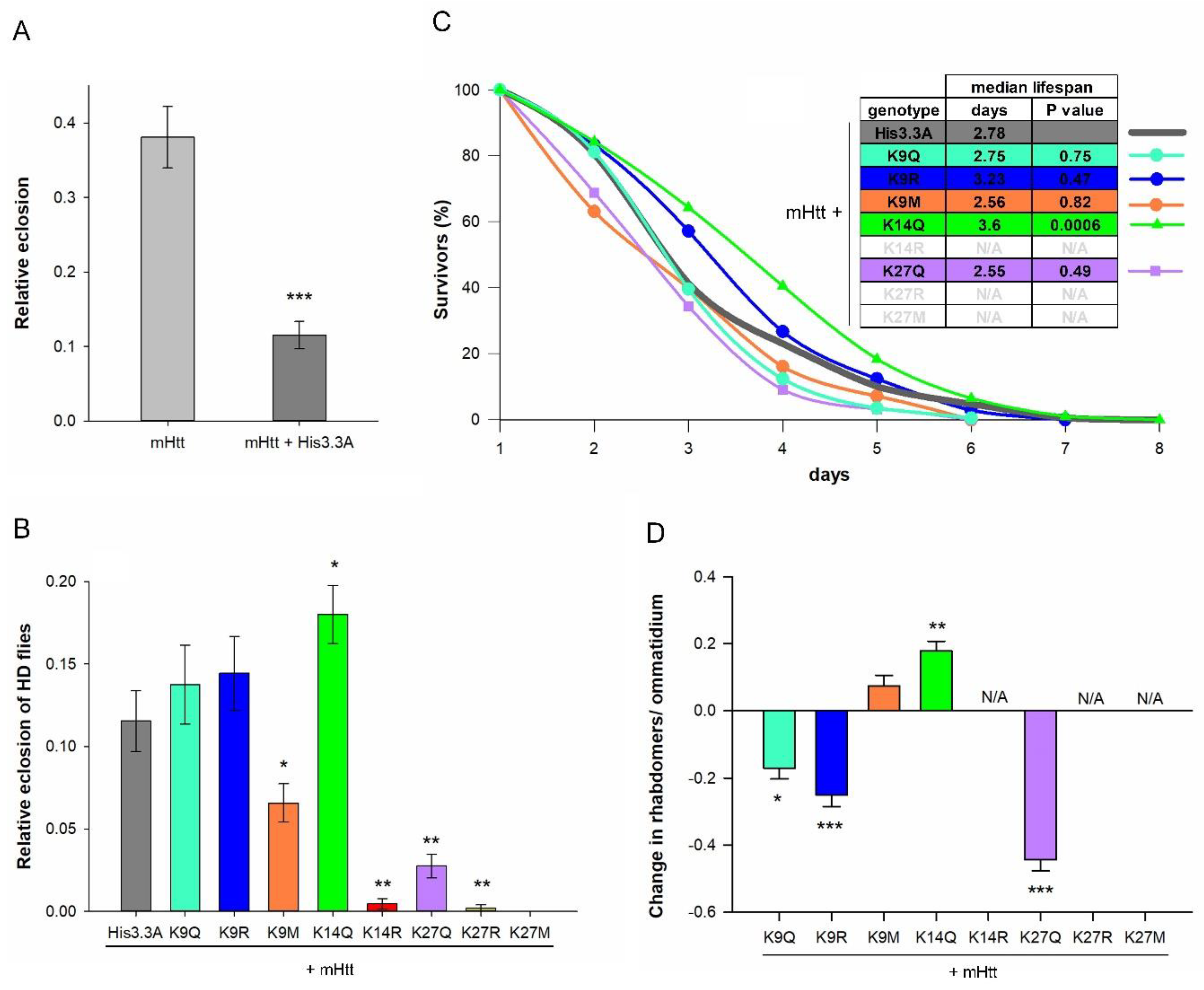
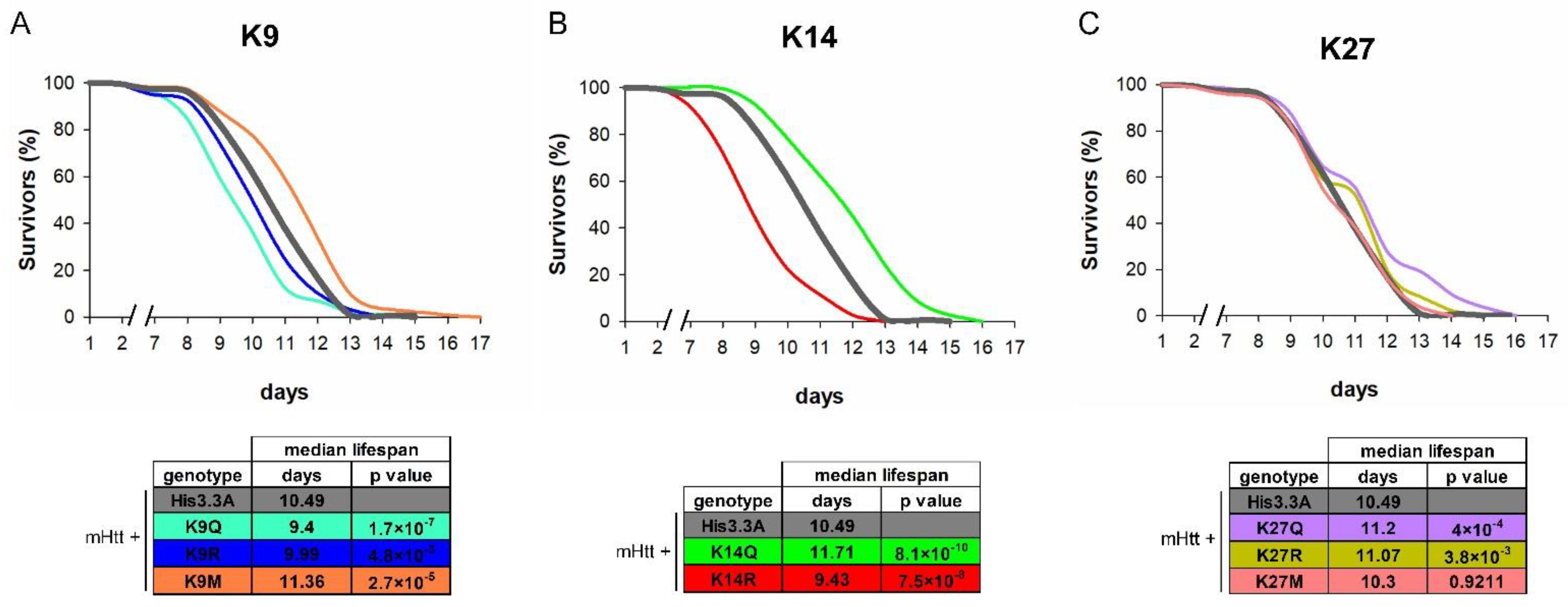
2.2. The Effects of H3.3-PTM Mutants on the Viability, Longevity, and Neurodegeneration of HD Flies
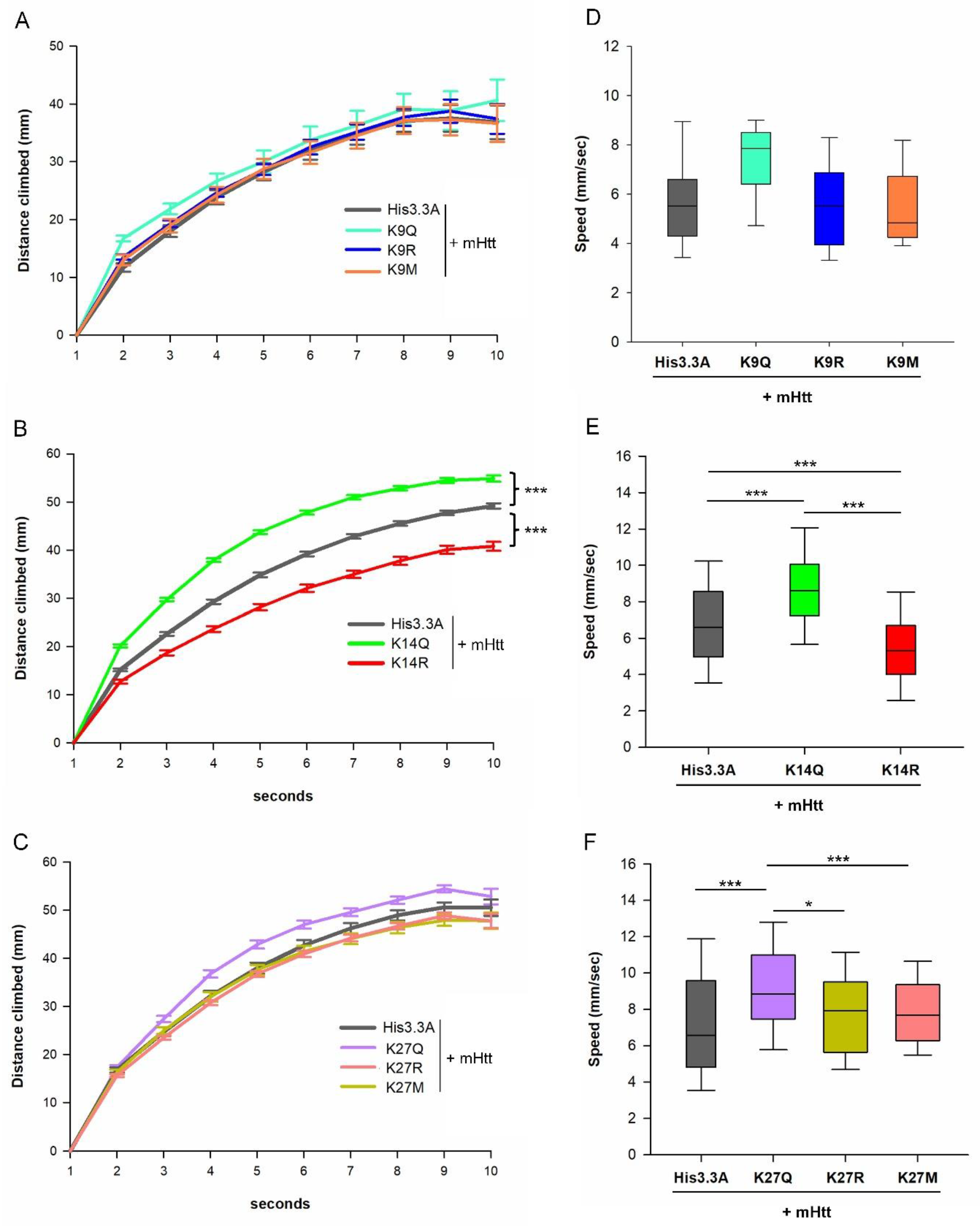
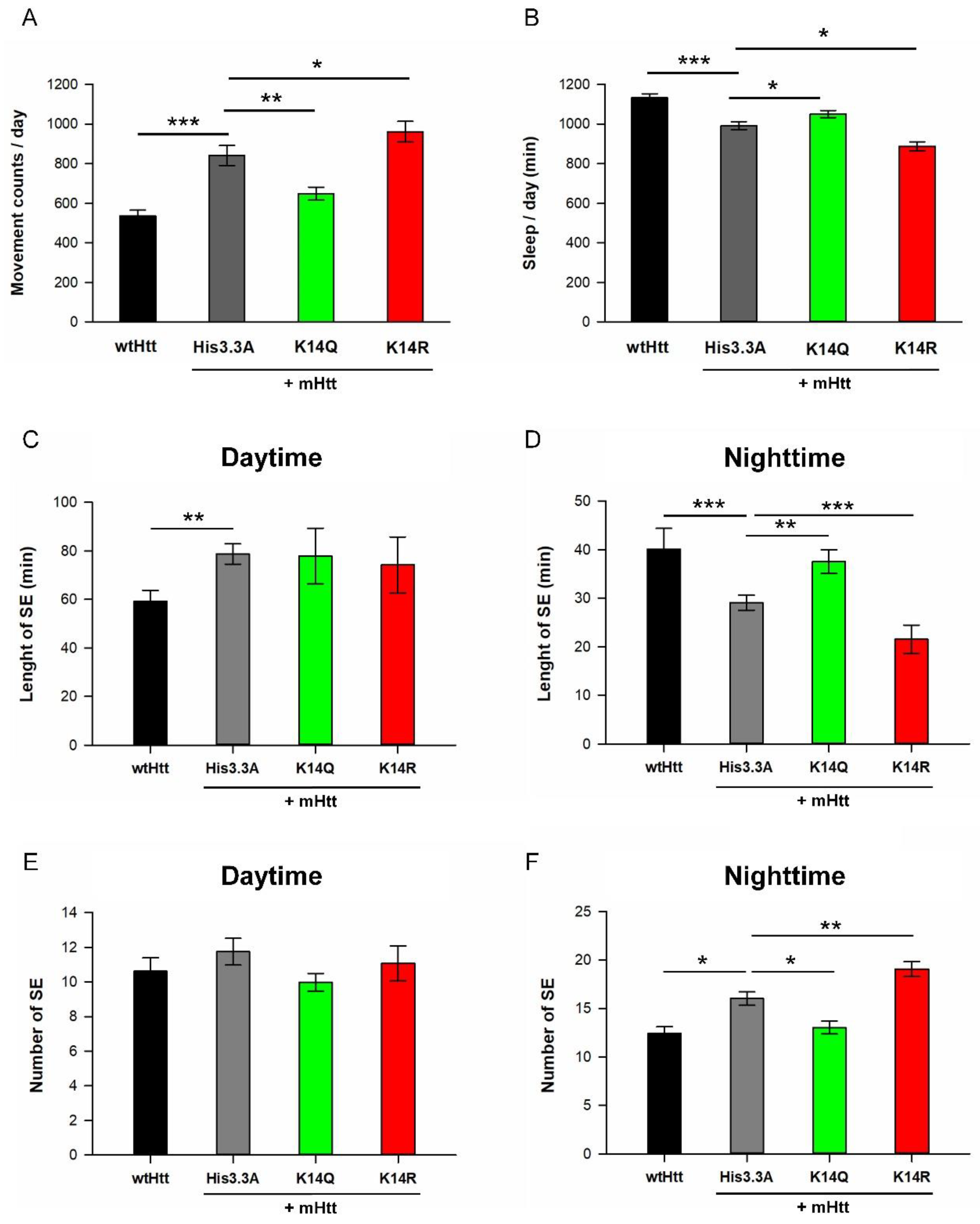
2.3. Adult Neuronal Expression of H3.3K14Q Improves Longevity and Mitigates Motor Dysfunction and Sleep Defects of HD Flies
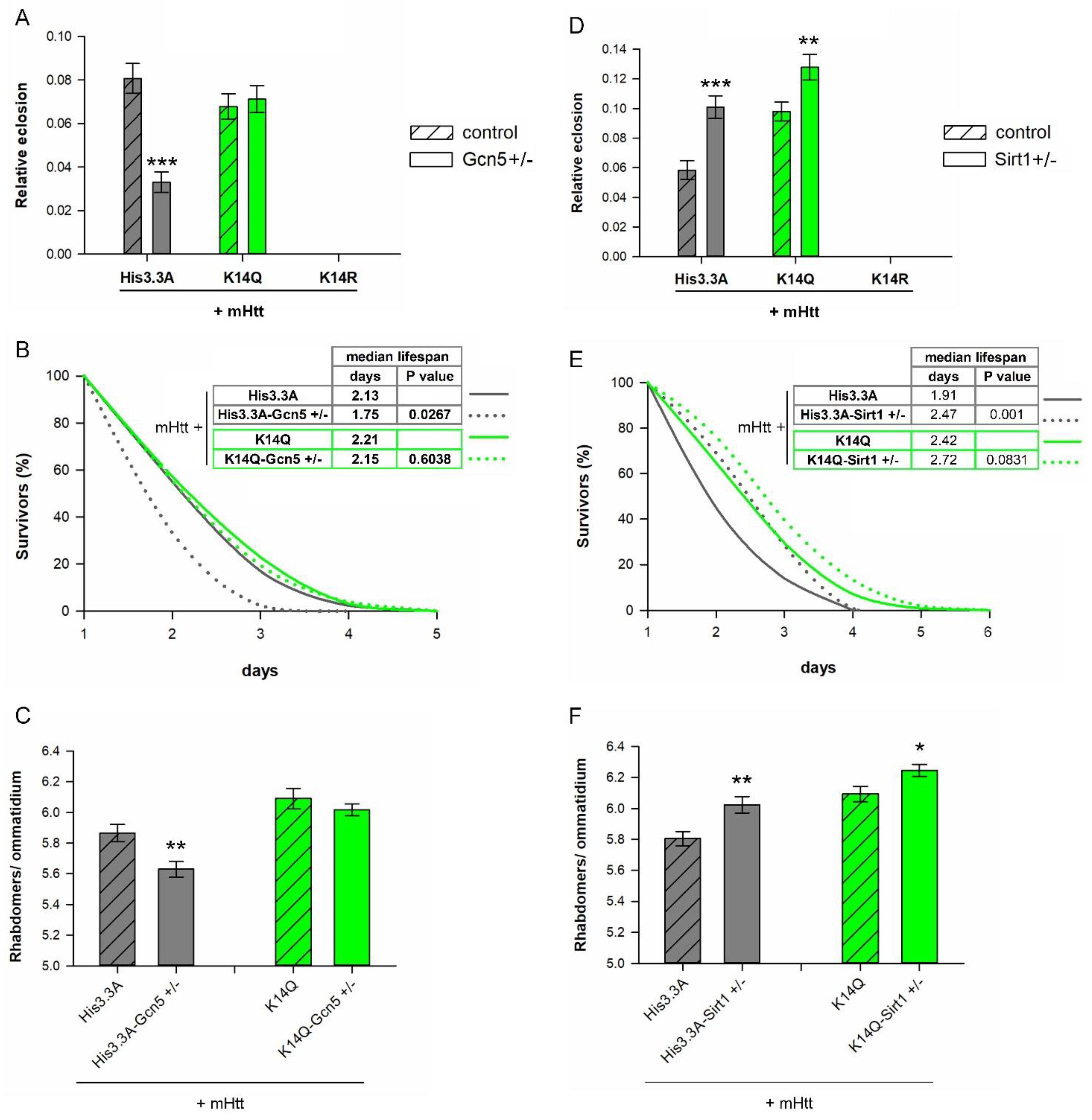
2.4. Negative Effects of Reduced GCN5 Are Averted in H3.3-K14Q-Expressing HD Flies
3. Discussion
4. Materials and Methods
4.1. Drosophila Melanogaster Stocks and Husbandry
4.2. Generating H3.3A PTM Mimetic Transgenic Flies
4.3. Immunoblotting
4.4. Immunohistochemistry
4.5. Histone Fractionation by Salt Elution
4.6. Validation of Transgene Expression
4.7. Drosophila Crosses and Eclosion Analysis
4.8. Longevity Analysis
4.9. Neurodegeneration Measurement
4.10. Climbing Assay
4.11. Activity Recording and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primer 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- McAdam, R.L.; Morton, A.; Gordon, S.L.; Alterman, J.F.; Khvorova, A.; Cousin, M.A.; Smillie, K.J. Loss of Huntingtin Function Slows Synaptic Vesicle Endocytosis in Striatal Neurons from the HttQ140/Q140 Mouse Model of Huntington’s Disease. Neurobiol. Dis. 2020, 134, 104637. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 2019, 14, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration. PLoS Genet. 2007, 3, e82. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hosp, F.; Frottin, F.; Ge, H.; Mann, M.; Hayer-Hartl, M.; Hartl, F.U. Soluble Oligomers of PolyQ-Expanded Huntingtin Target a Multiplicity of Key Cellular Factors. Mol. Cell 2016, 63, 951–964. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s Disease: Underlying Molecular Mechanisms and Emerging Concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef]
