
Identification and Characterization of Transcription Factors Involved in Geraniol Biosynthesis in Rosa chinensis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
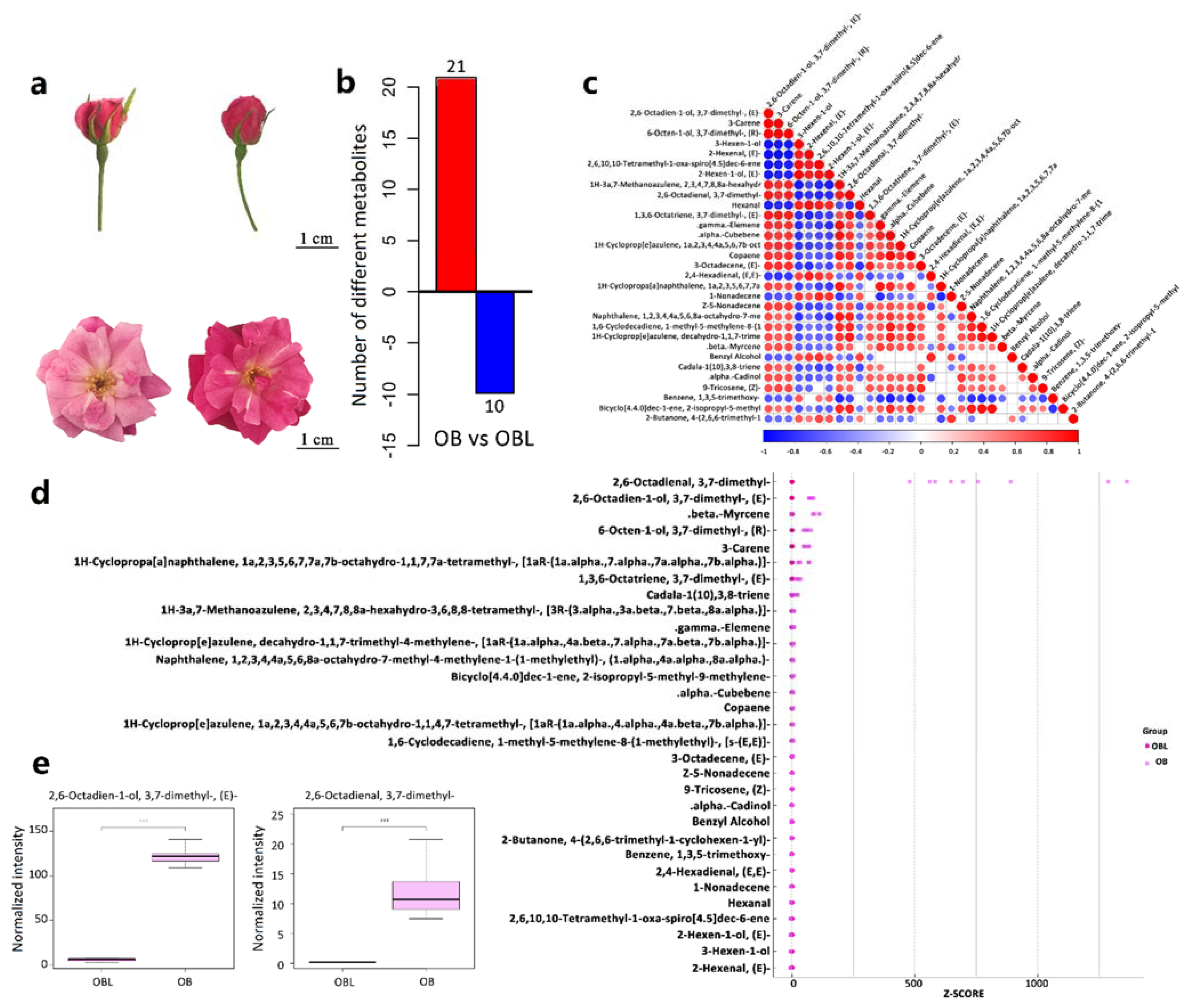
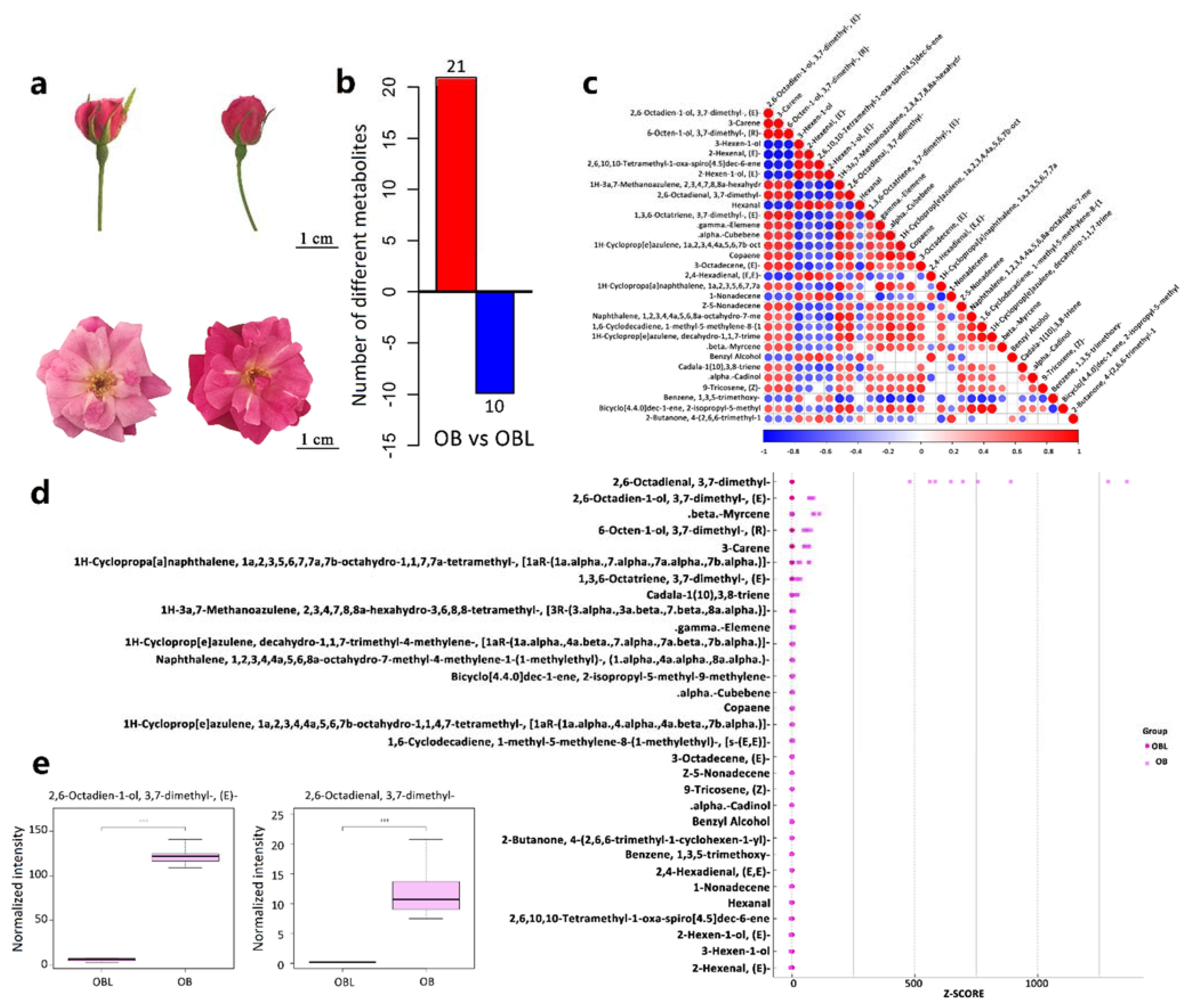
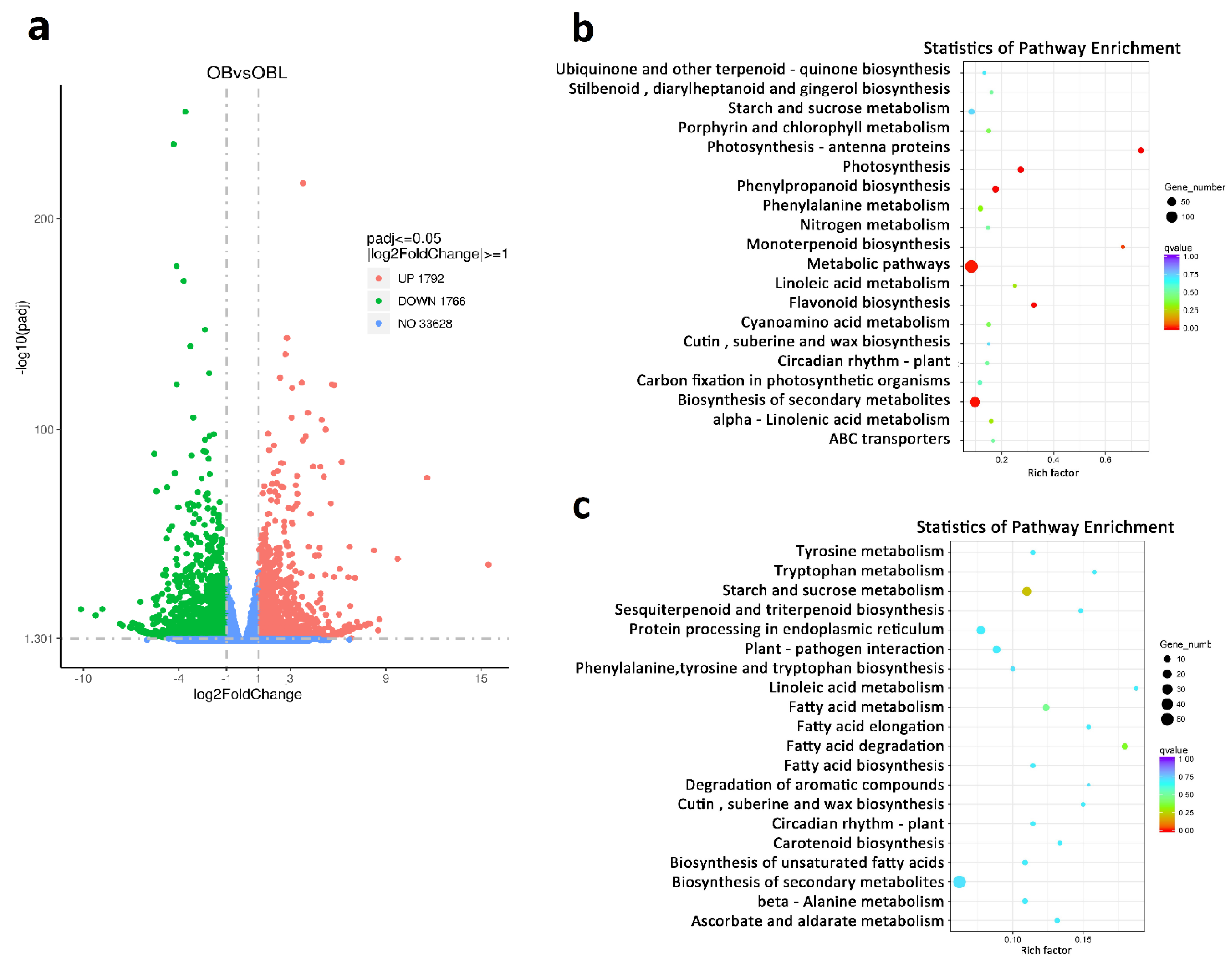
2.1. Geraniol Is the Most Significant Differentially Abundant Volatile in Petals between OB and OBL
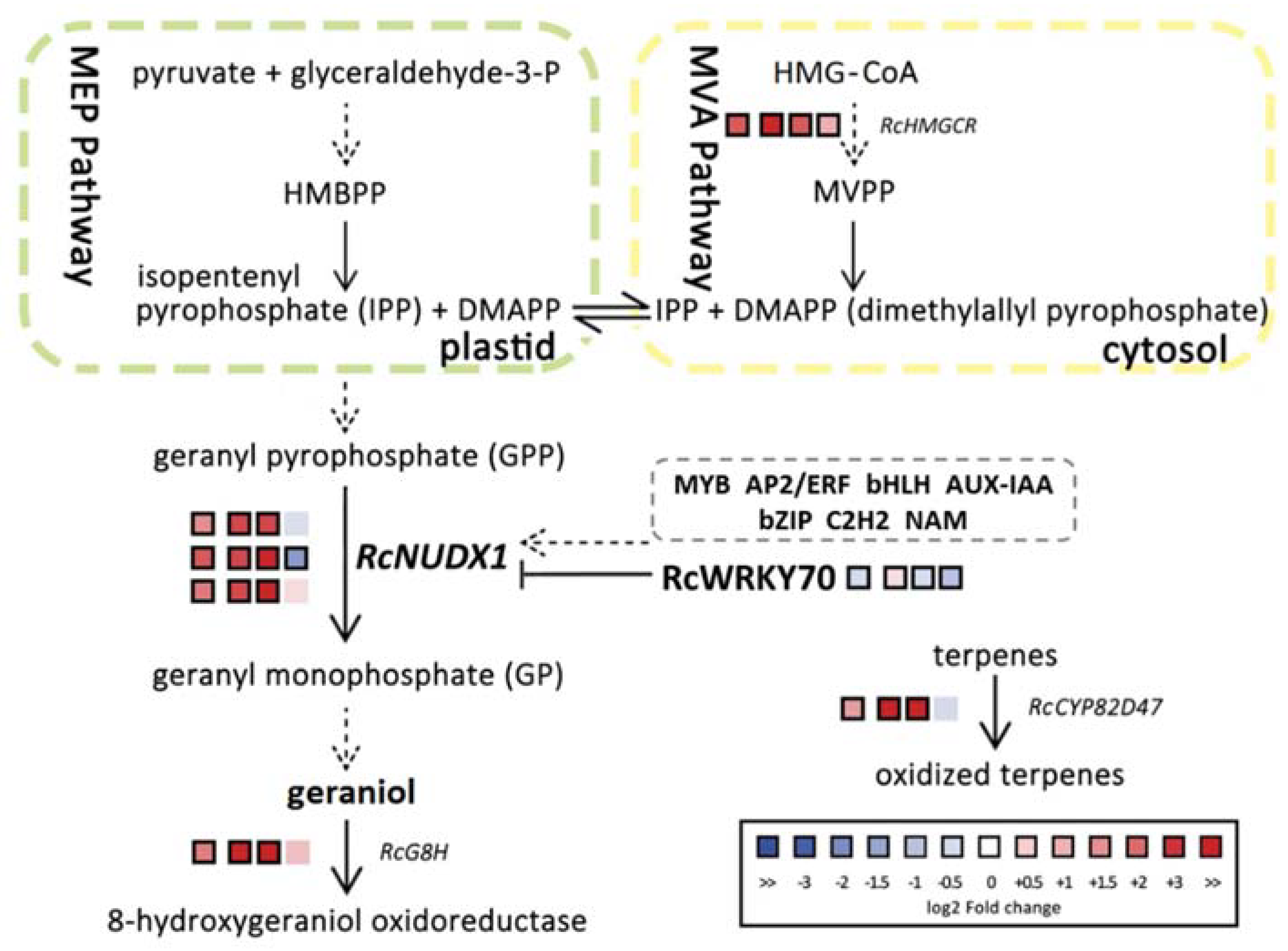
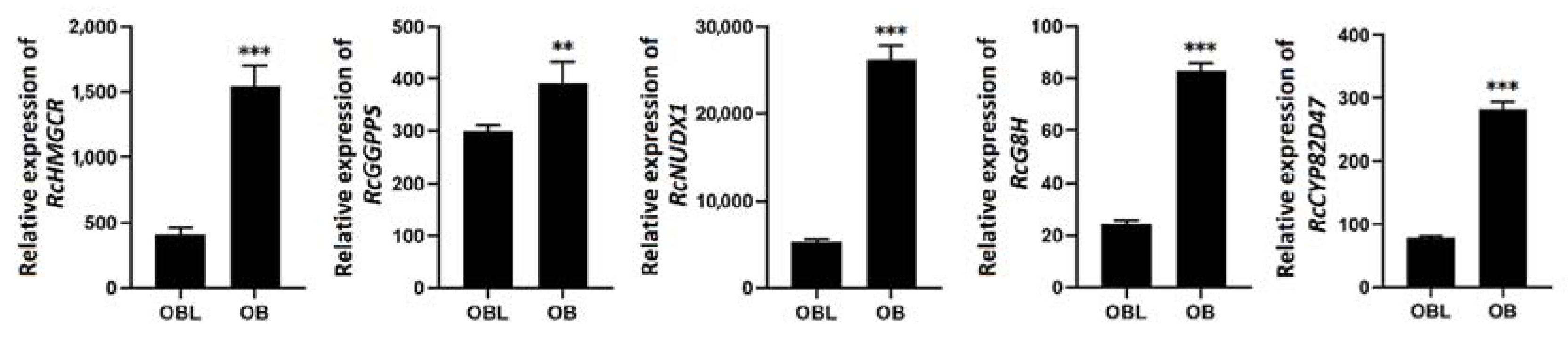
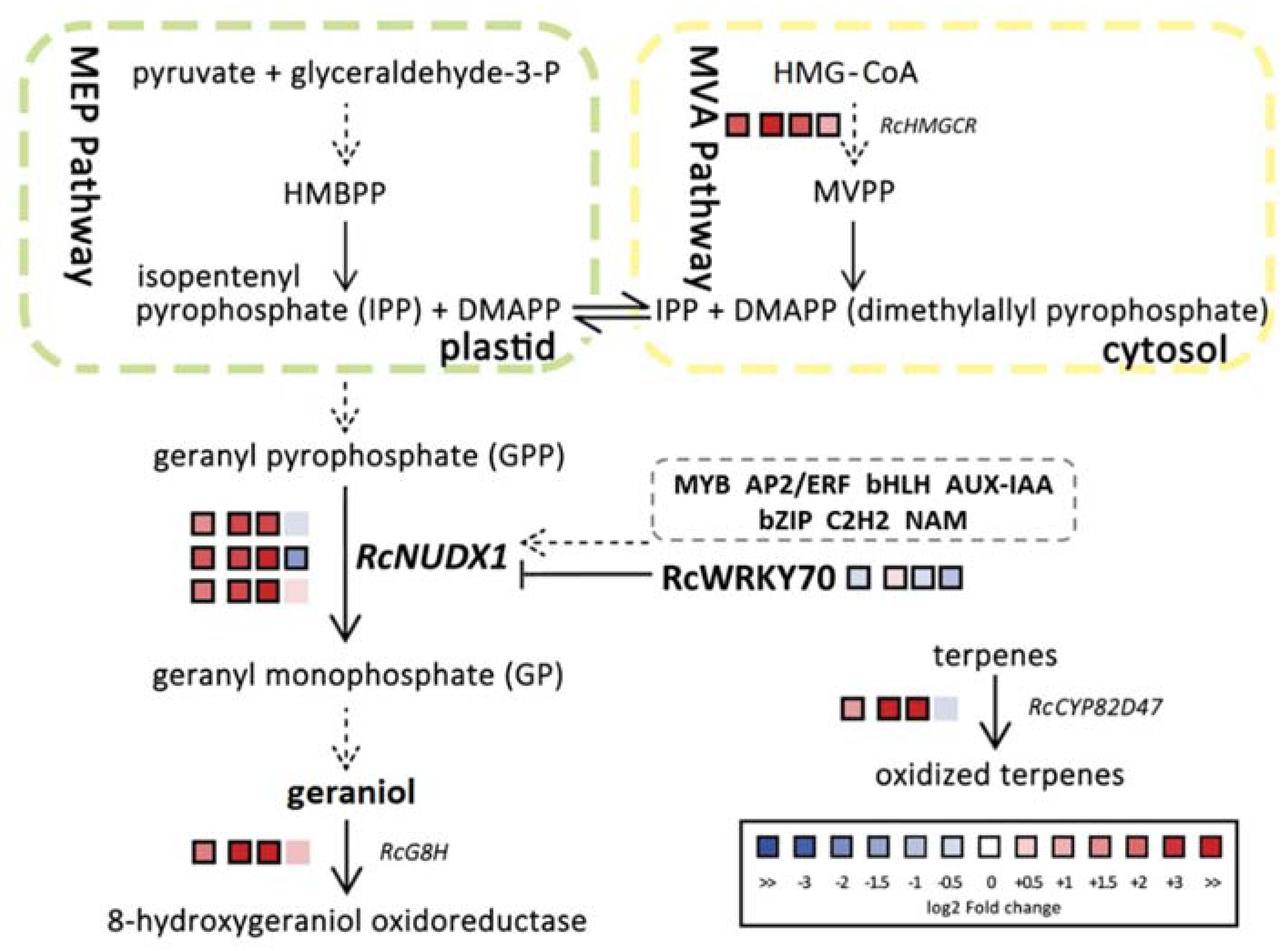
2.2. RcNUDX1 and Geraniol Biosynthesis
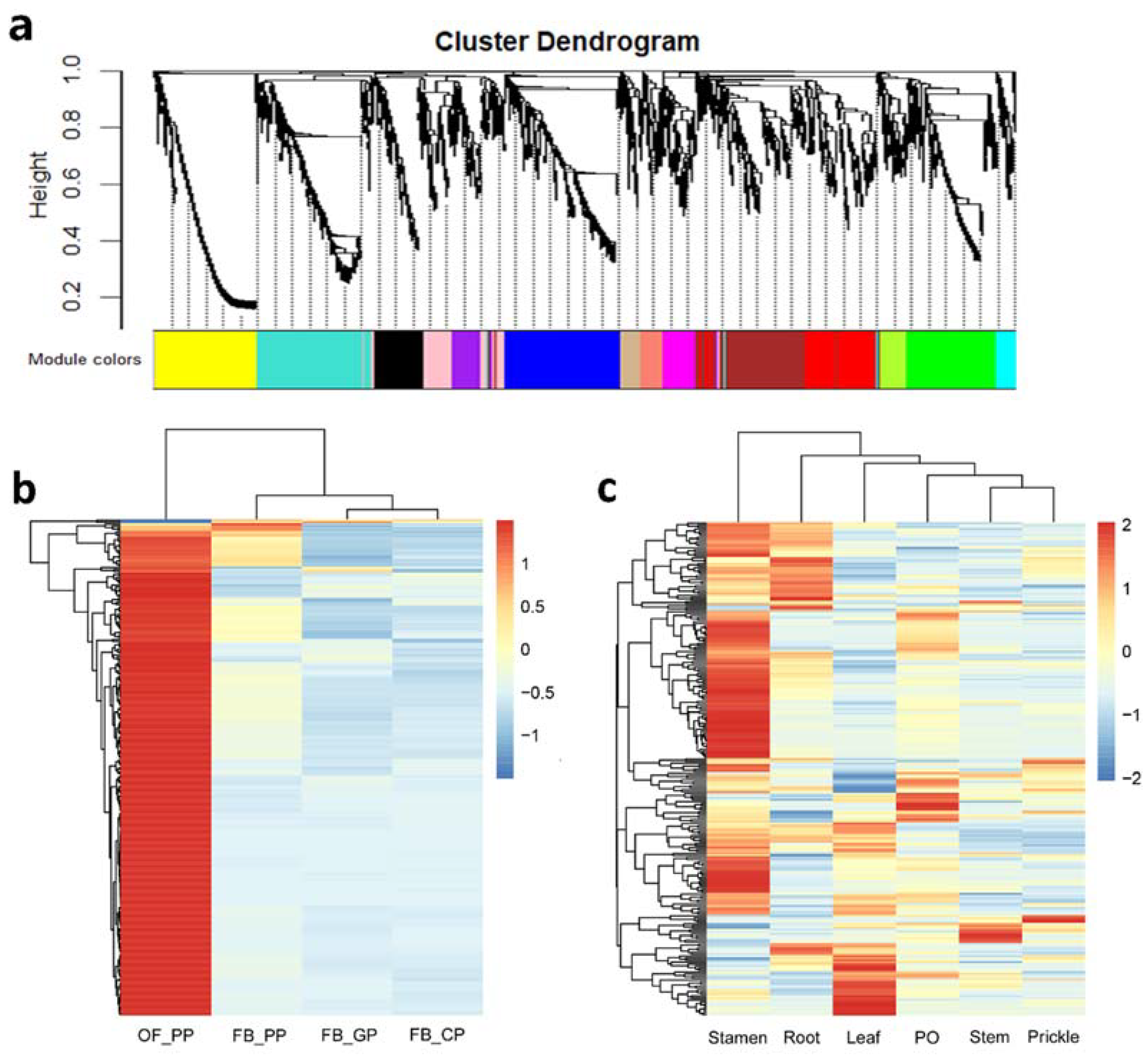
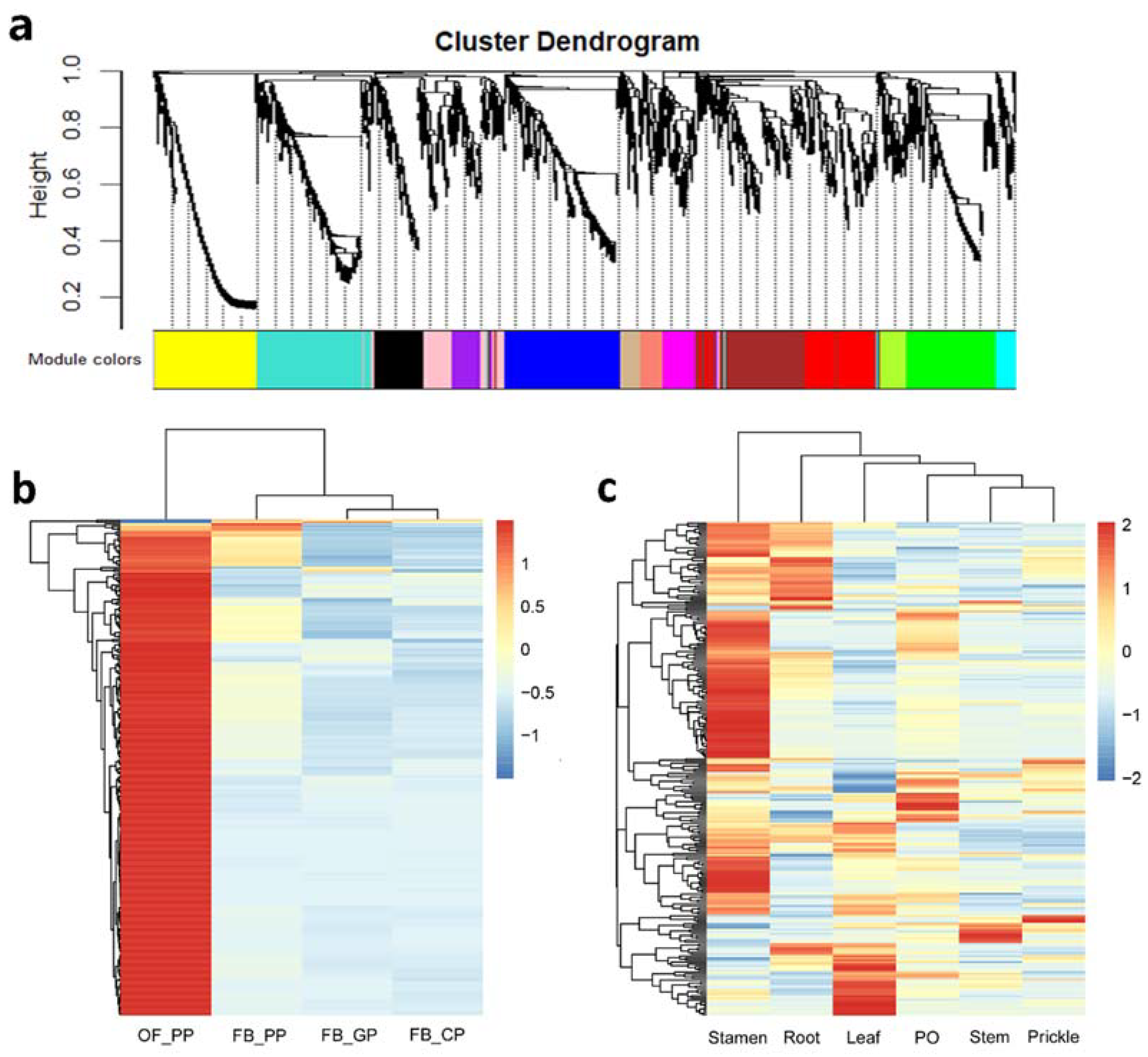
2.3. Prediction of RcNUDX1 Co-Expressed Genes Based on Weighted Gene Co-Expression Network Analysis
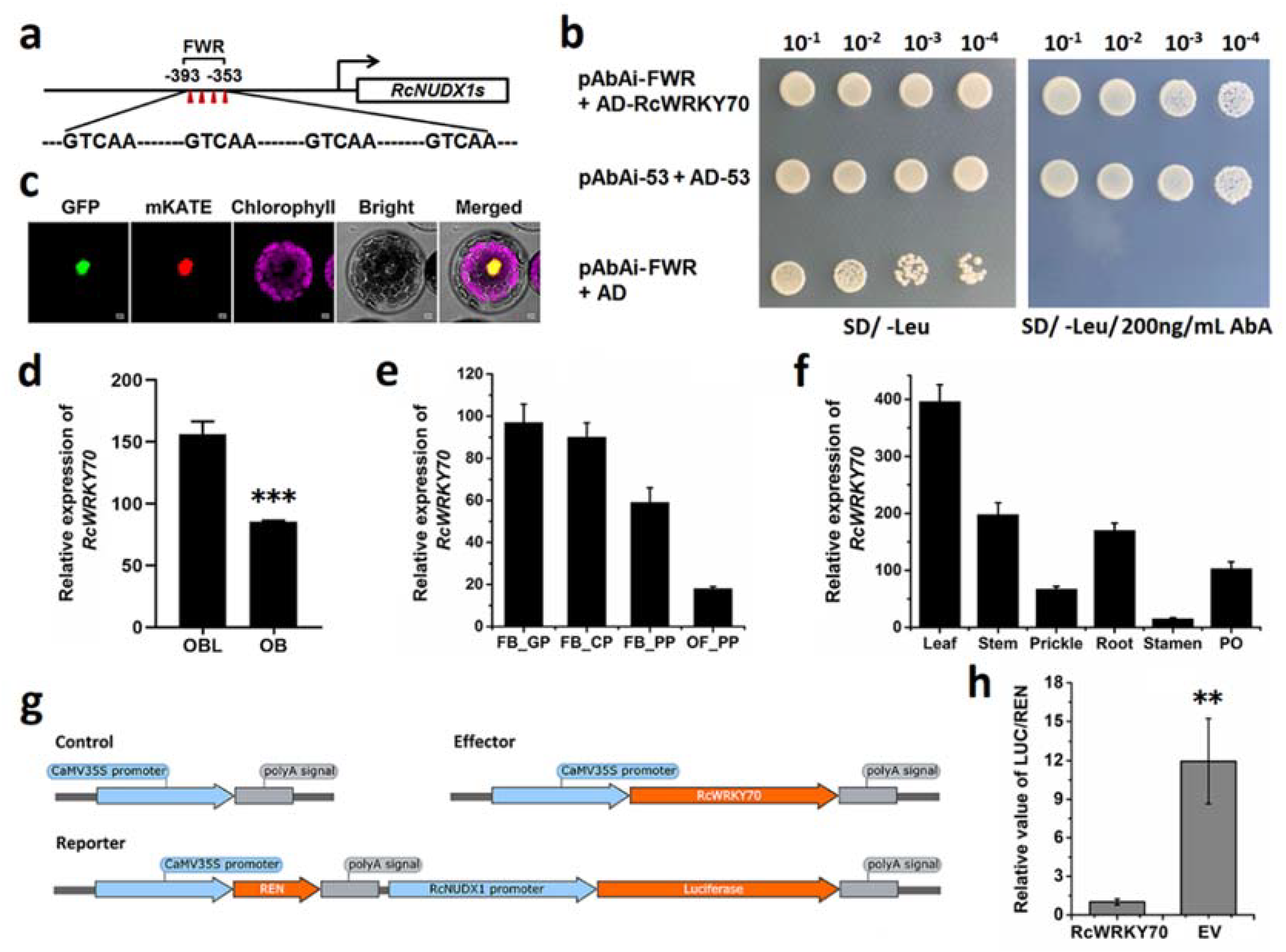
2.4. RcWPKY70 May Be a Transcriptional Repressor of RcNUDX1 Expression
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Gas Chromatography—Mass Spectrometry (GC–MS) Analysis
4.3. Transcriptome Deep Sequencing (RNA-Seq)
4.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.5. RT–qPCR Validation
4.6. Subcellular Localization
4.7. Cloning of the RcNUDX1 Promoter
4.8. Yeast One-Hybrid cDNA Library Creation
4.9. Construction of Bait Plasmids Construction and Self-Activation Test
4.10. Yeast One-Hybrid Screening
4.11. Dual–Luciferase (Dual–LUC) Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tholl, D. Biosynthesis and Biological Functions of Terpenoids in Plants. In Biotechnology of Isoprenoids; Schrader, J., Bohlmann, J., Eds.; Advances in Biochemical Engineering/Biotechnology; Springer International Publishing: Cham, Switzerland, 2015; Volume 148, pp. 63–106. ISBN 978-3-319-20106-1. [Google Scholar]
