2. Results and Discussion
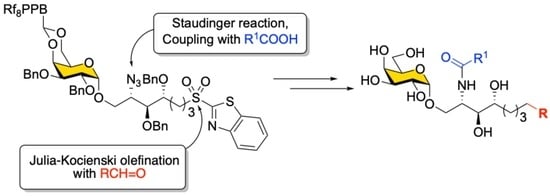
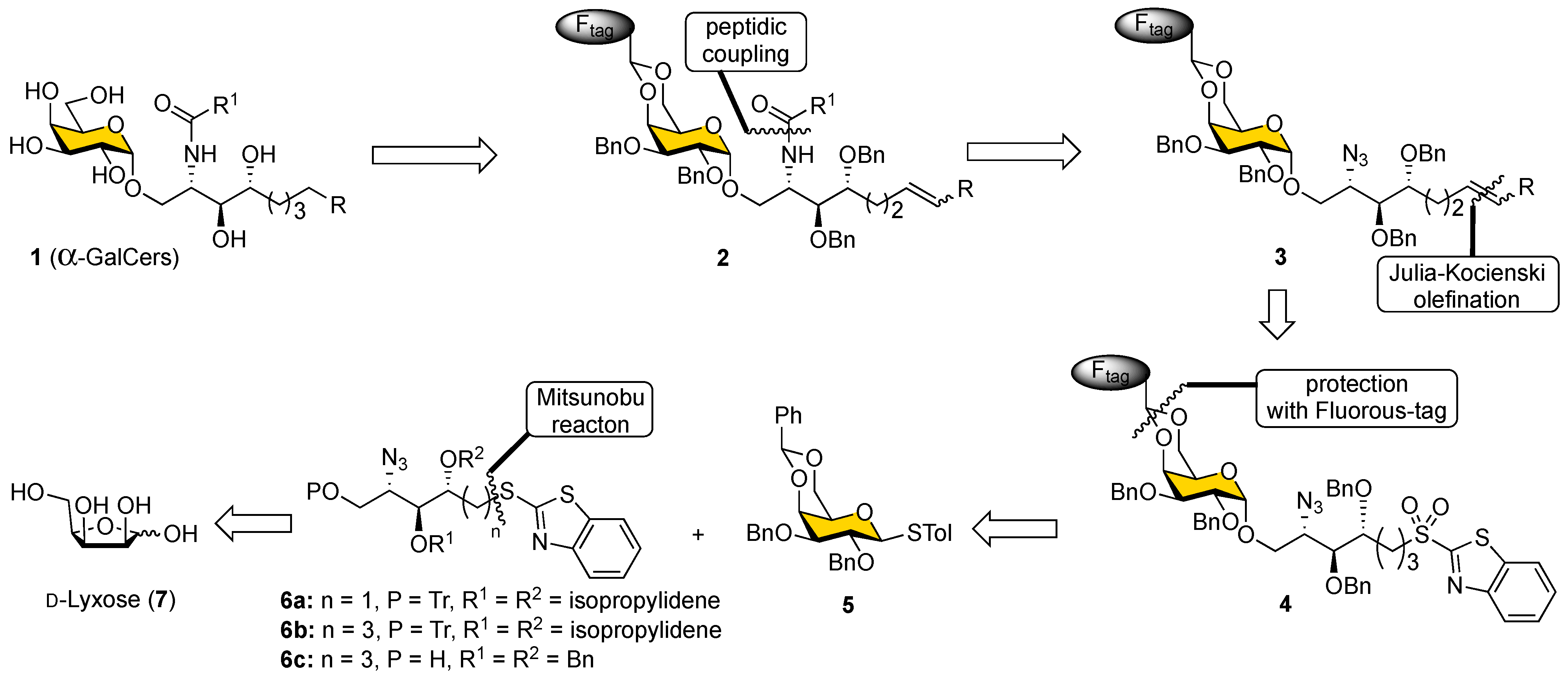
The retrosynthetic analysis for the synthesis of α-GalCer and its analogs (
1) with the fluorous-tag-assisted methodology is illustrated in
Scheme 1. The construction of the Cer portion was implemented in the late steps of the synthesis by introducing fatty acids through amide bond formation with the amine intermediate pre-masked as an azide (
3), whose olefinic linkage was formed by Julia–Kocienski olefination using sulfone
4 with aldehydes. Compound
4 was obtained by a stereoselective glycosylation reaction of the relatively simple alcohol template
6 and 4,6-
O-benzylidene-protected
p-tolyl-1-thio-β-
D-galactoside donor
5, whose cyclic acetal protection could be replaced by a fluorous tag later. Importantly, the presence of a cyclic 4,6-
O-benzylidene acetal protecting group on galactosyl donors ensured α-stereoselectivity in the glycosylation [
37]. On the other hand, the Psp chain precursor, benzothiazolyl sulfide (SB
T) derivative
6 was designed to be readily sourced from
D-lyxose-derived alcohol using the Mitsunobu reaction. The strategy outlined in
Scheme 1 is appealing for the generation of a library because the core structure
4 can be used to generate diverse analogs through the olefination reaction, and fatty acids can later be assembled in a modular fashion. Although the modified Julia–Kocienski olefination [
48] has been reported for α-
C-analogs of KRN7000 [
49], to the best of our knowledge, a merger of the Julia–Kocienski olefination and fluorous-tag-assisted purification for creating a small α-GalCer glycolipid library with a variety of lengths in both the Psp and the acyl chain has not been examined.
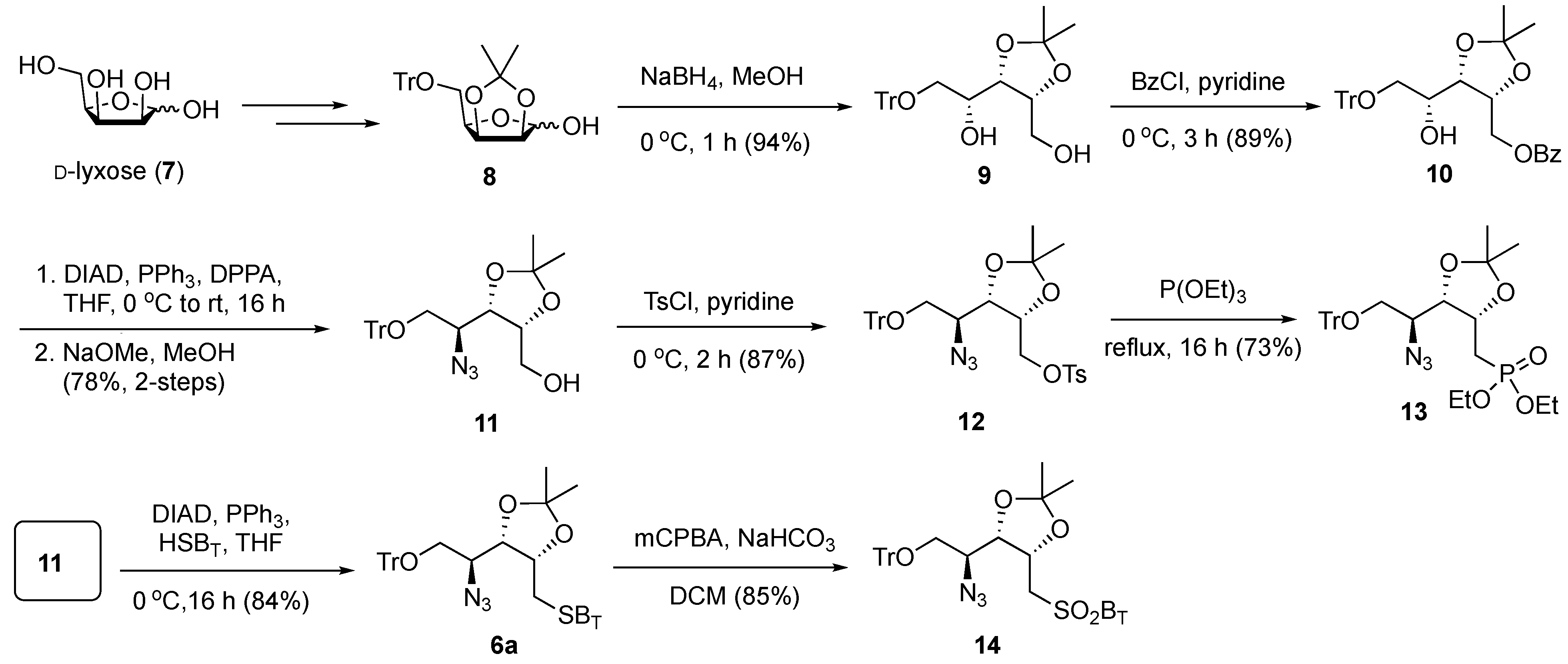
The synthesis of SB
T derivative
6a is shown in
Scheme 2. Commercially available D-lyxose (
7) was transformed to lactol
8, using our previously developed procedures [
40]. NaBH
4-mediated reduction of lactol
8 produced 1,4-diol intermediate
9 (94%), which was readily converted to benzoate ester
10 by a temperature-controlled regioselective benzoylation (benzoyl chloride, pyridine, 0 °C) at the primary hydroxyl group with high yield (89%). The secondary hydroxyl group in
10 was transformed to an azide by the Mitsunobu reaction with diphenylphosphoryl azide (DPPA) with complete inversion at the reacting carbon center, and the basic removal of the benzoate ester produced the alcohol
11 (78% over two steps). The primary alcohol in
11 was then transformed to the tosylate
12; however, attempts to achieve the S
N2 substitution with 2-mercaptobenzothiazole (HSB
T) under basic conditions were fruitless. Therefore, tosylate
12 was heated to reflux in neat triethyl phosphite to give 73% yield of the phosphonate derivative
13. Disappointingly, in a typical Horner–Wadsworth–Emmons olefination using undecanal and LiHMDS at −78 °C, the phosphonate
13 produced an intractable mixture of products. The Mitsunobu reaction of
11 with HSB
T gave the corresponding sulfide
6a with 84% yield. Oxidation of
6a to the corresponding benzothiazol-2-yl sulfone
14 was readily achieved upon treatment with
meta-chloroperoxybenzoic acid (
m-CPBA) in dichloromethane in the presence of NaHCO
3 to give
14 with 85% yield. While the strategy for synthesizing
6a was straightforward, in the initial examination into generating the Psp backbones, the sulfone
14 could not provide a satisfactory yield of Julia–Kocienski olefination (see
Table 1 below). Therefore, we explored the less sterically hindered sulfone
15 for the synthesis of Psp.