- Seredenina, T.; Luthi-Carter, R. What Have We Learned from Gene Expression Profiles in Huntington’s Disease? Neurobiol. Dis. 2012, 45, 83–98. [Google Scholar] [CrossRef]
- Neueder, A.; Bates, G.P. A Common Gene Expression Signature in Huntington’s Disease Patient Brain Regions. BMC Med. Genom. 2014, 7, 60. [Google Scholar] [CrossRef]
- Malla, B.; Guo, X.; Senger, G.; Chasapopoulou, Z.; Yildirim, F. A Systematic Review of Transcriptional Dysregulation in Huntington’s Disease Studied by RNA Sequencing. Front. Genet. 2021, 12, 1898. [Google Scholar] [CrossRef]
- Kuhn, A.; Goldstein, D.R.; Hodges, A.; Strand, A.D.; Sengstag, T.; Kooperberg, C.; Becanovic, K.; Pouladi, M.A.; Sathasivam, K.; Cha, J.-H.J.; et al. Mutant Huntingtin’s Effects on Striatal Gene Expression in Mice Recapitulate Changes Observed in Human Huntington’s Disease Brain and Do Not Differ with Mutant Huntingtin Length or Wild-Type Huntingtin Dosage. Hum. Mol. Genet. 2007, 16, 1845–1861. [Google Scholar] [CrossRef]
- Hervas-Corpion, I.; Guiretti, D.; Alcaraz-Iborra, M.; Olivares, R.; Campos-Caro, A.; Barco, A.; Valor, L.M. Early Alteration of Epigenetic-Related Transcription in Huntington’s Disease Mouse Models. Sci. Rep. 2018, 8, 9925. [Google Scholar] [CrossRef]
- Yildirim, F.; Ng, C.W.; Kappes, V.; Ehrenberger, T.; Rigby, S.K.; Stivanello, V.; Gipson, T.A.; Soltis, A.R.; Vanhoutte, P.; Caboche, J.; et al. Early Epigenomic and Transcriptional Changes Reveal Elk-1 Transcription Factor as a Therapeutic Target in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 24840–24851. [Google Scholar] [CrossRef]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s Disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar] [CrossRef]
