
Molecular and Functional Characterization of BDNF-Overexpressing Human Retinal Pigment Epithelial Cells Established by Sleeping Beauty Transposon-Mediated Gene Transfer

 , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
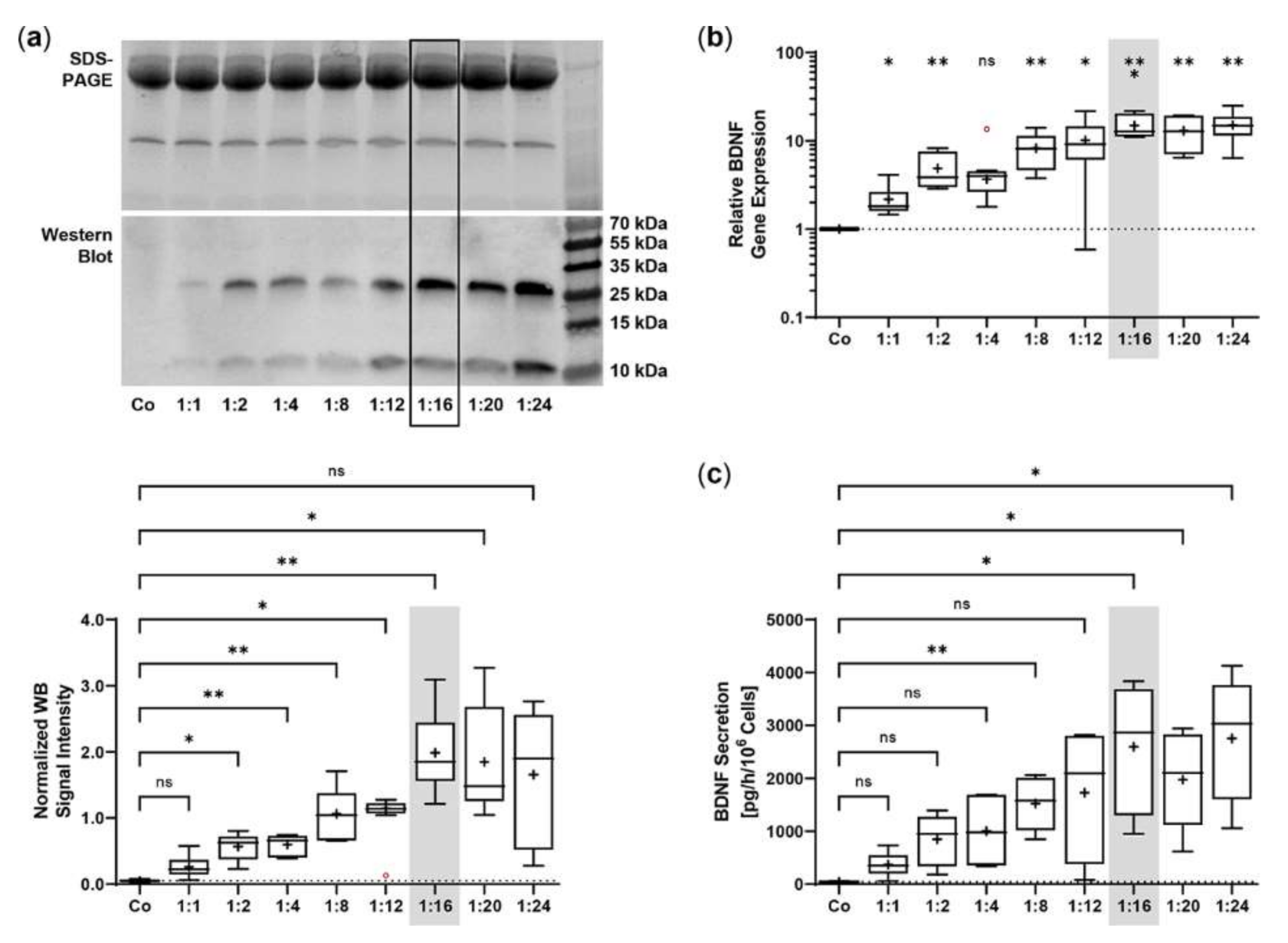
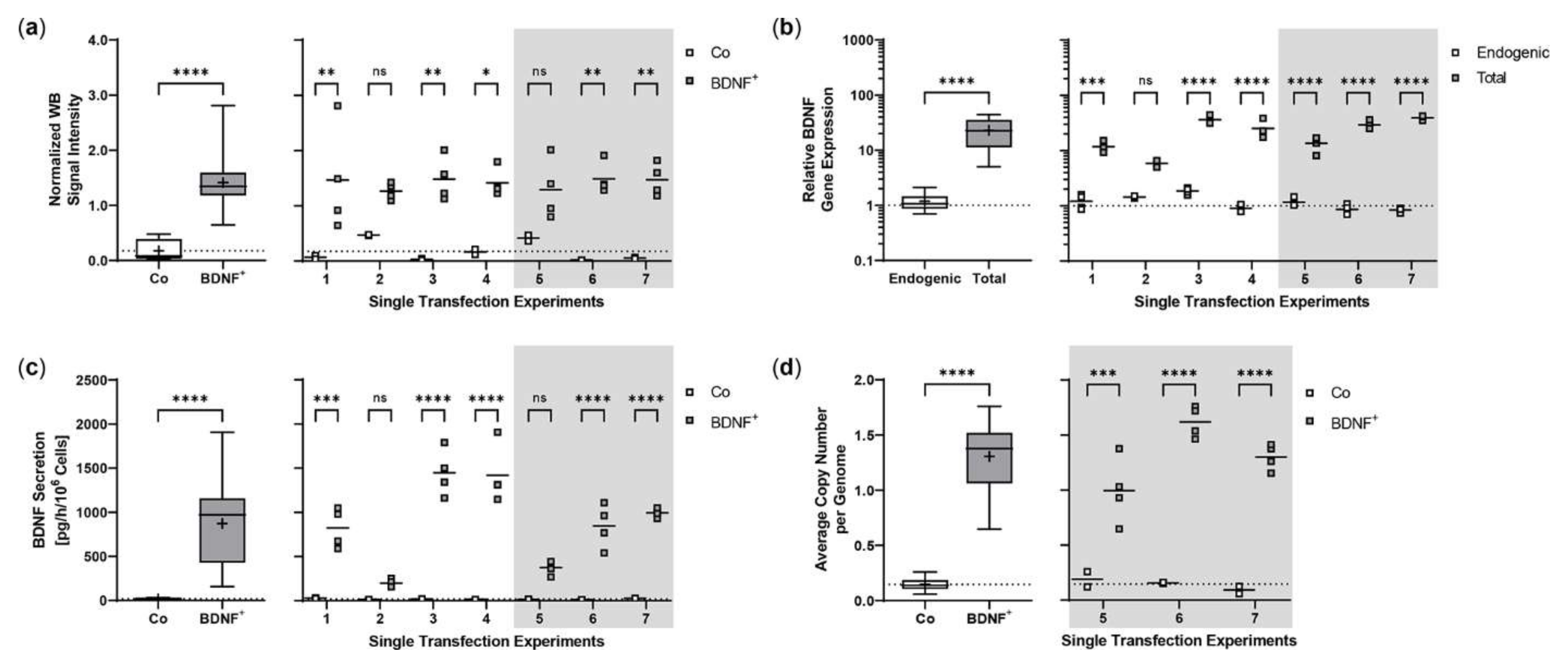
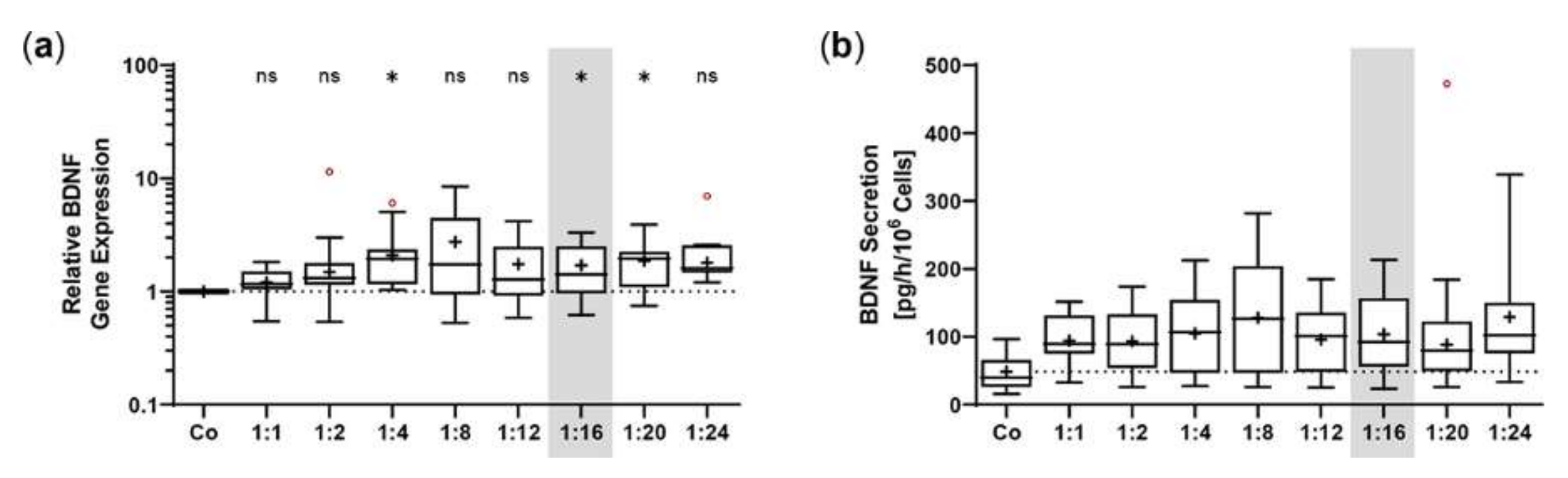
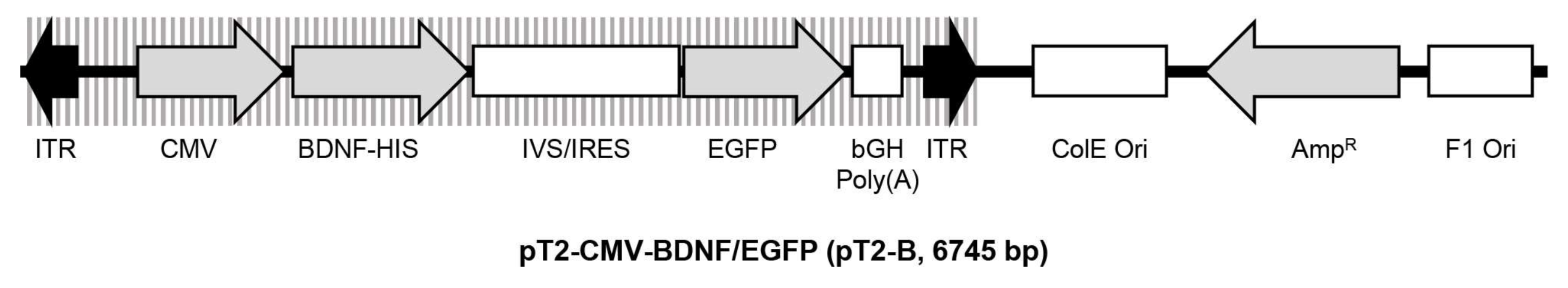
2.1. Sleeping Beauty Transposon-Mediated Delivery of the Human BDNF Gene into the ARPE-19 Cell Line
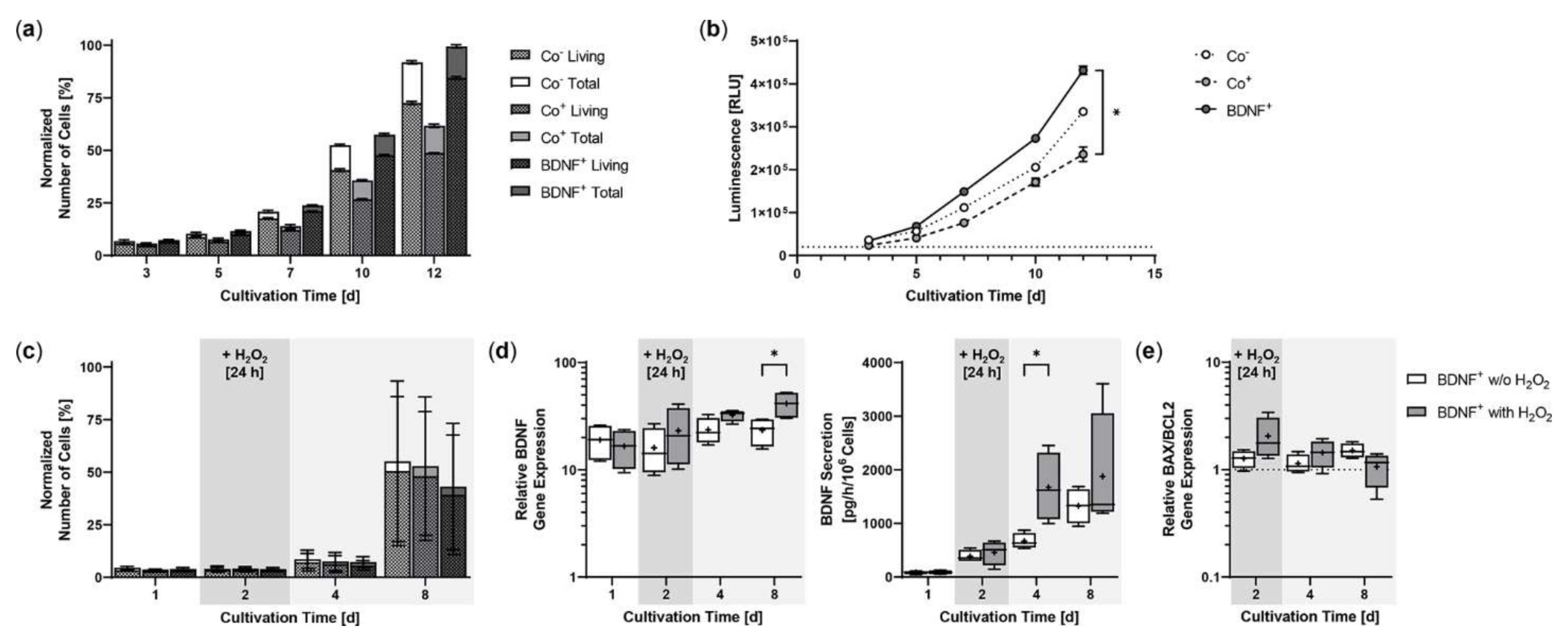
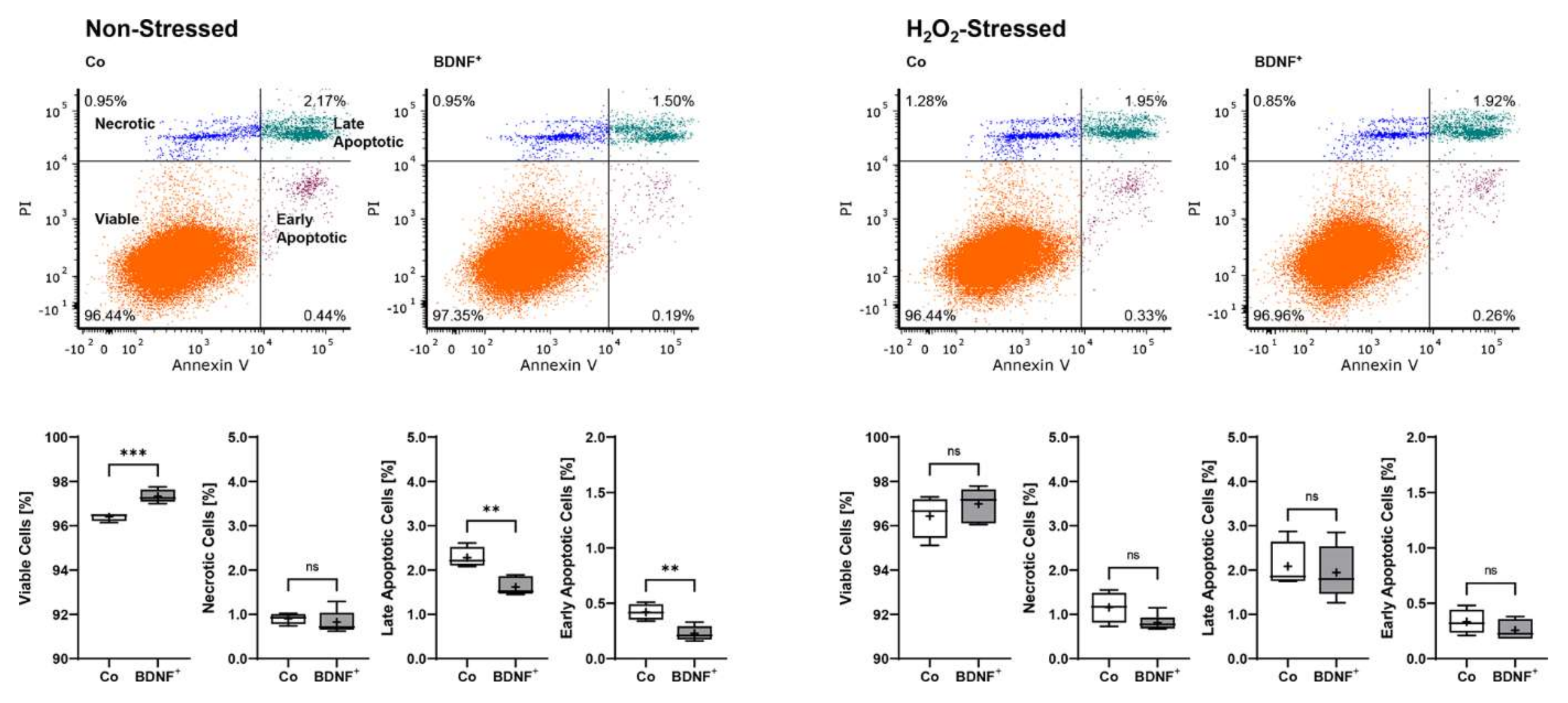
2.2. Proliferation and Cell Viability of BDNF-Transfected ARPE-19 Cell Cultures
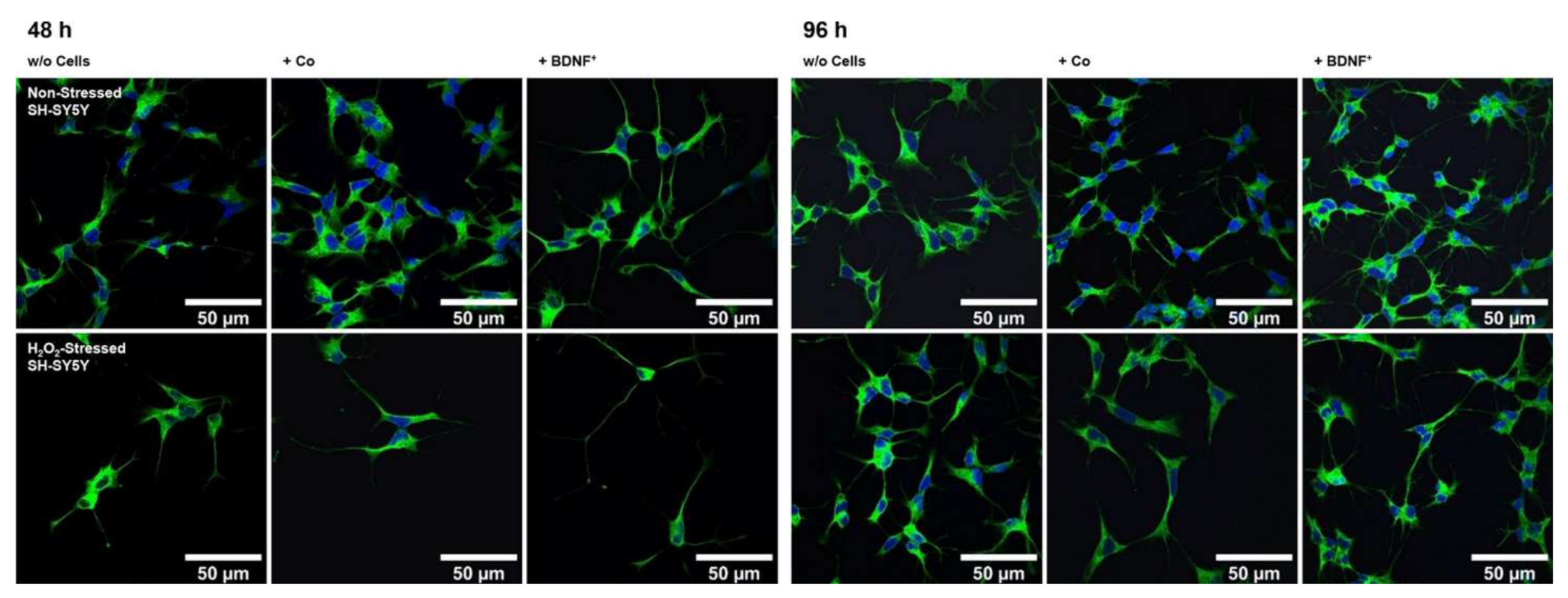
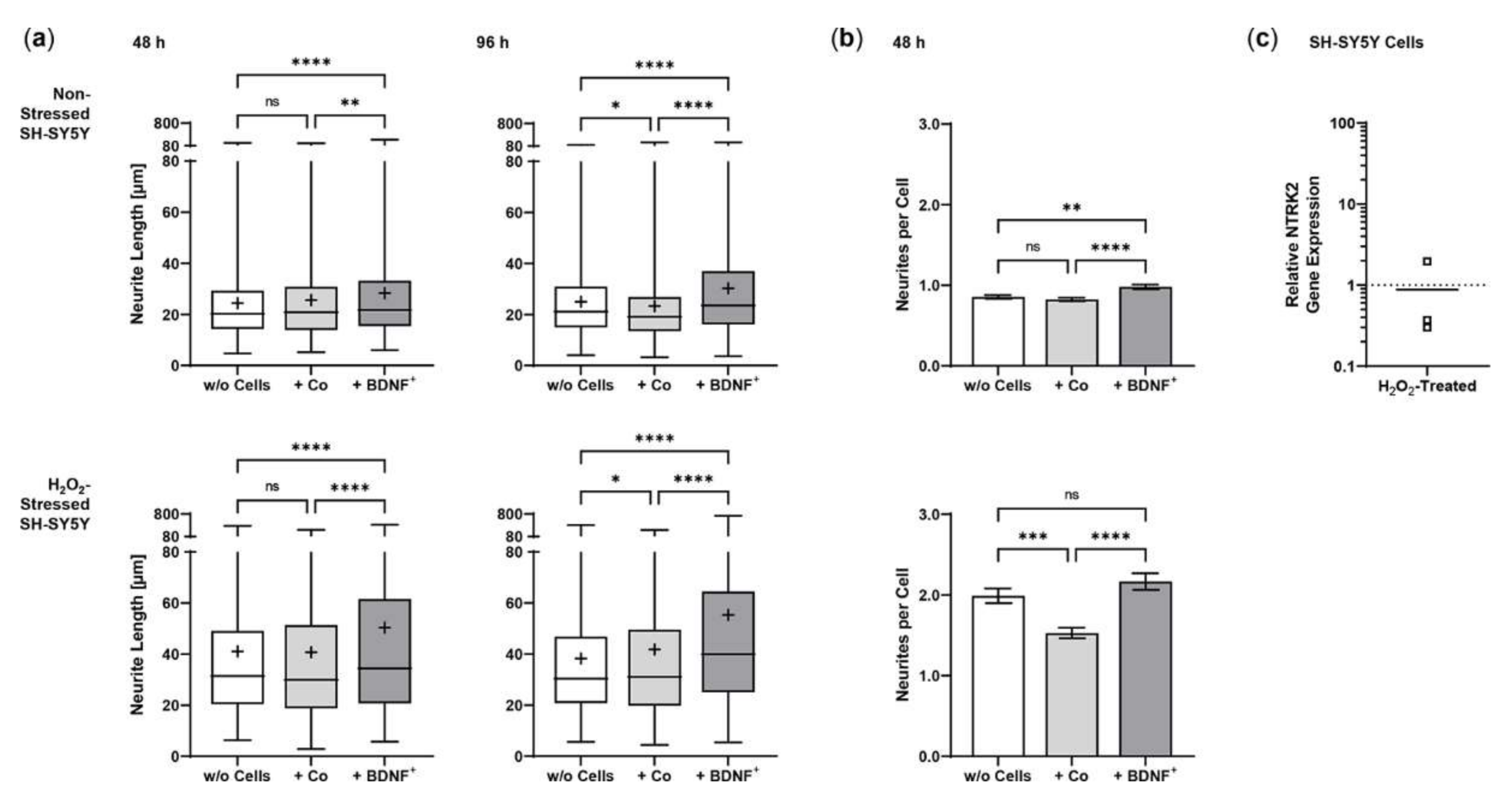
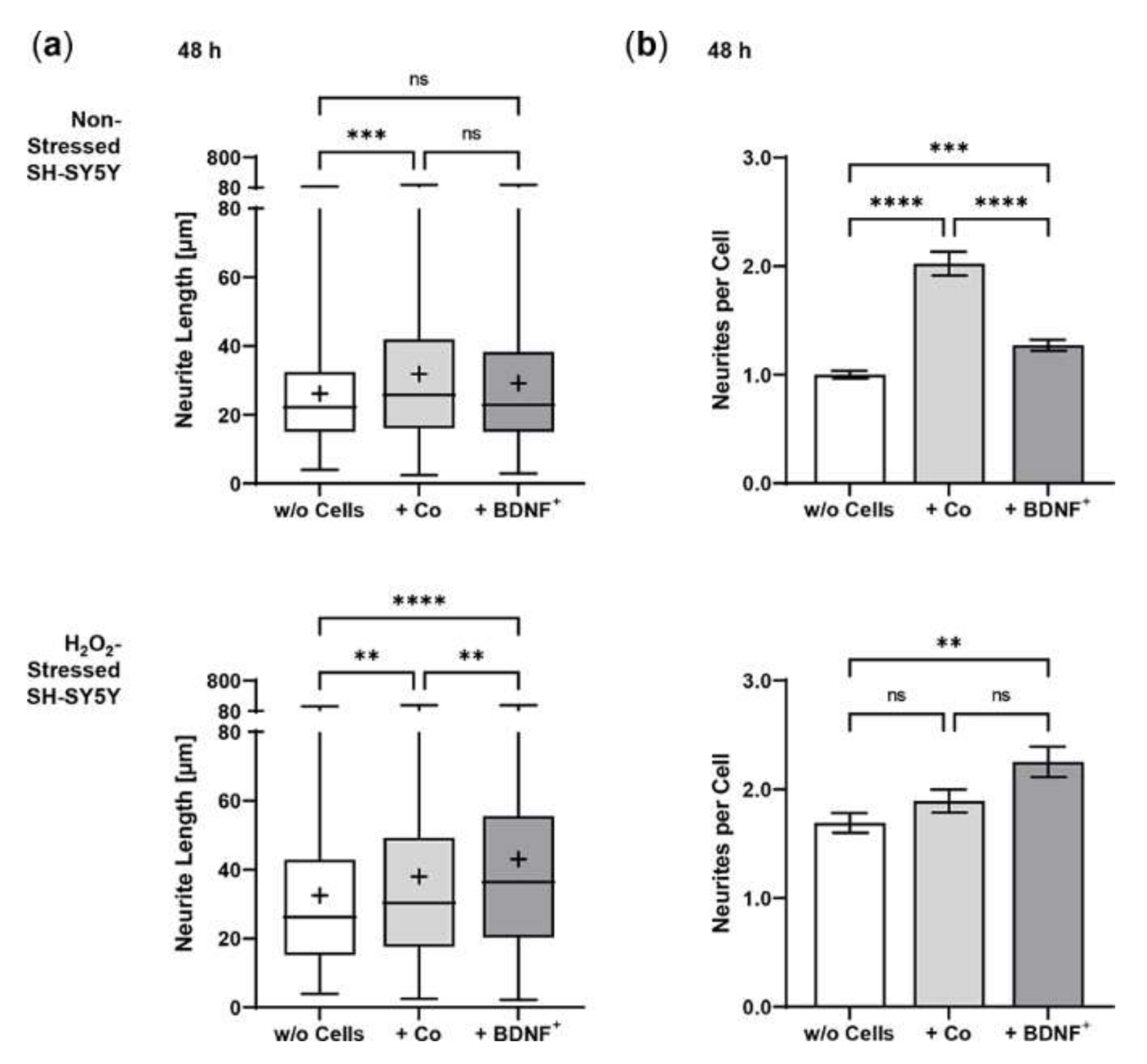
2.3. Neurite Outgrowth in SH-SY5Y Cells Co-Cultured with BDNF-Transfected ARPE-19 Cells
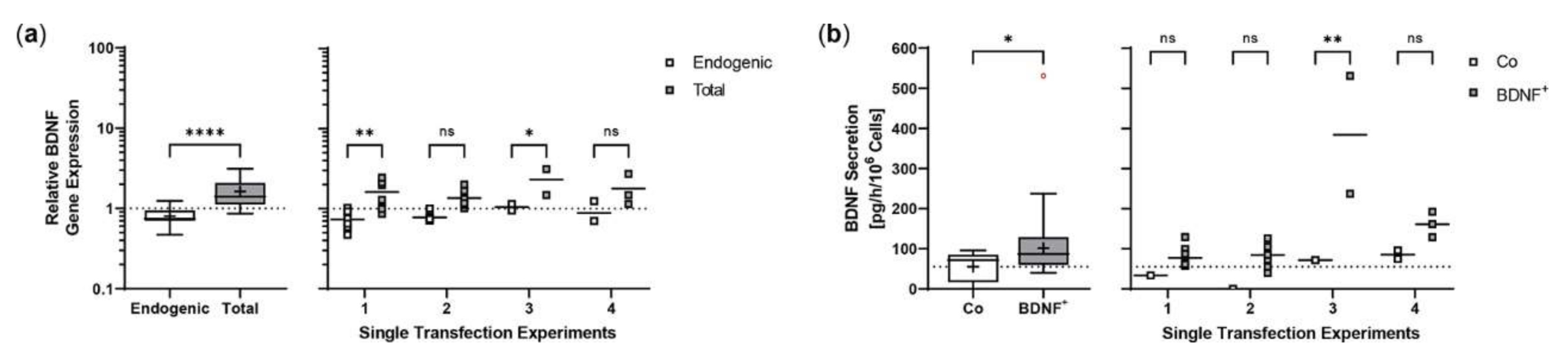
2.4. Sleeping Beauty Transposon-Mediated Delivery of the Human BDNF Gene into Primary Human RPE Cells
2.5. Neurite Outgrowth in SH-SY5Y Cells Co-Cultured with BDNF-Transfected Primary Human RPE Cells
3. Discussion
4. Materials and Methods
4.1. Plasmid Constructs
4.2. Cell Line Cultivation
4.3. Isolation and Cultivation of Primary Human RPE Cells
4.4. Electroporation of ARPE-19 Cells and Primary Human RPE Cells
4.5. SDS-PAGE and Western Blot Analysis
4.6. BDNF ELISA
4.7. Real-Time Quantitative PCR
4.8. Droplet Digital PCR
4.9. Cell Proliferation Assay
4.10. Annexin V/Propidium Iodide Assay
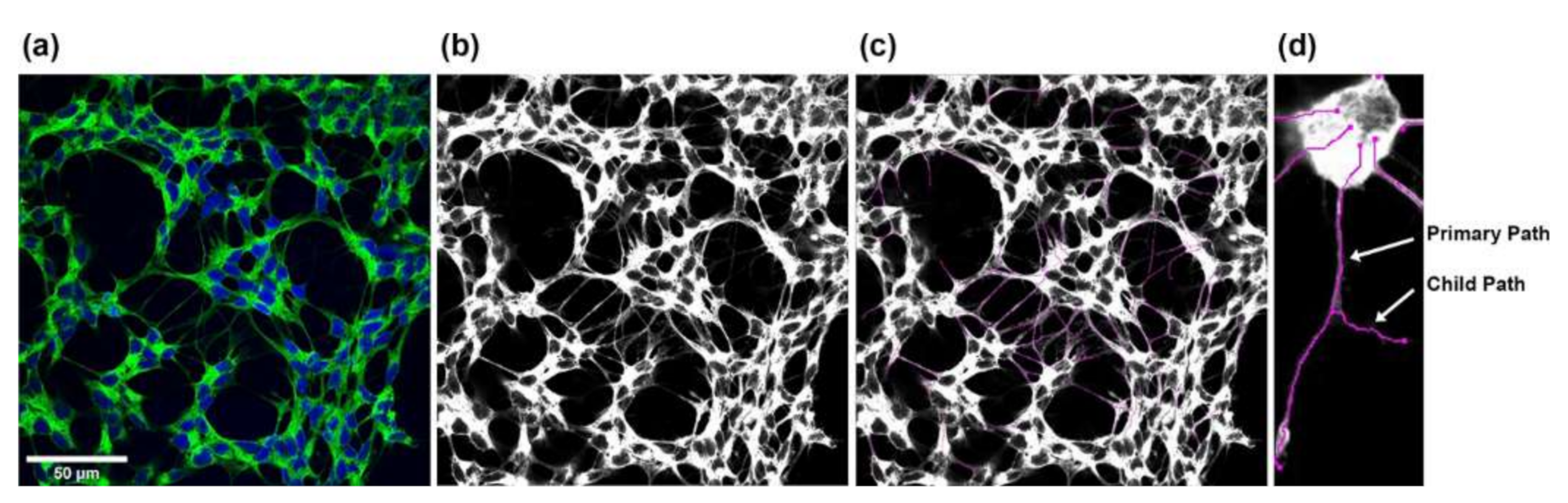
4.11. Co-Culture Model and Neurite Outgrowth Quantitation
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in Europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2020, 104, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Parravano, M.; Costanzo, E.; Scondotto, G.; Trifiro, G.; Virgili, G. Anti-VEGF and Other Novel Therapies for Neovascular Age-Related Macular Degeneration: An Update. BioDrugs 2021, 35, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Westby, K.; Csaky, K.G.; Mones, J.; Pearlman, J.A.; Patel, S.S.; Joondeph, B.C.; Randolph, J.; Masonson, H.; Rezaei, K.A. C5 Inhibitor Avacincaptad Pegol for Geographic Atrophy Due to Age-Related Macular Degeneration: A Randomized Pivotal Phase 2/3 Trial. Ophthalmology 2021, 128, 576–586. [Google Scholar] [CrossRef]
- Saad, L.; Washington, I. Can Vitamin A be Improved to Prevent Blindness due to Age-Related Macular Degeneration, Stargardt Disease and Other Retinal Dystrophies? Adv. Exp. Med. Biol. 2016, 854, 355–361. [Google Scholar] [CrossRef]
- Da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef]
- Kashani, A.H.; Uang, J.; Mert, M.; Rahhal, F.; Chan, C.; Avery, R.L.; Dugel, P.; Chen, S.; Lebkowski, J.; Clegg, D.O.; et al. Surgical Method for Implantation of a Biosynthetic Retinal Pigment Epithelium Monolayer for Geographic Atrophy: Experience from a Phase 1/2a Study. Ophthalmol. Retin. 2020, 4, 264–273. [Google Scholar] [CrossRef]