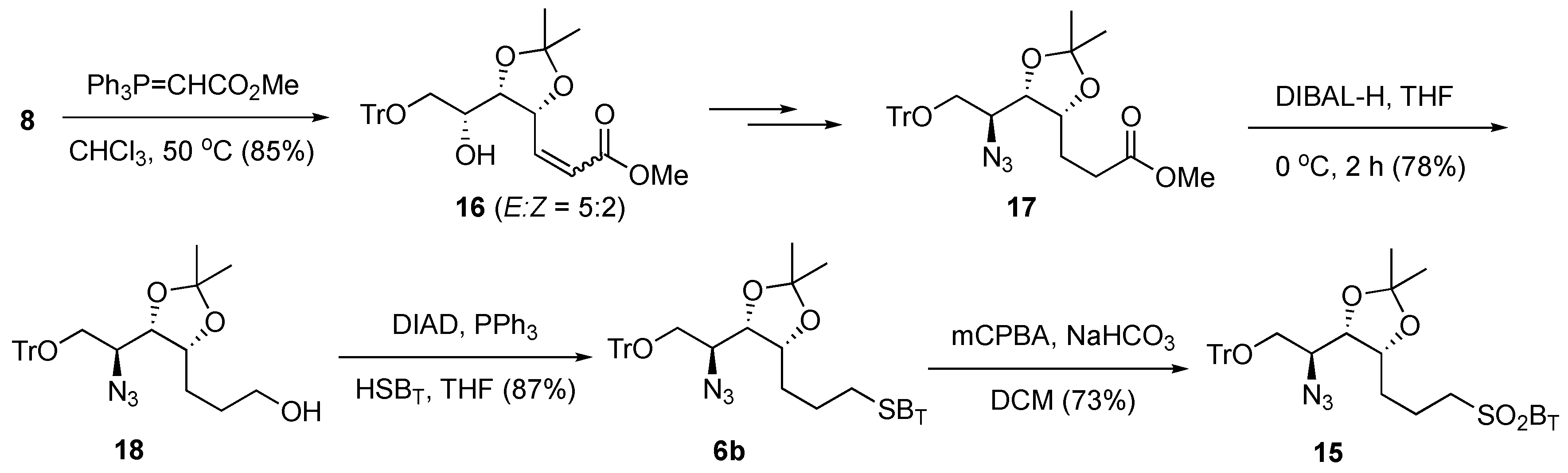
To reduce the steric hindrance,
6b, which bears a longer carbon chain, was prepared (
Scheme 3). In our previous study [
41], a Wittig reaction between
8 and a stabilized ylide Ph
3P = CHCO
2Me gave an undesired intramolecular cyclization adduct by Michael addition as the major product. Fortunately, after detailed investigation and careful optimization of the reaction conditions (
Table S1), we found that an 85% yield of
16 (
E/
Z = 5/2) was obtained by changing the reaction solvent to CHCl
3 and performing the reaction at 50 °C. The alkene (
16), without further purifications, was then routinely transformed to the saturated intermediate azide
17 by following established procedures [
41]. Subsequent reduction of the methyl ester of
17 with DIBAL-H at 0 °C gave alcohol
18 (78% yield), which, upon reaction with HSB
T, afforded the sulfide
6b with a yield of 87% (
Scheme 3). Sulfide
6b was then oxidized with
m-CPBA to provide the corresponding sulfone
15 with a good yield (73%).
For the initial evaluation of the synthesis of Psp backbone by Julia–Kocienski olefination, as shown in
Table 1, a relatively simple sulfone substrate
14 was selected as the target molecule for olefination. The bases and temperature were examined first using a carbonyl compound in a low-polarity solvent, THF (entries 1–6,
Table 1). Accordingly,
14 was deprotonated with a base (NaH, LHMDS, KHMDS, KOH, or DBU) followed by addition of undecanal, but this resulted in a disappointing yield of the desired olefin
19 (13% to 33%) and some aldol condensation products. The low yield of the olefination in truncated sulfone
14 was likely due to the steric hindrance imparted by the structurally rigid protecting group with a cyclic acetal moiety (acetonide) adjacent to α-carbon of sulfone, which resulted in the difficulty of proton abstraction by a base. As a result, large amounts of starting material (
14) remained unreacted, as determined by TLC.
Next, the Julia–Kocienski olefination reaction of a three-carbon-spacer sulfone
15 was investigated. As with the investigation of truncated sulfone
14, the use of NaH as a base at −78 °C in the olefination of
15 with undecanal gave a moderate yield (53%) of the corresponding
E-olefinic compound
20 (entry 7,
Table 1). The reaction progressed smoothly with an improved outcome, and we obtained a 78% yield (trace amounts of
Z-isomer formed, which was separated by simple column chromatography) with LiHMDS (entry 8,
Table 1). This indicated that steric crowding was considerably eased in sulfone
15, and that sufficient reactivity for efficient coupling could be retained.
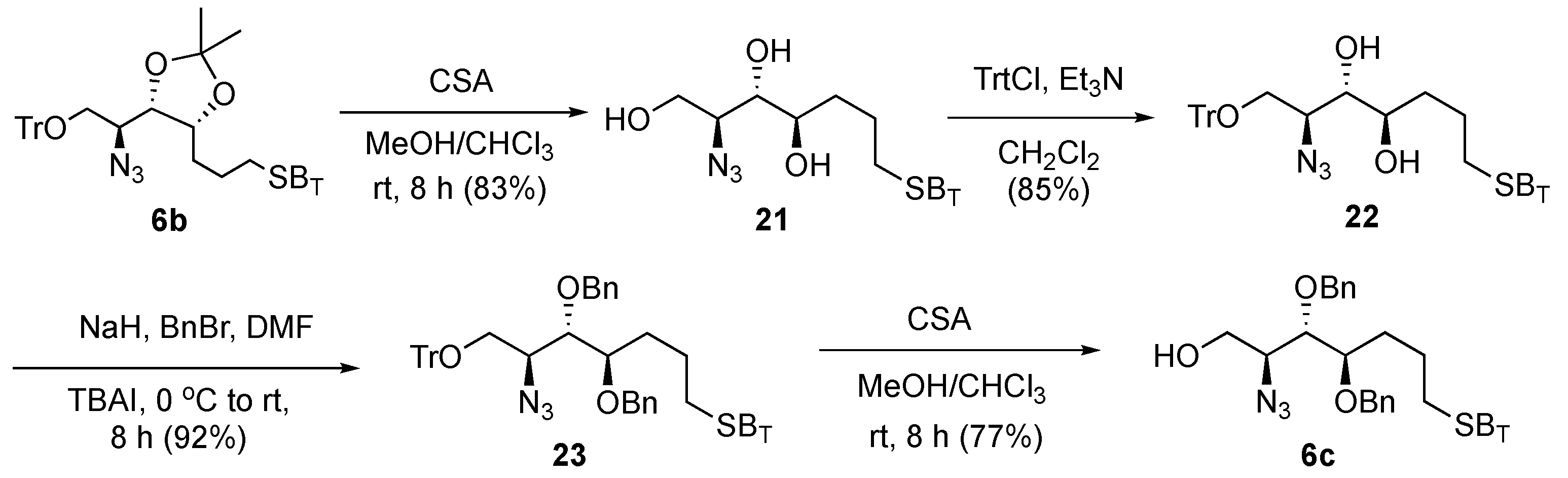
On the basis of the above observation, the acetonide protection in
6b was converted to an acyclic ether derivative
6c to further reduce the steric effect, enhance the yield of olefination, simplify the post-glycosylation deprotection steps, and increase the ease of purification (
Scheme 4). We anticipated that benzyl ethers would shorten one step of deprotection when compared with acetonide, as the former could be removed simultaneously with double bonds under hydrogenolytic debenzylation conditions. Thus, we accomplished cleavage of the acetonide protection in
6b with camphor sulfonic acid (CSA) in MeOH–CHCl
3 solution, generating the triol (
21) with concomitant removal of the trityl protecting group with 83% isolated yield. The trityl ether at the primary hydroxyl group in
21 was reinstalled by reacting with trityl chloride for 8 h to provide
22 (85%), which was then treated with BnBr and NaH to produce
23 with 92% yield. Finally, the trityl ether was removed under acidic conditions to afford the target acceptor
6c (77% yield).
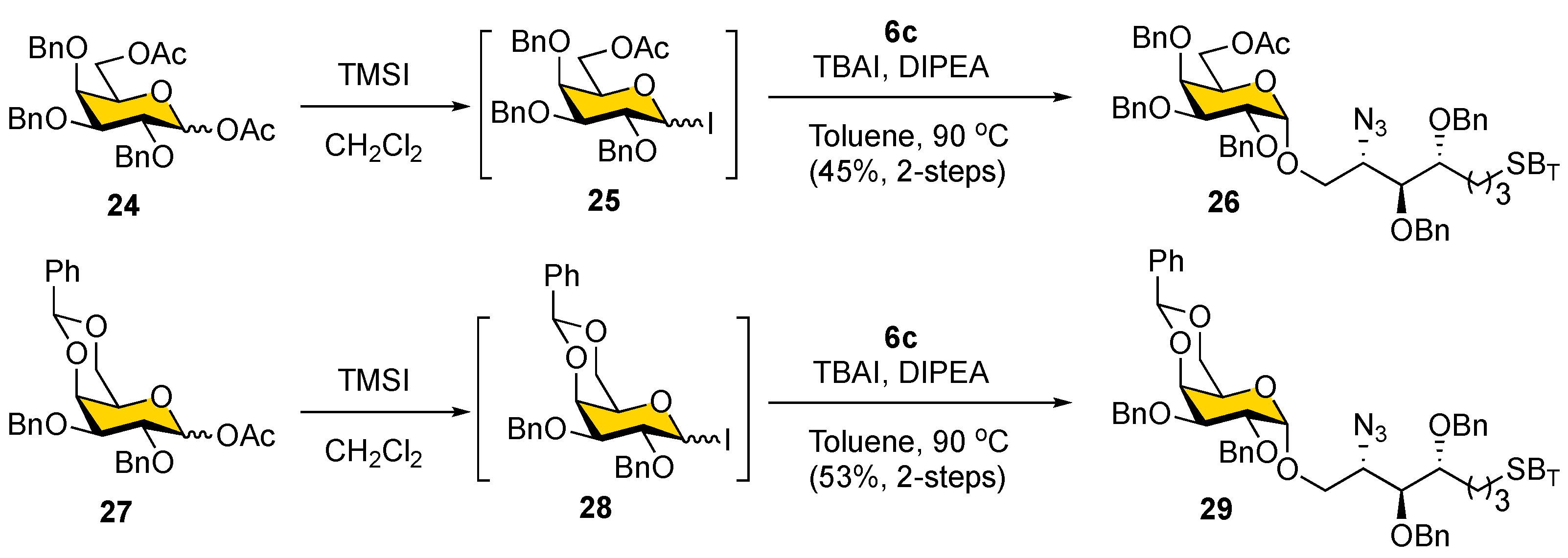
Before installing a fluorous tag, the α-galactosylation of the newly designed alcohol acceptor
6c was conducted (
Scheme 5). Stereoselective formation of α-galactopyranosyl linkages such as those present in α-GalCer is considered challenging because of the absence of neighboring group participation [
50]. The 1,2-
cis-glycosides can be formed stereospecifically under thermodynamic control conditions and by using C2 nonparticipating groups, typically benzyl ether [
51]. Galactosyl iodides are particularly attractive among commonly used glycosyl donors as they are known to undergo exclusive α-stereoselective glycosidation with electron-rich lipid acceptors; they have been adopted in the synthesis of KRN7000 analogs [
52]. Thus, the iodide donor (
25), generated in situ from
24 (1.2 equiv), was reacted with
6c at 90
°C in toluene, using TBAI as a promoter to provide 45% yield of the α-glycoside
26 (δ
H-1α = 4.90 ppm,
J = 3.5 Hz) after column chromatography. However, only a slightly higher yield of the α-glycoside
29 (δ
H-1α = 4.93 ppm,
J = 3.3 Hz) was obtained when a 4,6-
O-benzylidene-protected iodide donor (
28) was used in the glycosylation reaction (53%, two steps). It is noteworthy that a significant amount of hydrolyzed side product was observed in these galactosylation reactions, presumably because of the instability of iodide donors. Therefore, we decided to proceed with the galactosylation of
6c acceptor with the more stable 4,6-
O-benzylidene-protected thioglycoside donor
5 (see below).