- Degenhardt, J.; Köllner, T.G.; Gershenzon, J. Monoterpene and Sesquiterpene Synthases and the Origin of Terpene Skeletal Diversity in Plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-Ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene Synthases Are Widely Distributed in Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarvey, D.J.; Croteau, R. Terpenoid Metabolism. Plant Cell 1995, 7, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Suire, C.; Bouvier, F.; Backhaus, R.A.; Bégu, D.; Bonneu, M.; Camara, B. Cellular Localization of Isoprenoid Biosynthetic Enzymes in Marchantia Polymorpha. Uncovering a New Role of Oil Bodies. Plant Physiol. 2000, 124, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Denbow, C.J.; Lång, S.; Cramer, C.L. The N-Terminal Domain of Tomato 3-Hydroxy-3-Methylglutaryl-CoA Reductases. J. Biol. Chem. 1996, 271, 9710–9715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrie, C.; Murcha, M.W.; Millar, A.H.; Smith, S.M.; Whelan, J. Nine 3-Ketoacyl-CoA Thiolases (KATs) and Acetoacetyl-CoA Thiolases (ACATs) Encoded by Five Genes in Arabidopsis Thaliana Are Targeted Either to Peroxisomes or Cytosol but Not to Mitochondria. Plant Mol. Biol. 2007, 63, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chandler, S.F.; Brugliera, F. Genetic Modification in Floriculture. Biotechnol. Lett 2011, 33, 207–214. [Google Scholar] [CrossRef]
- Dudareva, N.; Cseke, L.; Blanc, V.M.; Pichersky, E. Evolution of Floral Scent in Clarkia: Novel Patterns of S-Linalool Synthase Gene Expression in the C. Breweri Flower. Plant Cell 1996, 8, 1137–1148. [Google Scholar] [CrossRef] [Green Version]
- Cairns, T.; American Rose Society. Modern Roses XI: The World Encyclopedia of Roses; Academic Press: San Diego, CA, USA, 2000; ISBN 978-0-12-155053-0. [Google Scholar]