- Allingham, M.J.; Mettu, P.S.; Cousins, S.W. Phase 1 Clinical Trial of Elamipretide in Intermediate Age-Related Macular Degeneration and High-Risk Drusen. Ophthalmol. Sci. 2022, 2, 100095. [Google Scholar] [CrossRef]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Phase 1 Clinical Trial of Elamipretide in Dry Age-Related Macular Degeneration and Noncentral Geographic Atrophy. Ophthalmol. Sci. 2022, 2, 100086. [Google Scholar] [CrossRef]
- Patel, U.; Boucher, M.; de Leseleuc, L.; Visintini, S. Voretigene Neparvovec: An Emerging Gene Therapy for the Treatment of Inherited Blindness. In CADTH Issues in Emerging Health Technologies; CADTH: Ottawa, ON, Canada, 2016; pp. 1–11. [Google Scholar]
- Maguire, A.M.; Bennett, J.; Aleman, E.M.; Leroy, B.P.; Aleman, T.S. Clinical Perspective: Treating RPE65-Associated Retinal Dystrophy. Mol. Ther. 2021, 29, 442–463. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.; Buchberger, A.; Holder, J.; Orhan, e.; Hughes, J. GT005, a gene therapy for the treatment of dry age-related macular degeneration (AMD). Investig. Ophthalmol. Vis. Sci. 2020, 61, 2295. [Google Scholar]
- Campochiaro, P.A.; Lauer, A.K.; Sohn, E.H.; Mir, T.A.; Naylor, S.; Anderton, M.C.; Kelleher, M.; Harrop, R.; Ellis, S.; Mitrophanous, K.A. Lentiviral Vector Gene Transfer of Endostatin/Angiostatin for Macular Degeneration (GEM) Study. Hum. Gene Ther. 2017, 28, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Nguyen, Q.D.; Shah, S.M.; Klein, M.L.; Holz, E.; Frank, R.N.; Saperstein, D.A.; Gupta, A.; Stout, J.T.; Macko, J.; et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: Results of a phase I clinical trial. Hum. Gene Ther. 2006, 17, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Tombran-Tink, J.; Johnson, L.V. Neuronal differentiation of retinoblastoma cells induced by medium conditioned by human RPE cells. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1700–1707. [Google Scholar]
- Tombran-Tink, J.; Chader, G.G.; Johnson, L.V. PEDF: A pigment epithelium-derived factor with potent neuronal differentiative activity. Exp. Eye Res. 1991, 53, 411–414. [Google Scholar] [CrossRef]
- Becerra, S.P.; Dass, C.R.; Yabe, T.; Crawford, S.E. Pigment epithelium-derived factor: Chemistry, structure, biology, and applications. J. Biomed. Biotechnol. 2012, 2012, 830975. [Google Scholar] [CrossRef]
- Thumann, G.; Stöcker, M.; Maltusch, C.; Salz, A.K.; Barth, S.; Walter, P.; Johnen, S. High efficiency non-viral transfection of retinal and iris pigment epithelial cells with pigment epithelium-derived factor. Gene Ther. 2010, 17, 181–189. [Google Scholar] [CrossRef]
- Johnen, S.; Djalali-Talab, Y.; Kazanskaya, O.; Möller, T.; Harmening, N.; Kropp, M.; Izsvak, Z.; Walter, P.; Thumann, G. Antiangiogenic and Neurogenic Activities of Sleeping Beauty-Mediated PEDF-Transfected RPE Cells In Vitro and In Vivo. Biomed. Res. Int. 2015, 2015, 863845. [Google Scholar] [CrossRef]
- Kuerten, D.; Johnen, S.; Harmening, N.; Souteyrand, G.; Walter, P.; Thumann, G. Transplantation of PEDF-transfected pigment epithelial cells inhibits corneal neovascularization in a rabbit model. Graefes. Arch. Clin. Exp. Ophthalmol. 2015, 253, 1061–1069. [Google Scholar] [CrossRef]