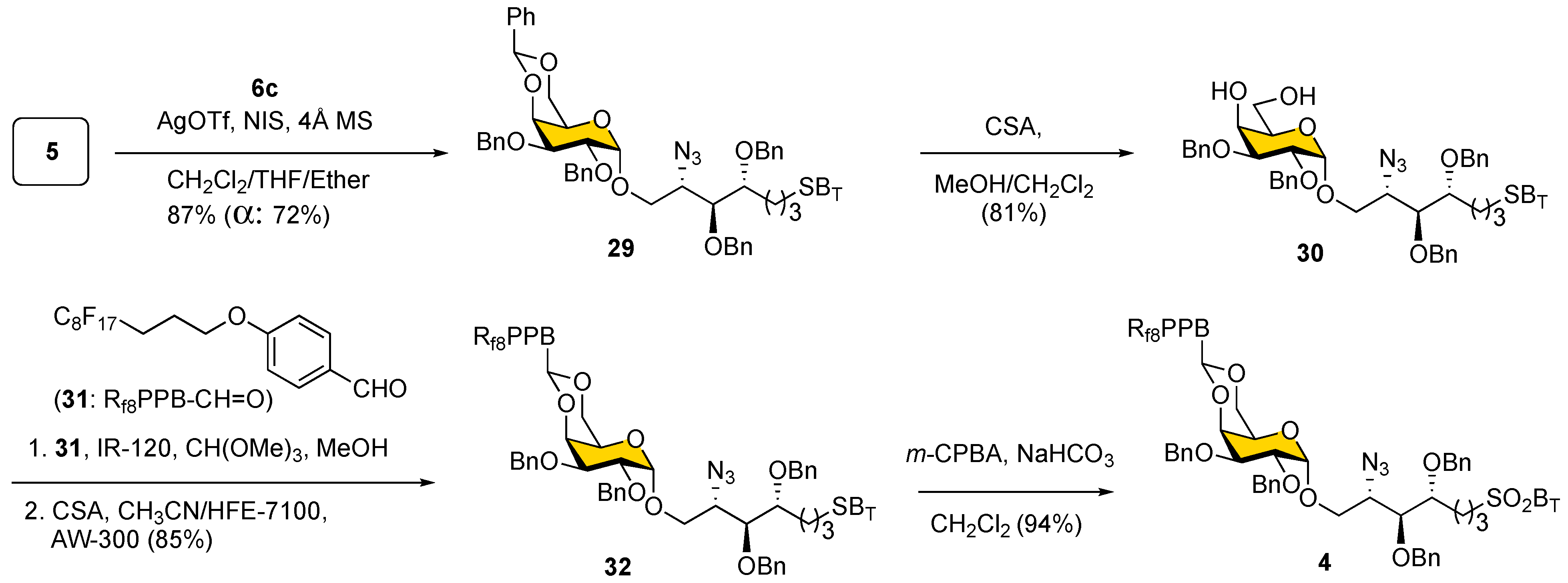
Thiogalactosyl
5, with its 4,6-
O-benzylidene protection, is a thermally stable donor and is known to favor the generation of α-glycosidic bonds [
37]. Thus, the glycosylation reaction between acceptor
6c and donor
5 was performed by employing
N-iodosuccinimide (NIS)/silver triflate as the promoter/activating reagent system in CH
2Cl
2/THF/ether at −30 °C to give
29 with 72% yield of α-anomer and a selectivity of 5:1 (α:β, total yield of 87%), as shown in
Scheme 6. It should be noted that the use of CH
2Cl
2 as solvent yields the product only with an α-anomer but with a slight decrease of yield to 60%. Hydrolysis of the 4,6-
O-benzylidene acetal in
29 was readily achieved by using CSA to afford 4,6-diol
30 (with 81% yield). The direct reaction between
30 and heavy fluorous tag perfluorinated benzaldehyde
31 in the presence of acid only provided a low product yield. Fortunately, the use of 1-dimethoxylmethyl 4-(1
H,1
H,2
H,2
H,3
H,3
H-perfluoroundecyl)oxybenzene, which was generated in situ from
31 using trimethyl orthoformate in the presence of catalytic amounts of
p-TsOH under conditions reported by the Takeuchi group [
53], gave a 63% yield of
Fbenzylidene acetal
32. The reaction yield can be further improved to 85% by the addition of molecular sieves (AW-300) in CH
3CN/HFE-7100 (1:1,
v/
v) as co-solvent. Oxidation of sulfide
32 under conditions (
m-CPBA, NaHCO
3) similar to those described above was straightforward in generating benzothiazolyl sulfone
4 with 94% yield. Notably, crude mixtures were quickly purified by an F-SPE cartridge with multiple washes using MeOH/H
2O (4:1) and 100% MeOH to elute out the desired fluorous-tagged glycosides (
32 and
4). Typically, one cycle of fluorous-assisted purification takes less than 20 min to afford a relatively pure product.
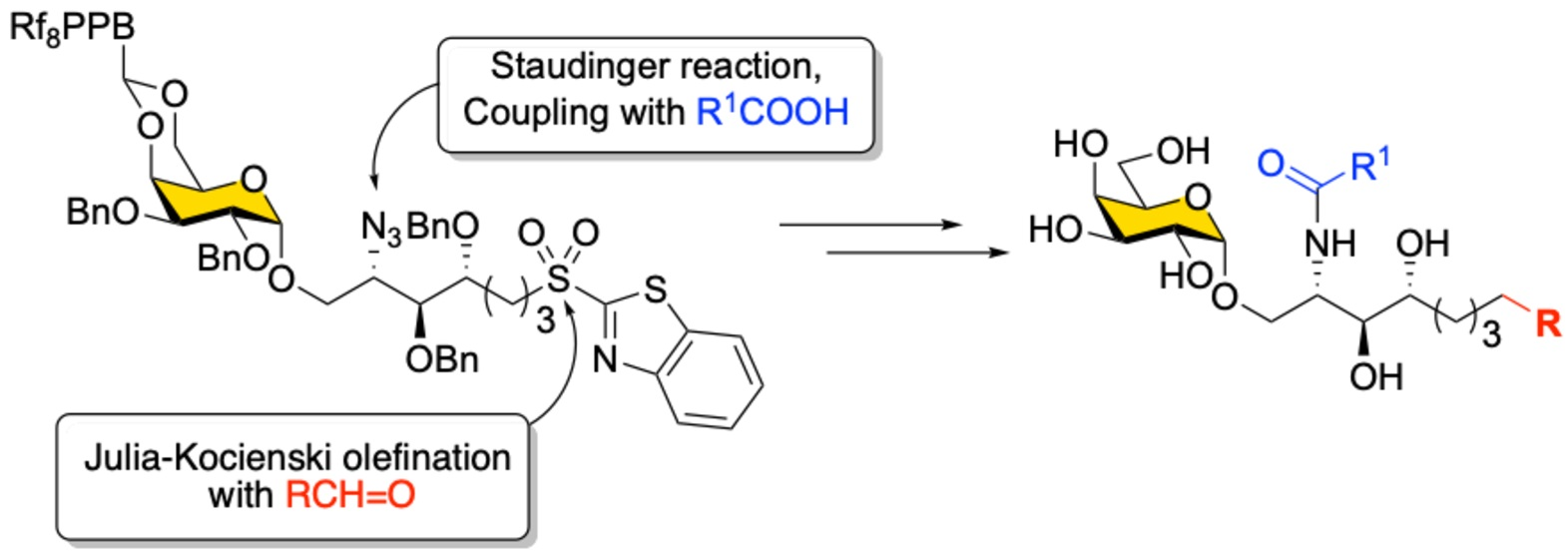
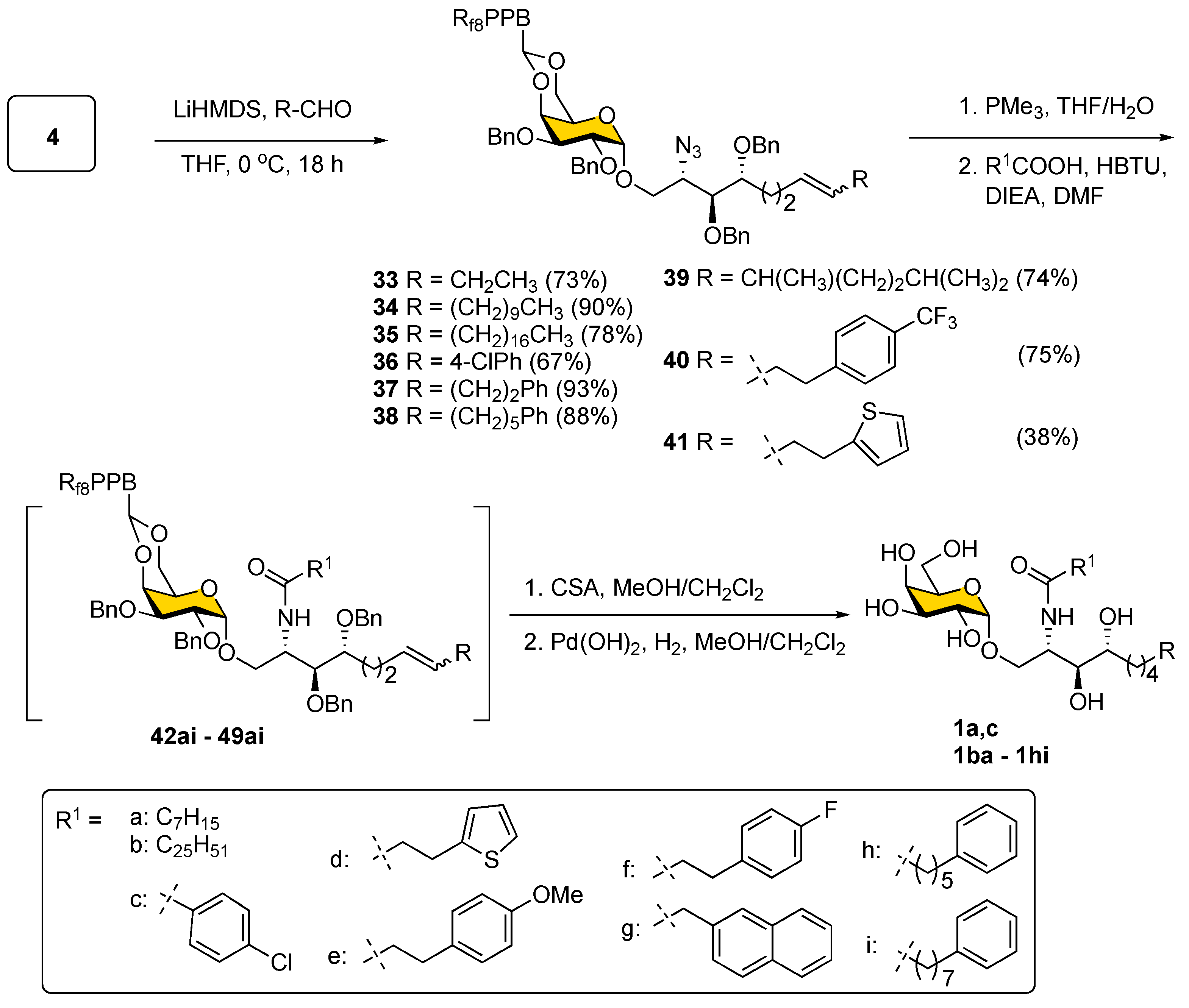
As shown in
Scheme 7, the Julia–Kocienski olefination was then applied to modify the Psp backbone on sulfone
4 by using various alkyl aldehydes to give olefins
33–
41 as a mixture of two geometrical isomers (predominantly
E-isomer) with yields of 67–93%, except for compound
41 (38%). Although the minor
Z-isomer could not be separated at this stage, this was unnecessary since the ensuing steps, including azide reduction and installation of the fatty acyl chain, progressed smoothly. After a quick passage through F-SPE, the final decoration was accomplished by the chemoselective reduction of the azide in
33–
41 to an amine via Staudinger reduction using PMe
3 in THF/H
2O followed by HBTU-mediated amide bond formation with nine different fatty acids (a–i) to produce α-GalCer analogs