- Guiretti, D.; Sempere, A.; Lopez-Atalaya, J.P.; Ferrer-Montiel, A.; Barco, A.; Valor, L.M. Specific Promoter Deacetylation of Histone H3 Is Conserved across Mouse Models of Huntington’s Disease in the Absence of Bulk Changes. Neurobiol. Dis. 2016, 89, 190–201. [Google Scholar] [CrossRef]
- McFarland, K.N.; Das, S.; Sun, T.T.; Leyfer, D.; Xia, E.; Sangrey, G.R.; Kuhn, A.; Luthi-Carter, R.; Clark, T.W.; Sadri-Vakili, G.; et al. Genome-Wide Histone Acetylation Is Altered in a Transgenic Mouse Model of Huntington’s Disease. PLoS ONE 2012, 7, e41423. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zsindely, N.; Faragó, A.; Marsh, J.L.; Bodai, L. Systematic Genetic Interaction Studies Identify Histone Demethylase Utx as Potential Target for Ameliorating Huntington’s Disease. Hum. Mol. Genet. 2018, 27, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.Z.; Greenwald, M.; et al. Histone Deacetylase Inhibitors Arrest Polyglutamine-Dependent Neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Cong, S.-Y.; Pepers, B.A.; Evert, B.O.; Rubinsztein, D.C.; Roos, R.A.C.; van Ommen, G.-J.B.; Dorsman, J.C. Mutant Huntingtin Represses CBP, but Not P300, by Binding and Protein Degradation. Mol. Cell. Neurosci. 2005, 30, 12–23. [Google Scholar] [CrossRef]
- Jiang, H.; Poirier, M.A.; Liang, Y.; Pei, Z.; Weiskittel, C.E.; Smith, W.W.; DeFranco, D.B.; Ross, C.A. Depletion of CBP Is Directly Linked with Cellular Toxicity Caused by Mutant Huntingtin. Neurobiol. Dis. 2006, 23, 543–551. [Google Scholar] [CrossRef]