- Hernandez, M.; Recalde, S.; Garcia-Garcia, L.; Bezunartea, J.; Miskey, C.; Johnen, S.; Diarra, S.; Sebe, A.; Rodriguez-Madoz, J.R.; Pouillot, S.; et al. Preclinical Evaluation of a Cell-Based Gene Therapy Using the Sleeping Beauty Transposon System in Choroidal Neovascularization. Mol. Ther. Methods Clin. Dev. 2019, 15, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Mates, L.; Chuah, M.K.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Johnen, S.; Izsvak, Z.; Stöcker, M.; Harmening, N.; Salz, A.K.; Walter, P.; Thumann, G. Sleeping Beauty transposon-mediated transfection of retinal and iris pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4787–4796. [Google Scholar] [CrossRef] [PubMed]
- Thumann, G.; Harmening, N.; Prat-Souteyrand, C.; Marie, C.; Pastor, M.; Sebe, A.; Miskey, C.; Hurst, L.D.; Diarra, S.; Kropp, M.; et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol. Ther. Nucleic Acids 2017, 6, 302–314. [Google Scholar] [CrossRef]
- Pastor, M.; Johnen, S.; Harmening, N.; Quiviger, M.; Pailloux, J.; Kropp, M.; Walter, P.; Ivics, Z.; Izsvak, Z.; Thumann, G.; et al. The Antibiotic-free pFAR4 Vector Paired with the Sleeping Beauty Transposon System Mediates Efficient Transgene Delivery in Human Cells. Mol. Ther. Nucleic Acids 2018, 11, 57–67. [Google Scholar] [CrossRef]
- Izsvak, Z.; Hackett, P.B.; Cooper, L.J.; Ivics, Z. Translating Sleeping Beauty transposition into cellular therapies: Victories and challenges. Bioessays 2010, 32, 756–767. [Google Scholar] [CrossRef]
- Levy, Y.S.; Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Therapeutic potential of neurotrophic factors in neurodegenerative diseases. BioDrugs 2005, 19, 97–127. [Google Scholar] [CrossRef]
- Ernfors, P.; Wetmore, C.; Olson, L.; Persson, H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron 1990, 5, 511–526. [Google Scholar] [CrossRef]
- Hofer, M.; Pagliusi, S.R.; Hohn, A.; Leibrock, J.; Barde, Y.A. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990, 9, 2459–2464. [Google Scholar] [CrossRef]
- Phillips, H.S.; Hains, J.M.; Laramee, G.R.; Rosenthal, A.; Winslow, J.W. Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science 1990, 250, 290–294. [Google Scholar] [CrossRef]
- Gall, C.M.; Gold, S.J.; Isackson, P.J.; Seroogy, K.B. Brain-derived neurotrophic factor and neurotrophin-3 mRNAs are expressed in ventral midbrain regions containing dopaminergic neurons. Mol. Cell. Neurosci. 1992, 3, 56–63. [Google Scholar] [CrossRef]
- Bennett, J.L.; Zeiler, S.R.; Jones, K.R. Patterned expression of BDNF and NT-3 in the retina and anterior segment of the developing mammalian eye. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2996–3005. [Google Scholar]
- Germana, A.; Sanchez-Ramos, C.; Guerrera, M.C.; Calavia, M.G.; Navarro, M.; Zichichi, R.; Garcia-Suarez, O.; Perez-Pinera, P.; Vega, J.A. Expression and cell localization of brain-derived neurotrophic factor and TrkB during zebrafish retinal development. J. Anat. 2010, 217, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Nawa, H.; Fukuchi, T.; Abe, H.; Takei, N. BDNF is upregulated by postnatal development and visual experience: Quantitative and immunohistochemical analyses of BDNF in the rat retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3211–3218. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Telegina, D.V.; Kolosova, N.G.; Kozhevnikova, O.S. Immunohistochemical localization of NGF, BDNF, and their receptors in a normal and AMD-like rat retina. BMC Med. Genom. 2019, 12, 48. [Google Scholar] [CrossRef]