42–
49 in the protected form with 82–97% yields (
Table S2) [
54]. Finally, acidic removal of 4,-6-
O-
Fbenzylidene acetal and catalytic hydrogenation with Pd(OH)
2 on carbon in MeOH/CH
2Cl
2 under an atmospheric pressure of H
2 was implemented to remove all the benzyl ethers and reduce the double bond in
42–
49 to afford a total of 61 members of an α-GalCer glycolipid library (ordinarily, the yield is higher than 70% over two steps for
42–
49) including KRN7000 (
1a) and its prototype
1c. The structures of all synthesized α-GalCer analogs are listed in
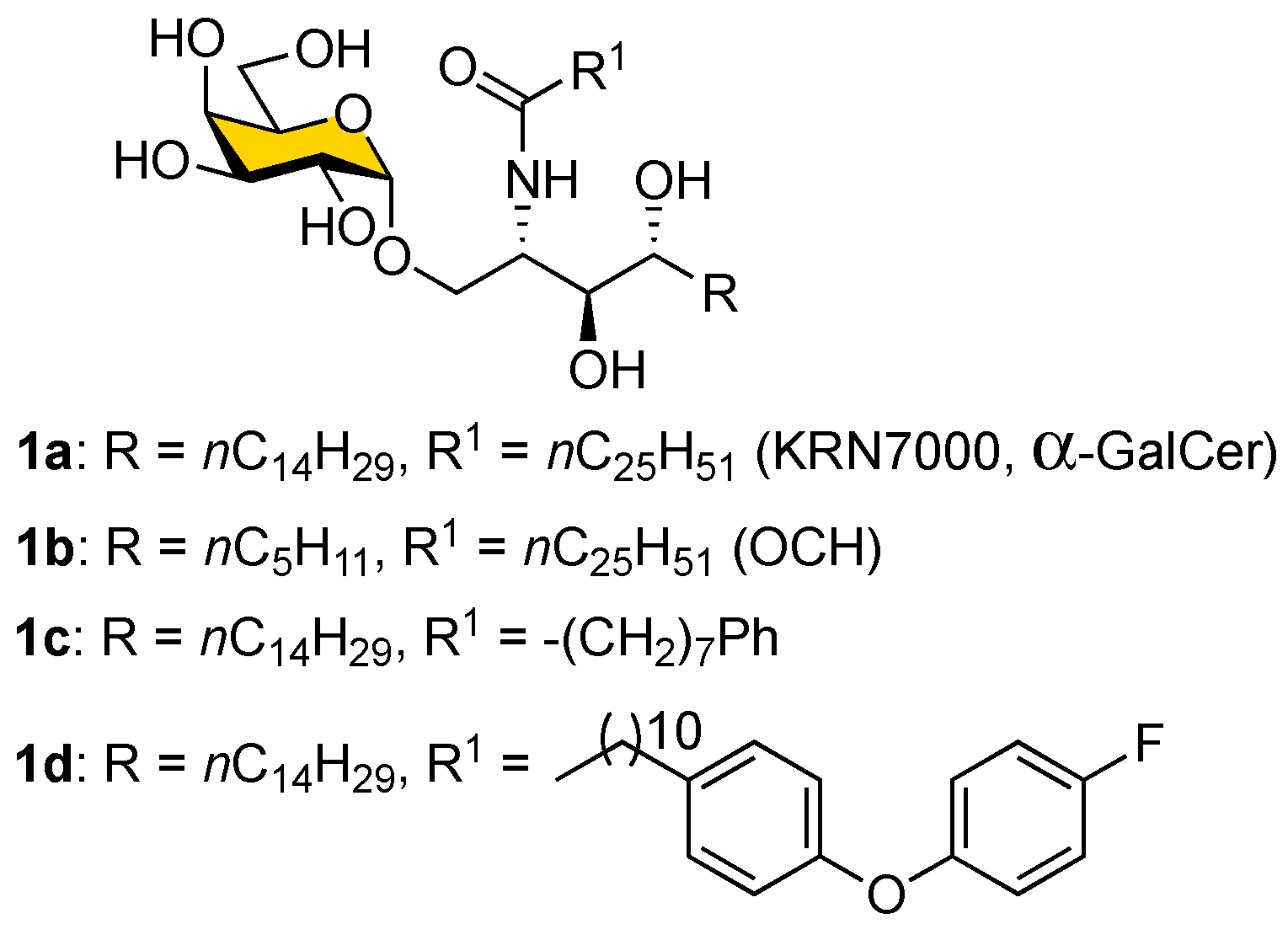
Table 2.
It is important to note that purification by F-SPE on FluroFlash® silica gel greatly facilitated the separation of fluorous tagged glycans from the non-fluorous mixture. The operational time for purification substantially decreased from hours to within 20 min when compared to traditional chromatographic purifications, resulting in a high purity of greater than 95% (as determined by 1H NMR analysis). However, products from the peptidic coupling require multiple purifications because of the presence of many reagents and other side products in the reaction mixture. Thus, the intermediates were not fully characterized, and only the final product spectra were collected and assigned. Importantly, after final cleavage, the fluorous tag was recycled with a recovery yield of 80% to 97%.
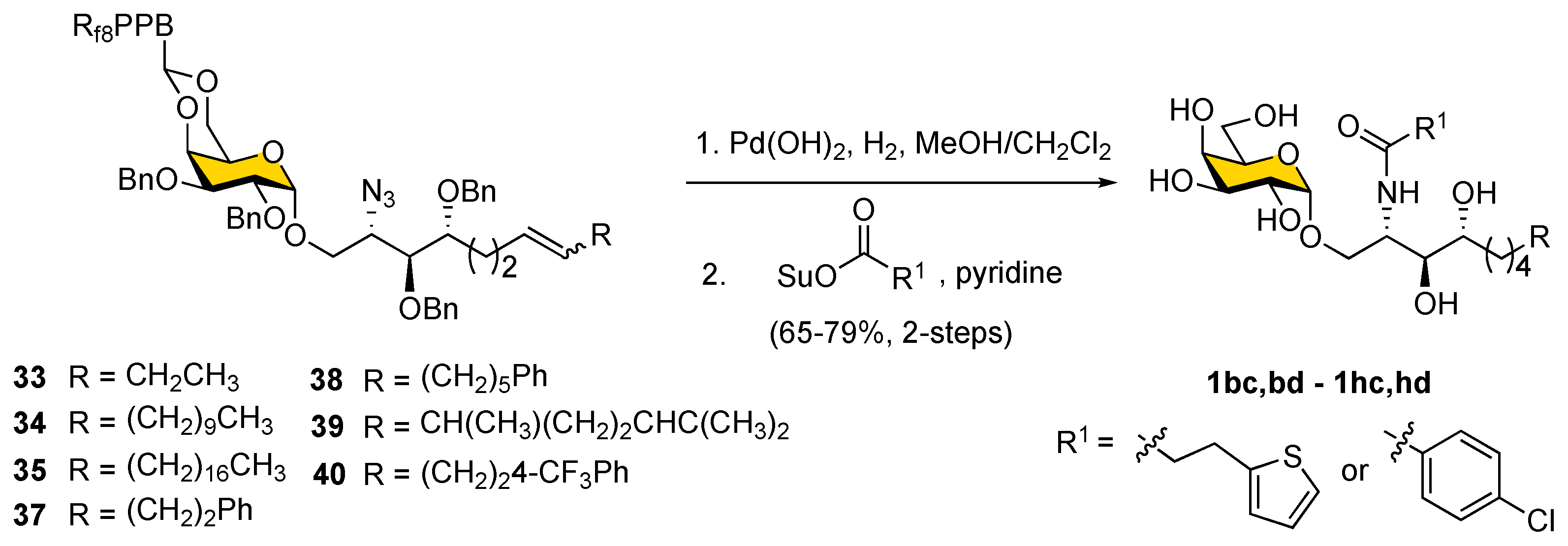
We noted in the case of
41 that the yield of olefination was low because of the rapid decomposition of 3-(thiophen-2-yl)-propanal. In the later deprotection step, it also proved difficult to achieve the selective removal of benzyl ether protecting groups under catalytic hydrogenolysis conditions in substrates containing the
para-chloro phenyl moiety,
36, since the
para-chloro phenyl moiety was reduced to an unsubstituted benzene ring. To circumvent this problem, an alternative strategy was applied by conducting global deprotection first and then installing the fatty acyl chain, as shown in
Scheme 8. Thus, a chlorophenyl or thiophene moiety could be incorporated into the fatty acid. Accordingly, compounds
33–
35 and
37–
40 were subjected to catalytic hydrogenolysis using Pearlman’s catalyst to simultaneously reduce azide, double bond, benzyl ether, and 4,6-
O-benzylidene protecting groups in one step. The resulting amines were then coupled with 3-(thiophen-2-yl)-propanoic acid
N-succinimidyl ester and 4-chlorobenzoic acid
N-succinimidyl ester to provide corresponding α-GalCer analogs (
1bc,
bd–
1hc,
hd;
Table 1) with overall yields of 65–79% (for two steps).