- Barbaro, B.A.; Lukacsovich, T.; Agrawal, N.; Burke, J.; Bornemann, D.J.; Purcell, J.M.; Worthge, S.A.; Caricasole, A.; Weiss, A.; Song, W.; et al. Comparative Study of Naturally Occurring Huntingtin Fragments in Drosophila Points to Exon 1 as the Most Pathogenic Species in Huntington’s Disease. Hum. Mol. Genet. 2015, 24, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An Optimized Transgenesis System for Drosophila Using Germ-Line-Specific PhiC31 Integrases. Proc. Natl. Acad. Sci. USA 2007, 104, 3312–3317. [Google Scholar] [CrossRef]
- Song, W.; Smith, M.R.; Syed, A.; Lukacsovich, T.; Barbaro, B.A.; Purcell, J.; Bornemann, D.J.; Burke, J.; Marsh, J.L. Morphometric Analysis of Huntington’s Disease Neurodegeneration in Drosophila. In Tandem Repeats in Genes, Proteins, and Disease; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 41–57. ISBN 978-1-62703-437-1. [Google Scholar]
- del Valle Rodríguez, A.; Didiano, D.; Desplan, C. Power Tools for Gene Expression and Clonal Analysis in Drosophila. Nat. Methods 2011, 9, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Faragó, A.; Zsindely, N.; Bodai, L. Mutant Huntingtin Disturbs Circadian Clock Gene Expression and Sleep Patterns in Drosophila. Sci. Rep. 2019, 9, 7174. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-M.; Andrews, A.J. Quantitating the Specificity and Selectivity of Gcn5-Mediated Acetylation of Histone H3. PLoS ONE 2013, 8, e54896. [Google Scholar] [CrossRef]
- Bodai, L.; Pallos, J.; Thompson, L.M.; Marsh, J.L. Pcaf Modulates Polyglutamine Pathology in a Drosophila Model of Huntington’s Disease. Neurodegener. Dis. 2012, 9, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Syed, A.; Lukacsovich, T.; Purcell, J.; Barbaro, B.A.; Worthge, S.A.; Wei, S.R.; Pollio, G.; Magnoni, L.; Scali, C.; et al. A Potent and Selective Sirtuin 1 Inhibitor Alleviates Pathology in Multiple Animal and Cell Models of Huntington’s Disease. Hum. Mol. Genet. 2014, 23, 2995–3007. [Google Scholar] [CrossRef]