- Foucher, F.; Hibrand-Saint Oyant, L.; Hamama, L.; Sakr, S.; Nybom, H.; Baudino, S.; Caissard, J.P.; Byrne, D.M.; Smulder, J.M.S.; Desnoyé, B.; et al. Towards the rose genome sequence and its use in research and breeding. Acta Hortic. 2015, 2064, 167–175. [Google Scholar] [CrossRef]
- Joichi, A.; Yomogida, K.; Awano, K.; Ueda, Y. Volatile Components of Tea-Scented Modern Roses and Ancient Chinese Roses. Flavour Fragr. J. 2004, 20, 152–157. [Google Scholar] [CrossRef]
- Jin, Y.; Cui, H.; Yuan, X.; Liu, L.; Liu, X.; Wang, Y.; Ding, J.; Xiang, H.; Zhang, X.; Liu, J.; et al. Identification of the Main Aroma Compounds in Chinese Local Chicken High-Quality Meat. Food Chem. 2021, 359, 129930. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Taneja, S.C.; Shah, B.A.; Sethi, V.K.; Qazi, G.N. Lipase-Catalyzed Separation of Geometrical Isomers: Geraniol–Nerol. Chem. Lett. 2007, 36, 1110–1111. [Google Scholar] [CrossRef]
- Paroul, N.; Grzegozeski, L.P.; Chiaradia, V.; Treichel, H.; Cansian, R.L.; Oliveira, J.V.; de Oliveira, D. Solvent-Free Geranyl Oleate Production by Enzymatic Esterification. Bioprocess. Biosyst. Eng. 2011, 34, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ramya, M.; Kwon, O.K.; An, H.R.; Park, P.M.; Baek, Y.S.; Park, P.H. Floral Scent: Regulation and Role of MYB Transcription Factors. Phytochem. Lett. 2017, 19, 114–120. [Google Scholar] [CrossRef]