We used Vα14-expressing murine NK1.2 (mNK1.2) cells and mCD1d-expressing A20 (CD1d-A20) cells to evaluate the bioactivities of the synthetic α-GalCer analogs, including glycolipids of the
1b,
1c,
1d,
1e,
1f,
1g, and
1h series (
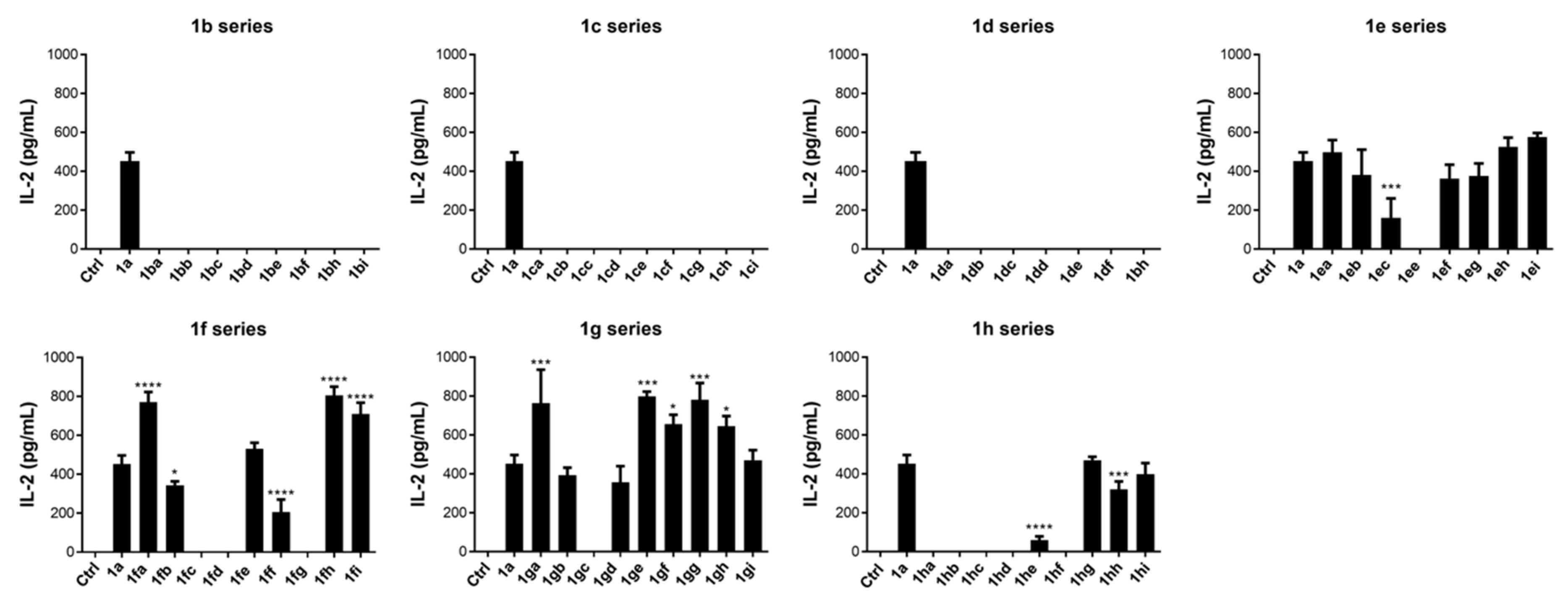
Table 2). As an initial biological study, ELISA assaying was used to determine the level of IL-2 secreted by murine NK1.2 cells upon stimulation by each glycolipid (1 μM). Fujimoto et al. also characterized IL-2 and INF-γ cytokine secretion level produced by the NKT cells upon stimulation by α-GalCer glycolipids ligands containing polar groups in the acyl chain [
55]. As shown in
Figure 2, glycolipids of the
1b,
1c, and
1d series were unable to stimulate NK1.2 cells. Each glycolipid in the
1e series (
1ea: 496.7 ± 63.7,
1eb: 380 ± 131.4,
1ef: 363.3 ± 70.8,
1eg: 375 ± 65,
1eh: 526.7 ± 46.5, and
1ei: 575 ± 21.8) had activity comparable to
1a (α-GalCer) except
1ec (
1ec: 161.7 ± 98.7) and
1ee (0,
p < 0.0001). Among the glycolipids in the
1f series,
1fa (770 ± 52.7),
1fh (805 ± 45), and
1fi (710 ± 57.7) were superior to
1a in stimulation of NKT cells. Five glycolipids [
1fb (343.3 ± 20.2),
1fc (0,
p < 0.0001),
1fd (0,
p < 0.0001),
1ff (205 ± 65.4), and
1fg (0,
p < 0.0001)] induced less IL-2 production than
1a. Meanwhile, glycolipid
1fe (531.7 ± 30.1) induced similar levels of IL-2 as
1a. Among the glycolipids in the
1g series, five [
1ga (763.3 ± 171.8),
1ge (798.3 ± 24.7),
1gf (656.7 ± 47.3),
1gg (780 ± 86.8), and
1gh (645 ± 52.2)] exhibited greater NKT cell stimulatory activity than
1a, but three showed comparable activities [
1gb (393.3 ± 38.8),
1gd (356.7 ± 83.3), and
1gi (470 ± 52.2)] and
1gc showed no activity (0,
p < 0.0001). Most of the glycolipids in the
1h series displayed no (
1ha,
1hb,
1hc,
1hd, and
1hf: 0,
p < 0.0001) or lower IL-2-stimulating activities [
1he (58.33 ± 20.8,
p < 0.0001) and
1hh (320 ± 40.9)] when compared with
1a, except for
1hg (470 ± 18) and
1hi (398.3 ± 56.9).
These results suggest that possessing a shorter [
1b series ((CH
2)
5CH
3)] or longer [
1c series ((CH
2)
20CH
3)] sphingosine chain than
1a [(CH
2)
13CH
3] might impair NKT-stimulatory activity, and that the Psp chain length in α-GalCer might be important for NKT cell activation. It has been reported that the lengths of the alkyl chain affect the stability of CD1d-glycolipid bound complexes, resulting in a modulated NKT cell TCR-binding affinity [
56]. The non-stimulating activity of the synthetic
1b series glycolipids, containing shorter lipid chain length, is likely due to the formation of less stable ternary CD1d-glycolipid-iNKT cell TCR complex [
57]. Notably,
1c series glycolipids possessing with 25 carbon in the sphingoid base chain abolished activity toward IL-2 cytokine production, possibly longer lipid chain cannot localize to the F’ pocket in the CD1d hydrophobic groove [
6]. Moreover, in Psp chains with a terminal phenyl ring, the short carbon chain length [(CH
2)
6Ph,
1e series] appear to have less NKT-stimulating activity than the (CH
2)
9Ph containing
1f series glycolipids. Shorter chain [(CH
2)
4Ph] derivatives in the
1d series showed no activity, demonstrating spacer length was too short for binding interaction in the hydrophobic F’ pocket. However, further analysis by molecular dynamic simulations were essential to support the conclusion. Although the analog
1ea with a truncated acyl chain (C
7H
15) showed activity comparable to that of
1a, the corresponding glycolipid
1fa showed much higher levels of IL-2 production. Introduction of a terminal phenyl ring with a suitable spacer length can dramatically enhance overall cytokine secretion [
17]. The results obtained from glycolipids
1eh, 1ei,
1fh and
1fi are consistent with this model. The 4-fluorophenyl ring attached to acyl chain containing analog also demonstrated stronger cytokine response relative to α-GalCer [
17]. Analog
1gf only induced a notable increase in IL-2 production among
1ef,
1ff,
1gf and
1hf α-GalCers. Notably, the addition of the CF
3 functional group (
1g series) to the phenyl group (
1e series) achieved increased bioactivity relative to the
1e series glycolipids. Furthermore, the IL-2 production levels of most of the
1g series glycolipids were equal or greater than that of α-GalCer (
1a). In addition, the carbon chain length of the acyl chains with a phenyl group might slightly affect their ability to activate NKT cells.
In this work, we primarily focused on the development of a robust synthetic route to lipid-modified α-GalCer glycolipids. Even though some of the synthetic glycolipids in the 1f and 1g series showed stronger IL-2 release relative to KRN7000 (1a), we are unable to identify TH1/TH2-selective cytokine responses with our α-GalCer analogs. Therefore, an in-depth study is required to determine the cytokine-biased glycolipid ligands. However, these efforts may provide useful guidelines for rational ligand design strategy in the context of CD1d-binding glycolipid ligands.