- Jing, H.; Lin, H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef]
- Gao, R.; Chakraborty, A.; Geater, C.; Pradhan, S.; Gordon, K.L.; Snowden, J.; Yuan, S.; Dickey, A.S.; Choudhary, S.; Ashizawa, T.; et al. Mutant Huntingtin Impairs PNKP and ATXN3, Disrupting DNA Repair and Transcription. eLife 2019, 8, e42988. [Google Scholar] [CrossRef]
- Beaver, M.; Bhatnagar, A.; Panikker, P.; Zhang, H.; Snook, R.; Parmar, V.; Vijayakumar, G.; Betini, N.; Akhter, S.; Elefant, F. Disruption of Tip60 HAT Mediated Neural Histone Acetylation Homeostasis Is an Early Common Event in Neurodegenerative Diseases. Sci. Rep. 2020, 10, 18265. [Google Scholar] [CrossRef]
- Taylor, J.P.; Taye, A.A.; Campbell, C.; Kazemi-Esfarjani, P.; Fischbeck, K.H.; Min, K.-T. Aberrant Histone Acetylation, Altered Transcription, and Retinal Degeneration in a Drosophila Model of Polyglutamine Disease Are Rescued by CREB-Binding Protein. Genes Dev. 2003, 17, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.S.; et al. Suberoylanilide Hydroxamic Acid, a Histone Deacetylase Inhibitor, Ameliorates Motor Deficits in a Mouse Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Hecklau, K.; Mueller, S.; Koch, S.P.; Mehkary, M.H.; Kilic, B.; Harms, C.; Boehm-Sturm, P.; Yildirim, F. The Effects of Selective Inhibition of Histone Deacetylase 1 and 3 in Huntington’s Disease Mice. Front. Mol. Neurosci. 2021, 14, 616886. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Reber, K.A.; Urbach, Y.K.; Schulze-Krebs, A.; Canneva, F.; Moceri, S.; Habermeyer, J.; Achoui, D.; Gupta, B.; Steindler, D.A.; et al. Early Postnatal Behavioral, Cellular, and Molecular Changes in Models of Huntington Disease Are Reversible by HDAC Inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E8765–E8774. [Google Scholar] [CrossRef]