- Foltran, R.B.; Diaz, S.L. BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 2016, 138, 204–221. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Kowianski, P.; Lietzau, G.; Czuba, E.; Waskow, M.; Steliga, A.; Morys, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Du, X.D.; Zhang, Y.; Yin, G.; Zhang, B.S.; Soares, J.C.; Zhang, X.Y. Association of low serum BDNF with depression in patients with Parkinson’s disease. Park. Relat. Disord. 2017, 41, 73–78. [Google Scholar] [CrossRef]
- Ola, M.S.; Nawaz, M.I.; El-Asrar, A.A.; Abouammoh, M.; Alhomida, A.S. Reduced levels of brain derived neurotrophic factor (BDNF) in the serum of diabetic retinopathy patients and in the retina of diabetic rats. Cell. Mol. Neurobiol. 2013, 33, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Inanc Tekin, M.; Sekeroglu, M.A.; Demirtas, C.; Tekin, K.; Doguizi, S.; Bayraktar, S.; Yilmazbas, P. Brain-Derived Neurotrophic Factor in Patients With Age-Related Macular Degeneration and Its Correlation With Retinal Layer Thicknesses. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Shpak, A.A.; Guekht, A.B.; Druzhkova, T.A.; Kozlova, K.I.; Gulyaeva, N.V. Brain-Derived Neurotrophic Factor in Patients with Primary Open-Angle Glaucoma and Age-related Cataract. Curr. Eye Res. 2018, 43, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Osborne, A.; Khatib, T.Z.; Songra, L.; Barber, A.C.; Hall, K.; Kong, G.Y.X.; Widdowson, P.S.; Martin, K.R. Neuroprotection of retinal ganglion cells by a novel gene therapy construct that achieves sustained enhancement of brain-derived neurotrophic factor/tropomyosin-related kinase receptor-B signaling. Cell Death Dis. 2018, 9, 1007. [Google Scholar] [CrossRef]
- Chiha, W.; Bartlett, C.A.; Petratos, S.; Fitzgerald, M.; Harvey, A.R. Intravitreal application of AAV-BDNF or mutant AAV-CRMP2 protects retinal ganglion cells and stabilizes axons and myelin after partial optic nerve injury. Exp. Neurol. 2020, 326, 113167. [Google Scholar] [CrossRef]
- Wojcik-Gryciuk, A.; Gajewska-Wozniak, O.; Kordecka, K.; Boguszewski, P.M.; Waleszczyk, W.; Skup, M. Neuroprotection of Retinal Ganglion Cells with AAV2-BDNF Pretreatment Restoring Normal TrkB Receptor Protein Levels in Glaucoma. Int. J. Mol. Sci. 2020, 21, 6262. [Google Scholar] [CrossRef]
- Grabundzija, I.; Irgang, M.; Mates, L.; Belay, E.; Matrai, J.; Gogol-Döring, A.; Kawakami, K.; Chen, W.; Ruiz, P.; Chuah, M.K.; et al. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 2010, 18, 1200–1209. [Google Scholar] [CrossRef]
- Galla, M.; Schambach, A.; Falk, C.S.; Maetzig, T.; Kuehle, J.; Lange, K.; Zychlinski, D.; Heinz, N.; Brugman, M.H.; Gohring, G.; et al. Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res. 2011, 39, 7147–7160. [Google Scholar] [CrossRef]