3. Materials and Methods
Materials and reagents. All reactions were performed in oven-dried glassware (120 °C) under a nitrogen atmosphere unless indicated otherwise. All chemicals were purchased as reagent grade and used without further purification. Dichloromethane (CH2Cl2) was distilled over calcium hydride. Tetrahydrofuran (THF) and ether were distilled over sodium metal/benzophenone ketyl radical. Anhydrous N,N-dimethylformamide (DMF) and methanol (MeOH) were purchased from Merck. Molecular sieves (MS) for glycosylation were MS 4Å (Aldrich) and activated by flame. 1H and 13C NMR spectra were recorded on either a Bruker AV-400 or AV-600 spectrometer operating at 400 or 600 MHz for 1H and 100 or 150 MHz for 13C, respectively. Chemical shifts (δ) are reported in ppm and referenced to the deuterated solvent used (chloroform-d (CDCl3), δ 7.24 and 77.0; methanol-d4 (CD3OD), δ 3.31 and 49.0; and acetone-d6 (CD3)2CO, 2.05, and 29.84 and 206.26), with coupling constants (J) reported in Hz. Two-dimensional (COSY) experiments were used to assist in assignment of the products. High-resolution mass spectra were recorded under ESI-TOF mass spectroscopy conditions. Analytical thin-layer chromatography (TLC) was performed on pre-coated plates (silica gel 60). Silica gel 60 (E. Merck) was employed for all flash-chromatography experiments. All fluorous-assisted purification was performed using FluoroFlash® SPE cartridge (Fluorous Tech. Inc., USA). The reactions were monitored by examination under UV light (254 nm) and by staining with p-anisaldehyde, ninhydrin, cerium molybdate, or potassium permanganate solutions.
Synthesis of α-GalCer glycolipids library (1). The azides (33–41) were converted to amines by Staudinger reduction, and the resultant amines were subsequently reacted with 9-different fatty acids. In a typical procedure, an azide (1.0 eq.) was dissolved in THF:H2O (1 mL, 1:1 v/v) to which a 1M solution of PMe3 in THF was added (2.5 eq.). After stirring at rt for 12 h or until the azide was completely consumed, the solution was evaporated under reduced pressure. The residue was subjected to a F-SPE cartridge to get the desired amine. A solution of amine (1 eq.) in anhydrous DMF (3 mL) was treated with acid (1.5 eq.), HBTU (1.2 eq.), and diisopropylethylamine (1.5 eq.) at 0 °C. The reaction mixture was stirred at rt for 16 h or until TLC indicated the disappearance of amine, then concentrated under reduced pressure and extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by a F-SPE cartridge (70% aqueous MeOH to 100% MeOH elution) to afford pure amides (42ai–49ai).
General procedure for the final deprotection of compounds 42ai–49ai. The fully unprotected glycolipid library was constructed by two deprotection steps: the hydrolysis of Fbenzylidene acetal was performed using CSA following a similar method as described for 30, and subjected to reductive hydrogenolysis to remove benzyl ethers and reduce unsaturation in phytosphingosine chain. In a typical experiment, CSA (1.0 eq.) was added to a solution of corresponding 4,6-O-Fbenzylidene derivative (1 eq.) in MeOH/CHCl3 (1/1, 2 mL) at rt with vigorous stirring. After 8 h, the reaction mixture was quenched with Et3N, concentrated under reduced pressure, and used without further purification. To a solution of above crude product in MeOH/CH2Cl2 (1/1, 2 mL), 10% Pd(OH)2 (10 w/w %) was added. The resultant mixture was degassed and saturated with a balloon filled with H2 gas and left stirring at rt for 8 h under a positive pressure of H2. The catalyst was removed by filtration through celite, rinsed with MeOH. The combined filtrates were evaporated in vacuo, and subjected to flash column chromatography on silica to afford pure α-GalCer glycolipids (1).
1ba. Rf 0.4 (CH2Cl2/MeOH = 6/1). = +36.89 (c = 2.34, MeOH/CH2Cl2 = 1/1). 1H NMR (600 MHz, CD3OD): δ 0.90 (t, J = 6.8 Hz, 6H), 1.29–1.43 (m, 20H), 2.23 (t, J = 7.6 Hz, 2H, H-1”), 3.55 (m, 1H), 3.60 (t, J = 6.1 Hz, 1H, H3′). 3.66–3.71 (m, 3H), 3.74 (dd, J = 3.1, 10.0 Hz, 1H), 3.79 (dd, J = 3.7, 10.0 Hz, 1H), 3.83 (t, J = 6.0 Hz, 1H), 3.88 (m, 2H), 4.21 (td, J = 4.7, 10.4 Hz, 1H), 4.87 (d, J = 3.6 Hz, 1H). 13C NMR (150 MHz, CD3OD): δ 9.4, 14.5, 23.8, 23.9, 27.1, 27.2, 30.3, 30.5, 30.6, 33.0, 33.2, 33.3, 37.4, 48.1, 62.9, 68.4, 70.4, 71.2, 71.6, 72.7, 73.1, 75.7, 101.3, 176.0. HRMS (ESI-TOF) m/z calcd for C24H47NO9Na [M+Na]+ 516.3143, found 516.3140.
Compound 4. To a solution of 32 (170 mg, 0.21 mmol) in CH2Cl2 (20 mL) was added solid NaHCO3 (88 mg, 1.05 mmol) and followed by addition of m-CPBA (36 mg, 0.63 mmol). The resulting solution was stirred at rt for 16 h. The solution was diluted with CH2Cl2, and quenched with satd Na2S2O3(aq.), washed with satd NaHCO3(aq.), and extracted by CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was dissolved in DMF (2 mL) and loaded onto F-SPE cartridge. The cartridge was successively washed with MeOH/H2O (4:1) to remove all non-fluorous materials and then 100% MeOH to elute out the desired pure sulfone 4 (163 mg, 94%) as a colorless oil. Rf 0.6 (n-hexanes/EtOAc = 2/1). = +27.61 (c = 1.5, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 1.61–1.98 (m, 4H), 2.04–2.11 (m, 2H), 2.23–2.36 (m, 2H), 3.39 (m, 2H), 3.53–3.57 (m, 2H), 3.61 (dt, J = 3.7, 7.5 Hz, 1H), 3.66 (dd, J = 6.2, 10.4 Hz, 1H), 3.73 (dd, J = 3.7, 6.8 Hz, 1H), 3.88 (dd, J = 1.5, 12.8 Hz, 1H), 3.94 (dd, J = 3.3, 10.4 Hz, 1H), 3.96 (dd, J = 3.1, 10.0 Hz, 1H), 4.01–4.06 (m, 1H), 4.02 (t, J = 6.1 Hz, 2H, FtagCH2CH2CH2O), 4.07 (dd, J = 3.4, 10.0 Hz, 1H), 4.16 (d, J = 3.1 Hz, 1H), 4.36 (d, J = 11.4 Hz, 1H), 4.53 (d, J = 11.4 Hz, 1H), 4.53 (d, J = 11.2 Hz, 1H), 4.59 (d, J = 11.2 Hz, 1H), 4.63 (d, J = 11.8 Hz, 1H), 4.72 (d, J = 12.3 Hz, 1H), 4.78 (d, J = 12.3 Hz, 1H), 4.85 (d, J = 11.8 Hz, 1H), 4.93 (d, J = 3.4 Hz, 1H), 5.40 (s, 1H), 6.85 (m, 2H), 7.18–7.42 (m, 22H), 7.60 (doublet of quintet, J = 1.5, 7.7 Hz, 2H), 7.99 (m, 1H), 8.17 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 18.8, 20.7, 28.1, 28.3, 54.7, 61.5, 63.2, 66.5, 68.3, 69.4, 72.1, 72.2, 73.8, 74.1, 74.6, 75.5, 76.0, 77.8, 78.5, 99.3, 101.1, 114.2 (2C), 122.5, 125.6, 127.7, 127.8 (2C), 128.4 (2C), 128.6 (4C), 131.0, 136.9, 137.8, 137.9, 138.8, 138.9, 152.9, 159.2, 165.9. HRMS (ESI-TOF) m/z calcd for C66H61F17N4O11S2Na [M+Na]+ 1495.3405, found 1495.3403.
Compound 6b. Diisopropyl azodicaboxylate (DIAD, 0.38 mL, 1.86 mmol) was added dropwise to a stirred solution of 18 (750 mg, 1.54 mmol), and PPh3 (470 mg, 1.86 mmol) in anhydrous THF (30 mL) at 0 °C under an atmosphere of nitrogen. Subsequently, 2-mercaptobenzothiazole (312 mg, 1.86 mmol) was added and the resulting solution was allowed to warm to rt over 5 h. EtOAc was then added, and the reaction mixture was sequentially washed with brine and water and then dried and concentrated in vacuo. Purification via flash silica gel column chromatography gave 6b as a yellow oil (850 mg, 87%. Rf 0.65 (n-hexanes/EtOAc = 4/1). = +5.83 (c = 3.5, CH2Cl2). 1H NMR (400 MHz, (CD3)2CO): δ 1.23 (s, 3H), 1.24 (s, 3H), 1.66–2.06 (m, 4H), 3.36 (dd, J = 7.3, 10.1 Hz, 1H), 3.49 (t, J = 7.2 Hz, 2H), 3.56 (dd, J = 2.5, 10.1 Hz, 1H), 3.66 (ddd, J = 2.5, 7.3, 9.1 Hz, 1H), 4.02 (dd, J = 5.7, 9.1 Hz, 1H), 4.24 (ddd, J = 3.4, 5.7, 10.1 Hz, 1H), 7.26–7. 54 (m, 15H); 13C NMR (100 MHz, CDCl3): δ 25.8, 26.6, 28.2, 28.6, 33.5, 60.6, 64.6, 75.9, 77.4, 87.5, 108.6, 121.1, 121.7, 124.4, 126.2, 127.3 (3C), 128.1 (6C), 128.9 (6C), 135.4, 143.9 (3C), 153.5, 167.1. HRMS (ESI-TOF) m/z calcd for C36H36N4O3S2Na [M+Na]+ 659.2121, found 659.2131.
Compound 6c. CSA (500 mg, 2.15 mmol) was added to the solution of 23 (1.67 g, 2.15 mmol) in MeOH/CHCl3 (1/1, 40 mL). After being stirred at rt for 8 h, the reaction mixture was quenched with Et3N and then extracted with EtOAc (3 × 20 mL). The combined extracts were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography to yield 6c (1.2 g, 77%). Rf 0.25 (n-hexanes/EtOAc = 4:1). = +11.33 (c = 2.67, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 1.76–2.00 (m, 4H), 2.41 (brs, 1H), 3.28 (t, J = 6.3 Hz, 2H), 3.65 (dd, J = 4.8, 9.6 Hz, 1H), 3.71 (m, 2H), 3.78 (dd, J = 5.3, 11.6Hz, 1H), 3.85 (dd, J = 4.6, 11.6 Hz, 1H), 4.54 (d, J = 11.3 Hz, 1H), 4.60 (d, J = 11.3 Hz, 1H), 4.66 (d, J = 11.3 Hz, 1H), 4.70 (d, J = 11.3 Hz, 1H), 7.25–7.41 (m, 12H), 7.73 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 25.0, 28.9, 33.4, 62.3, 63.2, 72.3, 73.7, 78.3, 79.7, 120.9, 121.4, 124.2, 126.0, 127.9, 128.0 (3C), 128.2 (2C), 128.5 (2C), 128.6 (2C), 135.2, 137.5, 137.6, 153.3, 168.8. HRMS (ESI-TOF) m/z calcd for C28H31N4O3S2 [M+H]+ 535.1832, found 535.1839.