- Narayan, P.; Reid, S.; Scotter, E.L.; McGregor, A.L.; Mehrabi, N.F.; Singh-Bains, M.K.; Glass, M.; Faull, R.L.M.; Snell, R.G.; Dragunow, M. Inconsistencies in Histone Acetylation Patterns among Different HD Model Systems and HD Post-Mortem Brains. Neurobiol. Dis. 2020, 146, 105092. [Google Scholar] [CrossRef]
- Alper, B.J.; Job, G.; Yadav, R.K.; Shanker, S.; Lowe, B.R.; Partridge, J.F. Sir2 Is Required for Clr4 to Initiate Centromeric Heterochromatin Assembly in Fission Yeast. EMBO J. 2013, 32, 2321–2335. [Google Scholar] [CrossRef]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 Acetylation Co-Occur at Many Gene Regulatory Elements, While H3K14ac Marks a Subset of Inactive Inducible Promoters in Mouse Embryonic Stem Cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef]
- Love, I.M.; Sekaric, P.; Shi, D.; Grossman, S.R.; Androphy, E.J. The Histone Acetyltransferase PCAF Regulates P21 Transcription through Stress-Induced Acetylation of Histone H3. Cell Cycle 2012, 11, 2458–2466. [Google Scholar] [CrossRef]
- Duan, M.-R.; Smerdon, M.J. Histone H3 Lysine 14 (H3K14) Acetylation Facilitates DNA Repair in a Positioned Nucleosome by Stabilizing the Binding of the Chromatin Remodeler RSC (Remodels Structure of Chromatin). J. Biol. Chem. 2014, 289, 8353–8363. [Google Scholar] [CrossRef]
- Johnsson, A.; Durand-Dubief, M.; Xue-Franzen, Y.; Ronnerblad, M.; Ekwall, K.; Wright, A. HAT-HDAC Interplay Modulates Global Histone H3K14 Acetylation in Gene-Coding Regions during Stress. EMBO Rep. 2009, 10, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-T.; Liu, G.; Leeds, P.; Chuang, D.-M. Combined Treatment with the Mood Stabilizers Lithium and Valproate Produces Multiple Beneficial Effects in Transgenic Mouse Models of Huntington’s Disease. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2011, 36, 2406–2421. [Google Scholar] [CrossRef] [PubMed]