- Hong, G.-J.; Xue, X.-Y.; Mao, Y.-B.; Wang, L.-J.; Chen, X.-Y. Arabidopsis MYC2 Interacts with DELLA Proteins in Regulating Sesquiterpene Synthase Gene Expression. Plant Cell 2012, 24, 2635–2648. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-X.; Li, J.-X.; Yang, C.-Q.; Hu, W.-L.; Wang, L.-J.; Chen, X.-Y. The Jasmonate-Responsive AP2/ERF Transcription Factors AaERF1 and AaERF2 Positively Regulate Artemisinin Biosynthesis in Artemisia annua L. Mol. Plant 2012, 5, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, H.; Li, F.; Chen, Z.; Li, X.; Zhu, L.; Wang, G.; Yu, J.; Huang, D.; Lang, Z. The Maize Transcription Factor EREB58 Mediates the Jasmonate-Induced Production of Sesquiterpene Volatiles. Plant J. 2015, 84, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, Y.; Shen, S.; Yin, X.; Klee, H.; Zhang, B.; Chen, K. Transcription Factor CitERF71 Activates the Terpene Synthase Gene CitTPS16 Involved in the Synthesis of E–Geraniol in Sweet Orange Fruit. J. Exp. Bot. 2017, 68, 4929–4938. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Gao, M.; Zhao, Y.; Chen, Y.; Wu, L.; Yin, H.; Yang, J.; Xiong, S.; Wang, S.; Wang, J.; et al. LcERF19, an AP2/ERF Transcription Factor from Litsea Cubeba, Positively Regulates Geranial and Neral Biosynthesis. Hortic. Res. 2022, 9, uhac093. [Google Scholar] [CrossRef]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C.; et al. The Rosa Genome Provides New Insights into the Domestication of Modern Roses. Nat. Genet. 2018, 50, 772–777. [Google Scholar] [CrossRef]
- Conart, C.; Saclier, N.; Foucher, F.; Goubert, C.; Rius-Bony, A.; Paramita, S.N.; Moja, S.; Thouroude, T.; Douady, C.; Sun, P.; et al. Duplication and Specialization of NUDX1 in Rosaceae Led to Geraniol Production in Rose Petals. Mol. Biol. Evol. 2022, 39, msac002. [Google Scholar] [CrossRef] [PubMed]
- Mączka, W.; Wińska, K.; Grabarczyk, M. One Hundred Faces of Geraniol. Molecules 2020, 25, 3303. [Google Scholar] [CrossRef] [PubMed]