Compound 15. To a solution of 6b (850 mg 1.27 mmol) in CH2Cl2 (20 mL) at rt was added solid NaHCO3 (534 mg, 6.36 mmol) and then mCPBA (660 mg, 3.82 mmol). The reaction mixture was stirred at rt for 16 h. The solution was diluted with CH2Cl2, and then washed with saturated aq. Na2S2O3 and saturated aqueous NaHCO3. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography to yield sulfone 15 (652 mg, 73%) as a colorless oil: Rf 0.3 (n-hexanes/EtOAc = 4/1); = +8.13 (c = 7, CH2Cl2): 1H NMR (400 MHz, (CD3)2CO): δ 1.19 (s, 3H), 1.21 (s, 3H), 1.61–1.71 (m, 1H), 1.80–2.02 (m, 3H), 3.34 (dd, J = 7.2, 10.0 Hz, 1H), 3.52 (dd, J = 2.3, 10.0 Hz, 1H), 3.57 (ddd, J = 2.3, 7.2, 9.2 Hz, 1H), 3.74 (t, J = 7.8 Hz, 2H), 3.99 (dd, J = 5.7, 9.2 Hz, 1H), 4.19 (ddd, J = 3.1, 5.7, 10.6 Hz, 1H), 7.25–7.37 (m, 9H), 7.48–7.52 (m, 6H), 7.68–7.76 (m, 2H), 8.23–8.32 (m, 2H); 13C NMR (100 MHz, (CD3)2CO): δ 19.9, 24.9, 27.3, 27.7, 53.9, 60.2, 64.4, 75.5, 76.9, 87.1, 108.2, 123.0, 125.0, 127.1 (3C), 127.8 (7C), 128.1, 128.6 (6C), 136.7, 143.9 (3C), 152.8, 166.7; HRMS (ESI-TOF) m/z calcd for C36H36N4O5S2Na [M+Na]+ 691.2019, found 691.2018.
Compound 18. DIBAL-H (1 M in THF, 4.84 mL, 4.84 mmol) was added to a solution of 17 (1 g, 1.94 mmol) in anhydrous THF (10 mL) at 0 °C dropwisely. The resulting mixture was stirred at 0 °C for 2 h under an atmosphere of nitrogen or until TLC indicated complete disappearance of starting materials. The reaction mixture was quenched with satd NH4Cl(aq.) and then diluted with EtOAc and aq. HCl (1.0 M) until a clear solution was obtained. The aqueous layer was extracted with EtOAc (3 × 20 mL) and the combined organic layers were washed with satd NaHCO3(aq.) and brine, dried over MgSO4, and concentrated under reduced pressure. The product (18, 738 mg, 78%) was isolated as a colorless oil after flash column chromatography on silica gel. Rf 0.2 (n-hexanes/EtOAc = 2/1); = +3.20 (c = 6, CH2Cl2). 1H NMR (400 MHz, (CD3)2CO): δ 1.22 (s, 3H), 1.24 (s, 3H), 1.51–1.64 (m, 2H), 1.72–1.77 (m, 2H), 3.35 (dd, J = 7.6, 10.0 Hz, 1H), 3.56 (dd, J = 2.4, 10.0 Hz, 1H), 3.58–3.64 (m, 3H), 3.66 (ddd, J = 2.4, 7.4, 9.0 Hz, 1H), 4.04 (dd, J = 5.5, 9.0 Hz, 1H), 4.19 (ddd, J = 2.7, 5.5, 9.4 Hz, 1H), 7.26–7.54 (m, 15H). 13C NMR (100 MHz, (CD3)2CO): δ 25.0, 25.8, 27.5, 29.8, 60.5, 61.4, 64.5, 75.8, 77.5, 87.0, 107.8, 127.1 (3C), 127.8 (6C), 128.6 (6C), 143.9 (3C). HRMS (ESI-TOF) m/z calcd for C29H33N3O4Na [M+Na]+ 510.2363, found 510.2365.
Compound 21. A solution of 6b (2 g, 3.14 mmol) in MeOH/CHCl3 (1/1, 50 mL) was treated with CSA (728 mg, 3.14 mmol), and the resulting mixture was stirred at rt for 8 h. The reaction mixture was quenched with Et3N and concentrated in vacuo. The residue was purified by flash silica gel column chromatography to yield compound 21 (934 mg, 83%). Rf 0.2 (n-hexanes/EtOAc = 1:1). = +16.83 (c = 3.0, CH2Cl2). 1H NMR (400 MHz, CD3OD): δ 1.55–2.14 (m, 4H), 3.38 (m, 2H), 3.54 (dd, J = 4.8, 6.8 Hz, 1H), 3.61 (m, 2H), 3.76 (dd, J = 7.9, 11.6 Hz, 1H), 3.92 (dd, J = 3.3, 11.6 Hz, 1H), 7.28–7.32 (dt, J = 1.0 , 7.6 Hz, 1H), 7.40–7.44 (dt, J = 1.0, 7.6 Hz, 1H), 7.80 (t, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CD3OD): δ 26.9, 33.0, 34.6, 62.6, 66.8, 72.4, 75.9, 122.1, 122.4, 125.6, 127.5, 136.2, 154.4, 169.7; HRMS (ESI-TOF) m/z calcd for C14H18N4O3S2Na [M+Na]+ 377.0713, found 377.0718.
Compound 22. Et3N (1.1 mL, 8.0 mmol) was added dropwise to a solution containing triol 21 (2.3 g, 7.05 mmol) and trityl chloride (2.16 g, 7.75 mmol) in anhydrous CH2Cl2 (60 mL) at rt under an atmosphere of N2. After being stirred at rt for 8 h, the solution was concentrated in vacuo, and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with brine, dried over MgSO4, and evaporated under reduced pressure. The residue was purified by flash silica gel column chromatography to get mono-tritylated ether 22 (3.57 g, 85%). Rf 0.25 (n-hexanes/EtOAc = 2:1). = +5.83 (c = 6.2, CH2Cl2): 1H NMR (400 MHz, CDCl3): δ 1.52–2.0 (m, 4H), 2.93 (brs, 1H), 3.20 (dt, J = 7.7, 13.8 Hz, 1H), 3.38 (dt, J = 7.5, 13.8 Hz, 1H), 3.44 (dd, J = 5.3, 9.9 Hz, 1H), 3.67 (m, 4H, H-4, H-6, H7), 3.79 (brs, 1H), 7.19–7.50 (m, 17H), 7.71 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 25.6, 29.9, 32.3, 62.3, 63.5, 70.8, 74.1, 87.5, 120.8, 120.9, 124.2, 126.0, 127.1 (3C), 127.8 (6C), 128.4 (6C), 134.7, 143.2 (3C), 152.5, 167.7. HRMS (ESI-TOF) m/z calcd for C33H32N4O3S2Na [M+Na]+ 619.1808, found 619.1809.
Compound 23. The diol 22 (1.25 g, 2.09 mmol) and TBAI (232 mg, 0.63 mmol.) were dissolved in DMF (20 mL) and cooled to 0 °C. A 60% dispersion of NaH in mineral oil (251 mg, 10.47 mmol) was added followed by addition of benzyl bromide (0.75 mL, 6.28 mmol). The reaction was warmed to rt with stirring over 8 h, quenched with MeOH and then concentrated in vacuo. The residue was extracted with EtOAc (3 × 20 mL), and the combined extracts were washed with water, brine, dried over MgSO4, and evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel to obtain 23 (1.5 g, 92%) as colorless oil. Rf 0.2 (n-hexanes/EtOAc = 9:1). = +3.52 (c = 2.53, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 1.68–1.94 (m, 4H), 3.25 (t, J = 7.0 Hz, 2H), 3.38 (dd, J = 8.1, 10.0 Hz, 1H), 3.45 (dd, J = 2.8, 10.0 Hz, 1H), 3.54–3.60 (m, 2H), 3.78 (ddd, J = 2.8, 5.4, 8.1 Hz, 1H), 4.04 (s, 2H), 4.46 (d, J = 11.2 Hz, 1H), 4.57 (d, J = 11.2 Hz, 1H), 7.08–7.45 (m, 27H), 7.73 (d, J = 7.9 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 24.9, 28.6, 33.5, 64.1, 64.1, 71.9, 73.5, 78.5, 78.6, 87.2, 120.8, 121.4, 124.0, 125.9, 127.0 (3C), 127.6, 127.6, 127.8 (8C), 127.9 (2C), 128.2 (2C), 128.3 (2C), 128.6 (6C), 135.1, 137.6, 137.9, 143.6 (3C), 153.2, 166.9. HRMS (ESI-TOF) m/z calcd for C47H44N4O3S2Na [M+Na]+ 799.2747, found 799.2750.
Compound 29. A stirred suspension of p-tolylthiogalactoside donor 5 (1.0 g, 1.6 mmol), acceptor 6c (1.4 g, 2.4 mmol), and activated MS 4Å (2.0 g) in Et2O-THF–CH2Cl2 (3:1:1, 30 mL) was cooled to −30 °C and then NIS (360 mg, 1.6 mmol) was added. After being stirred for 10 min at ambient temperature, AgOTf (21 mg, 0.081 mmol) was added. The solution was stirred for an additional 1 h and then warmed to −10 °C. The reaction mixture was quenched with Et3N (2 mL), and then filtered through a pad of celite. The filtrate was poured into satd NaHCO3(aq.) and extracted with EtOAc (3 × 20 mL). The combined extracts were washed with brine, dried over MgSO4, and evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel to give product 29 (1.3 g, 72%, α-anomer) as a colorless oil. Rf 0.25 (n-hexanes/EtOAc = 4:1). = +18.93 (c = 5.5, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 1.69–1.90 (m, 4H), 3.24 (t, J = 6.7 Hz, 2H), 3.52 (m, 1H), 3.64–3.70 (m, 3H), 3.72 (dd, J = 4.0, 5.7 Hz, 1H), 3.80 (d, J = 12.5 Hz, 1H), 3.92–4.02 (m, 3H), 4.06 (dd, J = 3.4, 10.0 Hz, 1H), 4.11 (d, J = 3.2 Hz, 1H), 4.44 (d, J = 11.4 Hz, 1H), 4.54 (d, J = 11.4 Hz, 1H), 4.61 (m, 2H), 4.63 (d, J = 11.8 Hz, 1H), 4.71 (d, J = 12.3 Hz, 1H), 4.77 (d, J = 12.3 Hz, 1H), 4.83 (d, J = 11.8 Hz, 1H), 4.93 (d, J = 3.3 Hz, 1H), 5.38 (s, 1H), 7.18–7.48 (m, 27H), 7.71 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 25.2, 28.9, 33.6, 61.8, 63.1, 68.3, 69.4, 72.0, 72.2, 73.7, 73.9, 74.7, 75.5, 75.9, 78.4, 78.7, 99.2, 101.2, 121.1, 121.6, 124.3, 126.2, 126.5, 127.70, 127.73, 127.8 (2C), 127.92 (2C), 127.95 (2C), 128.0, 128.1 (2C), 128.2 (2C), 128.3 (2C), 128.4 (2C), 128.5 (2C), 128.6 (4C), 129.0, 135.3, 138.0 (2C), 138.9 (2C), 153.4, 167.1. HRMS (ESI-TOF) m/z calcd for C55H56N4O8S2Na [M+Na]+ 987.3437, found 987.3439.