- Valor, L.M.; Guiretti, D.; Lopez-Atalaya, J.P.; Barco, A. Genomic Landscape of Transcriptional and Epigenetic Dysregulation in Early Onset Polyglutamine Disease. J. Neurosci. 2013, 33, 10471–10482. [Google Scholar] [CrossRef] [PubMed]
- Torres-Zelada, E.F.; Weake, V.M. The Gcn5 Complexes in Drosophila as a Model for Metazoa. Biochim. Biophys. Acta BBA—Gene Regul. Mech. 2021, 1864, 194610. [Google Scholar] [CrossRef]
- Jin, Q.; Yu, L.-R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.-E.; Wang, C.; Brindle, P.K.; Dent, S.Y.R.; Ge, K. Distinct Roles of GCN5/PCAF-Mediated H3K9ac and CBP/P300-Mediated H3K18/27ac in Nuclear Receptor Transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, S.; Xia, Y.; Lu, Y.; Xiao, S.; Cao, Y.; Zhuang, S.; Tan, X.; Fu, Q.; Xie, L.; et al. Loss of GCN5 Leads to Increased Neuronal Apoptosis by Upregulating E2F1- and Egr-1-Dependent BH3-Only Protein Bim. Cell Death Dis. 2018, 8, e2570. [Google Scholar] [CrossRef]
- Naia, L.; Rego, A.C. Sirtuins: Double Players in Huntington’s Disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 2183–2194. [Google Scholar] [CrossRef]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of Specific HDACs and Sirtuins Suppresses Pathogenesis in a Drosophila Model of Huntington’s Disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef]
- Hödl, M.; Basler, K. Transcription in the Absence of Histone H3.3. Curr. Biol. 2009, 19, 1221–1226. [Google Scholar] [CrossRef]
- Henn, L.; Szabó, A.; Imre, L.; Román, Á.; Ábrahám, A.; Vedelek, B.; Nánási, P.; Boros, I.M. Alternative Linker Histone Permits Fast Paced Nuclear Divisions in Early Drosophila Embryo. Nucleic Acids Res. 2020, 48, 9007–9018. [Google Scholar] [CrossRef]