- Gogol-Döring, A.; Ammar, I.; Gupta, S.; Bunse, M.; Miskey, C.; Chen, W.; Uckert, W.; Schulz, T.F.; Izsvak, Z.; Ivics, Z. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4(+) T Cells. Mol. Ther. 2016, 24, 592–606. [Google Scholar] [CrossRef]
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z.; et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019, 37, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Djuzenova, C.S.; Zimmermann, U.; Frank, H.; Sukhorukov, V.L.; Richter, E.; Fuhr, G. Effect of medium conductivity and composition on the uptake of propidium iodide into electropermeabilized myeloma cells. Biochim. Biophys. Acta 1996, 1284, 143–152. [Google Scholar] [CrossRef]
- Yao, S.; Rana, S.; Liu, D.; Wise, G.E. Electroporation optimization to deliver plasmid DNA into dental follicle cells. Biotechnol. J. 2009, 4, 1488–1496. [Google Scholar] [CrossRef]
- Sherba, J.J.; Hogquist, S.; Lin, H.; Shan, J.W.; Shreiber, D.I.; Zahn, J.D. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Sci. Rep. 2020, 10, 3053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiu, S.; Zhang, X.; Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Gibot, L.; Montigny, A.; Baaziz, H.; Fourquaux, I.; Audebert, M.; Rols, M.P. Calcium Delivery by Electroporation Induces In Vitro Cell Death through Mitochondrial Dysfunction without DNA Damages. Cancers 2020, 12, 425. [Google Scholar] [CrossRef] [PubMed]
- Polajzer, T.; Jarm, T.; Miklavcic, D. Analysis of damage-associated molecular pattern molecules due to electroporation of cells in vitro. Radiol. Oncol. 2020, 54, 317–328. [Google Scholar] [CrossRef]
- Wiencke, A.K.; Kiilgaard, J.F.; Nicolini, J.; Bundgaard, M.; Ropke, C.; La Cour, M. Growth of cultured porcine retinal pigment epithelial cells. Acta Ophthalmol. Scand. 2003, 81, 170–176. [Google Scholar] [CrossRef]
- Chopra, S.; Ruzgys, P.; Maciulevičius, M.; Jakutavičiūtė, M.; Šatkauskas, S. Investigation of Plasmid DNA Delivery and Cell Viability Dynamics for Optimal Cell Electrotransfection In Vitro. Appl. Sci. 2020, 10, 6070. [Google Scholar] [CrossRef]
- Kreiss, P.; Cameron, B.; Rangara, R.; Mailhe, P.; Aguerre-Charriol, O.; Airiau, M.; Scherman, D.; Crouzet, J.; Pitard, B. Plasmid DNA size does not affect the physicochemical properties of lipoplexes but modulates gene transfer efficiency. Nucleic Acids Res. 1999, 27, 3792–3798. [Google Scholar] [CrossRef]
- Chabot, S.; Orio, J.; Schmeer, M.; Schleef, M.; Golzio, M.; Teissie, J. Minicircle DNA electrotransfer for efficient tissue-targeted gene delivery. Gene Ther. 2013, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Schulze, J.; Warwas, D.P.; Ehlert, N.; Lenarz, T.; Warnecke, A.; Behrens, P. Long-term delivery of brain-derived neurotrophic factor (BDNF) from nanoporous silica nanoparticles improves the survival of spiral ganglion neurons in vitro. PLoS ONE 2018, 13, e0194778. [Google Scholar] [CrossRef] [PubMed]