- Papachristos, D.P.; Karamanoli, K.I.; Stamopoulos, D.C.; Menkissoglu-Spiroudi, U. The Relationship between the Chemical Composition of Three Essential Oils and Their Insecticidal Activity AgainstAcanthoscelides Obtectus(Say). Pest. Manag. Sci. 2004, 60, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Krist, S.; Buchbauer, G. Antimicrobial Effect of Vapours of Geraniol, (R)-(–)-Linalool, Terpineol,γ-Terpinene and 1,8-Cineole on Airborne Microbes Using an Airwasher. Flavour Fragr. J. 2007, 22, 435–437. [Google Scholar] [CrossRef]
- de Cássia da Silveira e Sá, R.; Andrade, L.; de Sousa, D. A Review on Anti-Inflammatory Activity of Monoterpenes. Molecules 2013, 18, 1227–1254. [Google Scholar] [CrossRef] [PubMed]
- Nagegowda, D.A.; Gupta, P. Advances in Biosynthesis, Regulation, and Metabolic Engineering of Plant Specialized Terpenoids. Plant Sci. 2020, 294, 110457. [Google Scholar] [CrossRef]
- Magnard, J.-L.; Roccia, A.; Caissard, J.-C.; Vergne, P.; Sun, P.; Hecquet, R.; Dubois, A.; Hibrand-Saint Oyant, L.; Jullien, F.; Nicolè, F.; et al. Biosynthesis of Monoterpene Scent Compounds in Roses. Science 2015, 349, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Bach, T.J. Hydroxymethylglutaryl-CoA Reductase, a Key Enzyme in Phytosterol Synthesis? Lipids 1986, 21, 82–88. [Google Scholar] [CrossRef]
- Lange, B.M.; Ghassemian, M. Genome organization in Arabidopsis thaliana: A survey for genes involved in isoprenoid and chlorophyll metabolism. Plant Mol. Biol. 2003, 51, 925–948. [Google Scholar] [CrossRef]