- Han, S.K.; Lee, D.; Lee, H.; Kim, D.; Son, H.G.; Yang, J.-S.; Lee, S.-J.V.; Kim, S. OASIS 2: Online Application for Survival Analysis 2 with Features for the Analysis of Maximal Lifespan and Healthspan in Aging Research. Oncotarget 2016, 7, 56147–56152. [Google Scholar] [CrossRef] [PubMed]
- Gilestro, G.F.; Cirelli, C. PySolo: A Complete Suite for Sleep Analysis in Drosophila. Bioinformatics 2009, 25, 1466–1467. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faragó, A.; Zsindely, N.; Farkas, A.; Neller, A.; Siági, F.; Szabó, M.R.; Csont, T.; Bodai, L. Acetylation State of Lysine 14 of Histone H3.3 Affects Mutant Huntingtin Induced Pathogenesis. Int. J. Mol. Sci. 2022, 23, 15173. https://doi.org/10.3390/ijms232315173
Faragó A, Zsindely N, Farkas A, Neller A, Siági F, Szabó MR, Csont T, Bodai L. Acetylation State of Lysine 14 of Histone H3.3 Affects Mutant Huntingtin Induced Pathogenesis. International Journal of Molecular Sciences. 2022; 23(23):15173. https://doi.org/10.3390/ijms232315173
Chicago/Turabian StyleFaragó, Anikó, Nóra Zsindely, Anita Farkas, Alexandra Neller, Fruzsina Siági, Márton Richárd Szabó, Tamás Csont, and László Bodai. 2022. "Acetylation State of Lysine 14 of Histone H3.3 Affects Mutant Huntingtin Induced Pathogenesis" International Journal of Molecular Sciences 23, no. 23: 15173. https://doi.org/10.3390/ijms232315173
APA StyleFaragó, A., Zsindely, N., Farkas, A., Neller, A., Siági, F., Szabó, M. R., Csont, T., & Bodai, L. (2022). Acetylation State of Lysine 14 of Histone H3.3 Affects Mutant Huntingtin Induced Pathogenesis. International Journal of Molecular Sciences, 23(23), 15173. https://doi.org/10.3390/ijms232315173