- Geissler, C.; Blumenstock, M.; Gabrielpillai, J.; Guchlerner, L.; Stover, T.; Diensthuber, M. Simultaneous treatment with pentoxifylline does not adversely affect the neurotrophic effects of brain-derived neurotrophic factor on spiral ganglion neurons. Neuroreport 2021, 32, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Ru, J.; Ma, W.; Gao, Y.; Liang, Z.; Liu, J.; Guo, J.H.; Li, L.Y. BDNF promotes the growth of human neurons through crosstalk with the Wnt/beta-catenin signaling pathway via GSK-3beta. Neuropeptides 2015, 54, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.; Smith, T.N.; Wang, N.; Chua, J.Y.; Westbroek, E.; Wang, K.; Guzman, R. BDNF Pretreatment of Human Embryonic-Derived Neural Stem Cells Improves Cell Survival and Functional Recovery After Transplantation in Hypoxic-Ischemic Stroke. Cell Transpl. 2015, 24, 2449–2461. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Sung, D.K.; Chang, Y.S.; Sung, S.I.; Kim, Y.E.; Kim, H.J.; Lee, S.M.; Park, W.S. BDNF-Overexpressing Engineered Mesenchymal Stem Cells Enhances Their Therapeutic Efficacy against Severe Neonatal Hypoxic Ischemic Brain Injury. Int. J. Mol. Sci. 2021, 22, 11395. [Google Scholar] [CrossRef]
- Lim, W.; Bae, H.; Bazer, F.W.; Song, G. Brain-derived neurotrophic factor improves proliferation of endometrial epithelial cells by inhibition of endoplasmic reticulum stress during early pregnancy. J. Cell. Physiol. 2017, 232, 3641–3651. [Google Scholar] [CrossRef]
- Chen, S.; Wang, F.; Liu, Z.; Zhao, Y.; Jiang, Y.; Chen, L.; Li, C.; Zhou, X. Brain-derived neurotrophic factor promotes proliferation and progesterone synthesis in bovine granulosa cells. J. Cell. Physiol. 2019, 234, 8776–8787. [Google Scholar] [CrossRef]
- Paris, A.J.; Hayer, K.E.; Oved, J.H.; Avgousti, D.C.; Toulmin, S.A.; Zepp, J.A.; Zacharias, W.J.; Katzen, J.B.; Basil, M.C.; Kremp, M.M.; et al. STAT3-BDNF-TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat. Cell Biol. 2020, 22, 1197–1210. [Google Scholar] [CrossRef]
- Ogura, Y.; Sato, K.; Kawashima, K.; Kobayashi, N.; Imura, S.; Fujino, K.; Kawaguchi, H.; Nedachi, T. Subtoxic levels of hydrogen peroxide induce brain-derived neurotrophic factor expression to protect PC12 cells. BMC Res. Notes 2014, 7, 840. [Google Scholar] [CrossRef]
- Cabral de Guimaraes, T.A.; Daich Varela, M.; Georgiou, M.; Michaelides, M. Treatments for dry age-related macular degeneration: Therapeutic avenues, clinical trials and future directions. Br. J. Ophthalmol. 2022, 106, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Kashani, A.H. Stem cell-derived retinal pigment epithelium transplantation in age-related macular degeneration: Recent advances and challenges. Curr. Opin. Ophthalmol. 2022, 33, 211–218. [Google Scholar] [CrossRef]
- Domenici, L.; Origlia, N.; Falsini, B.; Cerri, E.; Barloscio, D.; Fabiani, C.; Sanso, M.; Giovannini, L. Rescue of retinal function by BDNF in a mouse model of glaucoma. PLoS ONE 2014, 9, e115579. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, H.; Yi, J.; Troy, J.B.; Zhang, H.F.; Liu, X. Long-Term Protection of Retinal Ganglion Cells and Visual Function by Brain-Derived Neurotrophic Factor in Mice With Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Hackett, S.F.; Friedman, Z.; Freund, J.; Schoenfeld, C.; Curtis, R.; DiStefano, P.S.; Campochiaro, P.A. A splice variant of trkB and brain-derived neurotrophic factor are co-expressed in retinal pigmented epithelial cells and promote differentiated characteristics. Brain Res. 1998, 789, 201–212. [Google Scholar] [CrossRef]