- Zvi, M.M.B.; Shklarman, E.; Masci, T.; Kalev, H.; Debener, T.; Shafir, S.; Ovadis, M.; Vainstein, A. PAP1 Transcription Factor Enhances Production of Phenylpropanoid and Terpenoid Scent Compounds in Rose Flowers. New Phytol. 2012, 195, 335–345. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.J.; Chen, X.; Wang, M.Y.; Matich, A.J.; Perez, R.L.; Allan, A.C.; Green, S.A.; Atkinson, R.G. Natural Variation in Monoterpene Synthesis in Kiwifruit: Transcriptional Regulation of Terpene Synthases by NAC and ETHYLENE-INSENSITIVE3-Like Transcription Factors. Plant Physiol. 2015, 167, 1243–1258. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Xiao, Z.; Yang, L.; Chen, Q.; Shao, L.; Liu, J.; Yu, Y. PhERF6, Interacting with EOBI, Negatively Regulates Fragrance Biosynthesis in Petunia Flowers. New Phytol. 2017, 215, 1490–1502. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-C.; Hung, Y.-C.; Tsai, W.-C.; Chen, W.-H.; Chen, H.-H. PbbHLH4 Regulates Floral Monoterpene Biosynthesis in Phalaenopsis Orchids. J. Exp. Bot. 2018, 69, 4363–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Wang, L.; Zhao, J.; Li, Y.; Huang, R.; Jiang, X.; Zhou, X.; Zhu, X.; He, Y.; He, Y.; et al. Genome-Wide Characterization of the C2H2 Zinc-Finger Genes in Cucumis Sativus and Functional Analyses of Four CsZFPs in Response to Stresses. BMC Plant Biol. 2020, 20, 359. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.A.; Mukhtar, M.S.; Blanco, F.; Boatwright, J.L.; Moreno, I.; Jordan, M.R.; Chen, Y.; Brandizzi, F.; Dong, X.; Orellana, A.; et al. IRE1/BZIP60-Mediated Unfolded Protein Response Plays Distinct Roles in Plant Immunity and Abiotic Stress Responses. PLoS ONE 2012, 7, e31944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meshi, T.; Iwabuchi, M. Plant Transcription Factors. Plant Cell Physiol 1995, 36, 1405–1420. [Google Scholar] [PubMed]