Compound 30. Compound 30 was prepared by following the similar method as described in the synthesis of 21 in a mixture of MeOH and CH2Cl2. Starting from 29 (500 mg, 0.52 mmol), pure compound 30 (318 mg) was isolated as a yellow oil in 81% yield. Rf 0.2 (n-hexanes/EtOAc = 1/1). = +22.38 (c = 5.51, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 1.74–1.99 (m, 4H), 2.71 (brs, 2H, OH), 3.26 (t, J = 6.70 Hz, 2H), 3.66–3.77 (m, 6H), 3.82 (dd, J = 5.1, 11.0 Hz, 1H), 3.87 (dd, J = 2.9, 10.0 Hz, 1H), 3.90 (dd, J = 2.8, 10.0 Hz, 1H), 3.98 (dd, J = 2.4, 10.2 Hz, 1H), 4.03–4.04 (m, 1H), 4.49 (d, J = 11.4 Hz, 1H), 4.57 (d, J = 11.4 Hz, 1H), 4.62 (d, J = 11.3 Hz, 1H), 4.66 (d, J = 10.4 Hz, 1H), 4.66 (d, J = 11.7 Hz, 1H), 4.68 (d, J = 10.3 Hz, 1H), 4.77 (d, J = 11.7 Hz, 1H), 4.78 (d, J = 11.3 Hz, 1H), 4.89 (d, J = 2.8 Hz, 1H), 7.22–7.41 (m, 22H), 7.71–7.74 (m, 1H), 7.82–7.84 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 25.1, 28.9, 33.6, 62.0, 63.0, 68.4, 69.1, 69.6, 72.1, 72.8, 73.3, 73.9, 75.7, 77.4, 78.7, 78.8, 98.5, 121.1, 121.6, 124.3, 126.2, 127.8, 127.9 (2C), 128.0, 128.1 (2C), 128.2 (2C), 128.3 (2C), 128.4 (2C), 128.5 (2C), 128.6 (2C), 128.7 (2C), 135.3, 138.0, 138.1 (2C), 138.5, 153.4, 167.1. HRMS (ESI-TOF) m/z calcd for C48H53N4O8S2 [M+H]+ 877.3305, found 877.3301.
Compound 32. To a solution of Fbenzaldehyde 31 (305 mg, 0.52 mmol) in MeOH (5 mL) was added trimethyl orthoformate (0.12 mL, 1.05 mmol), IR-120 (150 mg). The reaction was stirred at rt until TLC indicated completed disappearance of starting material. The reaction mixture was quenched with Et3N and then filtered. The filtrate was evaporated under reduced pressure and purified by flash column chromatography on silica gel to give Fbenzaldehyde dimethylacetal. To the above Fbenzaldehyde dimethylacetal in acetonitrile (5 mL) and HFE-7100 (5 mL) was added compound 30 (460 mg, 0.52 mmol), CSA (36 mg, 0.16 mmol), and AW-300 (500 mg). The reaction was stirred at rt until TLC indicated completed disappearance of starting material. The reaction was quenched with Et3N, and concentrated in vacuo. The residue was subjected to a F-SPE cartridge (FluoroFlash® Slica Gel, 40 μm) and eluted by 80% aq. MeOH and then 100% MeOH to afford pure 4,6-O-Fbenzylidene derivative 32 (643 mg, 85% yield). Rf 0.4 (n-hexanes/EtOAc = 3:1). = +23.53 (c = 2.33, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 1.84–2.04 (m, 4H), 2.18–2.46 (m, 4H), 3.38 (t, J = 6.9 Hz, 2H), 3.65 (m, 1H), 3.78–3.83 (m, 3H), 3.86 (dd, J = 4.5, 5.6 Hz, 1H), 3.93 (dd, J = 1.4, 12.6 Hz, 1H), 4.07–4.13 (m, 3H), 4.15 (t, J = 6.00 Hz, 2H, FtagCH2CH2CH2O), 4.18 (dd, J = 3.4, 10.1 Hz, 1H), 4.24 (d, J = 2.8 Hz, 1H), 4.58 (d, J = 11.5 Hz, 1H), 4.68 (d, J = 11.5 Hz, 1H), 4.71 (d, J = 10.6 Hz, 1H), 4.75 (d, J = 10.6 Hz, 1H), 4.77 (d, J = 11.8 Hz, 1H), 4.84 (d, J = 12.3 Hz, 1H), 4.90 (d, J = 12.3 Hz, 1H), 4.97 (d, J = 11.8 Hz, 1H), 5.07 (d, J = 3.4 Hz, 1H), 5.47 (s, 1H), 6.98 (d, J = 8.7 Hz, 2H), 7.31–7.54 (m, 24H), 7.85 (d, J = 7.5 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H). 13C NMR (150 MHz, CDCl3): δ 20.7, 25.3, 28.1, 29.0, 33.7, 61.8, 63.1, 66.5, 68.4, 69.4, 72.1, 72.2, 73.7, 74.0, 74.7, 75.6, 76.0, 78.5, 78.8, 99.3, 101.1, 114.2, 121.1, 121.6, 124.4, 126.2, 127.6, 127.7, 127.8 (2C), 127.9 (3C), 128.0 (4C), 128.1, 128.2 (2C), 128.3 (2C), 128.4 (2C), 128.5 (2C), 128.6 (2C), 128.7 (2C), 131.1, 135.3, 138.0, 138.2, 138.9 (2C), 153.5, 159.2, 167.1. HRMS (ESI-TOF) m/z calcd for C66H61F17N4O9S2Na [M+Na]+ 1463.3506, found 1463.3511.
General procedure for the synthesis of alkenes (33–41) via Julia–Kocienski olefination reaction. In a typical procedure, a solution of sulfone 4 (820 mg, 0.56 mmol) in anhydrous THF (10 mL) was treated at 0 °C with LiHMDS (1.2 mL, 1.2 mmol, 1 M in THF) with vigorous stirring. After 15 min, a solution of aldehyde (80 mg, 0.83 mmol) in anhydrous THF (2 mL) was added dropwise at 0 °C. The reaction mixture was gradually warmed to rt and allowed to stir for 16 h under an atmosphere of nitrogen. The reaction mixture was quenched with satd NH4Cl(aq) solution, extracted with EtOAc (3 × 20 mL), washed once with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to a FluoroFlash® cartridge to afford pure alkene in 80% MeOH elution to afford 33 (540 mg, 73%). Rf 0.55 (n-hexanes/EtOAc = 2/1); = +27.93 (c = 2.5, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 0.97 (ddt, J = 0.6, 3.4, 7.5 Hz, 3H ), 1.59 (m, 1H), 1.76 (m, 1H), 1.96–2.16 (m, 6H), 2.32 (m, 2H), 3.58 (m, 1H), 3.66–3.77 (m, 4H), 3.89 (dt, J = 1.9, 12.6 Hz, 1H), 4.00–4.11 (m, 4H), 4.05 (t, J = 5.8 Hz, 2H, FtagCH2CH2CH2O), 4.16 (d, J = 3.1 Hz, 1H), 4.50 (dd, J = 2.3, 11.5 Hz, 1H), 4.59–4.63 (m,2H), 4.68 (d, J = 11.9 Hz, 1H), 4.69 (d, J = 11.3 Hz, 1H), 4.75 (d, J = 12.3 Hz, 1H), 4.82 (d, J = 12.3 Hz, 1H), 4.87 (d, J = 11.9 Hz, 1H), 4.98 (d, J = 3.2 Hz, 1H), 5.27–5.47 (m, 2H), 5.42 (s, 1H, benzylidene), 6.88 (d, J = 8.4 Hz, 2H), 7.22–7.34 (m, 18H) 7.40 (d, J = 7.3 Hz, 2H), 7.45 (d, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 14.1, 20.7, 23.2, 25.7, 28.6, 30.3, 61.9, 63.1, 66.5, 68.6, 69.4, 72.2, 72.3, 73.7, 73.9, 74.8, 75.6, 75.9, 78.9, 79.2, 99.3, 101.1, 114.2 (2C), 127.6, 127.7, 127.8, 127.9 (4C), 128.0 (2C), 128.1 (4C), 128.4 (2C), 128.5 (2C), 128.6 (4C), 128.7, 131.1, 132.5, 132.9, 138.2, 138.5, 138.9 (2C), 159.2; HRMS (ESI-TOF) m/z calcd for [M+Na]+ 1338.4107, found 1338.4102.
Bioactivity assay
A20 cells (2 ×104) overexpressing mCD1d were loaded with chemically synthesized glycolipids (1 μM) and then cultured with mNK1.2 cells (2 × 104). Upon stimulation by glycolipids, mNK1.2 cells secreted IL-2 into culture medium. Three days later, supernatants were collected to determine the level of IL-2 by enzyme-linked immunosorbent assay (ELISA).
ELISA
Mouse IL-2 in cell culture supernatants was determined by DuoSet ELISA Development System according to the manufacturer’s procedures (R&D System, Minneapolis, MN, USA). Briefly, supernatant and standards were serially diluted, added into each well, and incubated for 2 h at room temperature (RT). After washing with the wash buffer, 100 uL of the detection antibody was added to each well and incubated for 2 h at RT. After washing, each well was incubated with 100 uL of diluted streptavidin-HRP for 20 min at RT, followed by incubated with 100 uL of substrate solution for 20 min at RT. The reaction was stopped by adding stop solution and read at 450 nm with SpectraMax M2 (Molecular Device, San Jose, CA, USA).


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