- Liu, Z.Z.; Zhu, L.Q.; Eide, F.F. Critical role of TrkB and brain-derived neurotrophic factor in the differentiation and survival of retinal pigment epithelium. J. Neurosci. 1997, 17, 8749–8755. [Google Scholar] [CrossRef] [PubMed]
- Inana, G.; Murat, C.; An, W.; Yao, X.; Harris, I.R.; Cao, J. RPE phagocytic function declines in age-related macular degeneration and is rescued by human umbilical tissue derived cells. J. Transl. Med. 2018, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Yenice, E.; Sengun, A.; Soyugelen Demirok, G.; Turacli, E. Ganglion cell complex thickness in nonexudative age-related macular degeneration. Eye 2015, 29, 1076–1080. [Google Scholar] [CrossRef]
- Sullivan, R.K.; Woldemussie, E.; Pow, D.V. Dendritic and synaptic plasticity of neurons in the human age-related macular degeneration retina. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2782–2791. [Google Scholar] [CrossRef]
- Chen, D.W.; Foldvari, M. In vitro bioassay model for screening non-viral neurotrophic factor gene delivery systems for glaucoma treatment. Drug Deliv. Transl. Res. 2016, 6, 676–685. [Google Scholar] [CrossRef]
- Rahimmi, A.; Peluso, I.; Rajabi, A.; Hassanzadeh, K. miR-185 and SEPT5 Genes May Contribute to Parkinson’s Disease Pathophysiology. Oxidative Med. Cell. Longev. 2019, 2019, 5019815. [Google Scholar] [CrossRef]
- Arslan, M.E.; Turkez, H.; Mardinoglu, A. In vitro neuroprotective effects of farnesene sesquiterpene on alzheimer’s disease model of differentiated neuroblastoma cell line. Int. J. Neurosci. 2021, 131, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y. Tauroursodeoxycholic Acid Alleviates H2O2-Induced Oxidative Stress and Apoptosis via Suppressing Endoplasmic Reticulum Stress in Neonatal Rat Cardiomyocytes. Dose-Response 2018, 16, 1559325818782631. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Chen, P.Y.; Way, T.D.; Cheng, C.L.; Huang, Y.P.; Hsia, T.C.; Chou, Y.C.; Peng, S.F. Pre-Treatment of Pterostilbene Enhances H2O2-induced Cell Apoptosis Through Caspase-dependent Pathway in Human Keratinocyte Cells. In Vivo 2021, 35, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Encinas, M.; Iglesias, M.; Llecha, N.; Comella, J.X. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J. Neurochem. 1999, 73, 1409–1421. [Google Scholar] [CrossRef]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Liu, S.; Guo, W.; Zhou, H.; Tang, L.; Feng, S.; Zhong, J.H.; Zhou, X.F. proBDNF inhibits the proliferation and migration of OLN93 oligodendrocytes. Mol. Med. Rep. 2018, 18, 3809–3817. [Google Scholar] [CrossRef]
- Yang, B.; Wang, L.; Nie, Y.; Wei, W.; Xiong, W. proBDNF expression induces apoptosis and inhibits synaptic regeneration by regulating the RhoA-JNK pathway in an in vitro post-stroke depression model. Transl. Psychiatry 2021, 11, 578. [Google Scholar] [CrossRef]
- Frank, L.; Ventimiglia, R.; Anderson, K.; Lindsay, R.M.; Rudge, J.S. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur. J. Neurosci. 1996, 8, 1220–1230. [Google Scholar] [CrossRef]