- Schluttenhofer, C.; Yuan, L. Regulation of Specialized Metabolism by WRKY Transcription Factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Muthuramalingam, M.; Zeng, X.; Iyer, N.J.; Klein, P.; Mahalingam, R. A GCC-Box Motif in the Promoter of Nudix Hydrolase 7 (AtNUDT7) Gene Plays a Role in Ozone Response of Arabidopsis Ecotypes. Genomics 2015, 105, 31–38. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY Transcription Factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The Role of WRKY Transcription Factors in Plant Abiotic Stresses. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Chi, Y.; Yang, Y.; Zhou, Y.; Zhou, J.; Fan, B.; Yu, J.-Q.; Chen, Z. Protein–Protein Interactions in the Regulation of WRKY Transcription Factors. Mol. Plant 2013, 6, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.; Wu, Y.; Zhou, J.; Lian, M.; Li, C.; Xu, Y.; Liu, S.; Wang, C.; Meng, Q. The Study of Fingerprint Characteristics of Dayi Pu-Erh Tea Using a Fully Automatic HS-SPME/GC-MS and Combined Chemometrics Method. PLoS ONE 2014, 9, e116428. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Han, Y.; Yong, X.; Yu, J.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. Identification of Candidate Adaxial–Abaxial-Related Genes Regulating Petal Expansion During Flower Opening in Rosa Chinensis “Old Blush”. Front. Plant Sci. 2019, 10, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Han, Y.; Wan, H.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. Comparative RNA-Seq Analysis of Transcriptome Dynamics during Petal Development in Rosa Chinensis. Sci. Rep. 2017, 7, 43382. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An Improvement of the 2ˆ(−Delta Delta CT) Method for Quantitative Real-Time Polymerase Chain Reaction Data Analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Abel, S.; Theologis, A. Transient Transformation of Arabidopsis Leaf Protoplasts: A Versatile Experimental System to Study Gene Expression. Plant J. 1994, 5, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhao, T.; Deng, L.; Lv, D.; Zhang, X.; Pan, X.; Xu, J.; Long, G. Visualizing the Essential Role of Complete Virion Assembly Machinery in Efficient Hepatitis C Virus Cell-to-Cell Transmission by a Viral Infection-Activated Split-Intein-Mediated Reporter System. J. Virol. 2017, 91, e01720-16. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Liu, X.; Peng, Y.; Li, Q.; Han, Y. Identification and Characterization of Transcription Factors Involved in Geraniol Biosynthesis in Rosa chinensis. Int. J. Mol. Sci. 2022, 23, 14684. https://doi.org/10.3390/ijms232314684
Yu J, Liu X, Peng Y, Li Q, Han Y. Identification and Characterization of Transcription Factors Involved in Geraniol Biosynthesis in Rosa chinensis. International Journal of Molecular Sciences. 2022; 23(23):14684. https://doi.org/10.3390/ijms232314684
Chicago/Turabian StyleYu, Jiayao, Xiaoyu Liu, Yifang Peng, Qi Li, and Yu Han. 2022. "Identification and Characterization of Transcription Factors Involved in Geraniol Biosynthesis in Rosa chinensis" International Journal of Molecular Sciences 23, no. 23: 14684. https://doi.org/10.3390/ijms232314684
APA StyleYu, J., Liu, X., Peng, Y., Li, Q., & Han, Y. (2022). Identification and Characterization of Transcription Factors Involved in Geraniol Biosynthesis in Rosa chinensis. International Journal of Molecular Sciences, 23(23), 14684. https://doi.org/10.3390/ijms232314684