- Knusel, B.; Gao, H.; Okazaki, T.; Yoshida, T.; Mori, N.; Hefti, F.; Kaplan, D.R. Ligand-induced down-regulation of Trk messenger RNA, protein and tyrosine phosphorylation in rat cortical neurons. Neuroscience 1997, 78, 851–862. [Google Scholar] [CrossRef]
- Chen, H.; Weber, A.J. Brain-derived neurotrophic factor reduces TrkB protein and mRNA in the normal retina and following optic nerve crush in adult rats. Brain Res. 2004, 1011, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, M.; Tur, M.K.; Sasse, S.; Krussmann, A.; Barth, S.; Engert, A. Secretion of functional anti-CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein. Expr. Purif. 2003, 28, 211–219. [Google Scholar] [CrossRef]
- Johnen, S.; Harmening, N.; Marie, C.; Scherman, D.; Izsvak, Z.; Ivics, Z.; Walter, P.; Thumann, G. Electroporation-Based Genetic Modification of Primary Human Pigment Epithelial Cells using the Sleeping Beauty Transposon System. J. Vis. Exp. 2021, 168, e61987. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Thomson, A.C.; Schuhmann, T.; de Graaf, T.A.; Sack, A.T.; Rutten, B.P.F.; Kenis, G. The Effects of Serum Removal on Gene Expression and Morphological Plasticity Markers in Differentiated SH-SY5Y Cells. Cell. Mol. Neurobiol. 2021, 42, 1829–1839. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Longair, M.H.; Baker, D.A.; Armstrong, J.D. Simple Neurite Tracer: Open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 2011, 27, 2453–2454. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattern, L.; Otten, K.; Miskey, C.; Fuest, M.; Izsvák, Z.; Ivics, Z.; Walter, P.; Thumann, G.; Johnen, S. Molecular and Functional Characterization of BDNF-Overexpressing Human Retinal Pigment Epithelial Cells Established by Sleeping Beauty Transposon-Mediated Gene Transfer. Int. J. Mol. Sci. 2022, 23, 12982. https://doi.org/10.3390/ijms232112982
Mattern L, Otten K, Miskey C, Fuest M, Izsvák Z, Ivics Z, Walter P, Thumann G, Johnen S. Molecular and Functional Characterization of BDNF-Overexpressing Human Retinal Pigment Epithelial Cells Established by Sleeping Beauty Transposon-Mediated Gene Transfer. International Journal of Molecular Sciences. 2022; 23(21):12982. https://doi.org/10.3390/ijms232112982
Chicago/Turabian StyleMattern, Larissa, Katrin Otten, Csaba Miskey, Matthias Fuest, Zsuzsanna Izsvák, Zoltán Ivics, Peter Walter, Gabriele Thumann, and Sandra Johnen. 2022. "Molecular and Functional Characterization of BDNF-Overexpressing Human Retinal Pigment Epithelial Cells Established by Sleeping Beauty Transposon-Mediated Gene Transfer" International Journal of Molecular Sciences 23, no. 21: 12982. https://doi.org/10.3390/ijms232112982
APA StyleMattern, L., Otten, K., Miskey, C., Fuest, M., Izsvák, Z., Ivics, Z., Walter, P., Thumann, G., & Johnen, S. (2022). Molecular and Functional Characterization of BDNF-Overexpressing Human Retinal Pigment Epithelial Cells Established by Sleeping Beauty Transposon-Mediated Gene Transfer. International Journal of Molecular Sciences, 23(21), 12982. https://doi.org/10.3390/ijms232112982