
A Dpagt1 Missense Variant Causes Degenerative Retinopathy without Myasthenic Syndrome in Mice

 , , , ,
, , , ,  , and
, and 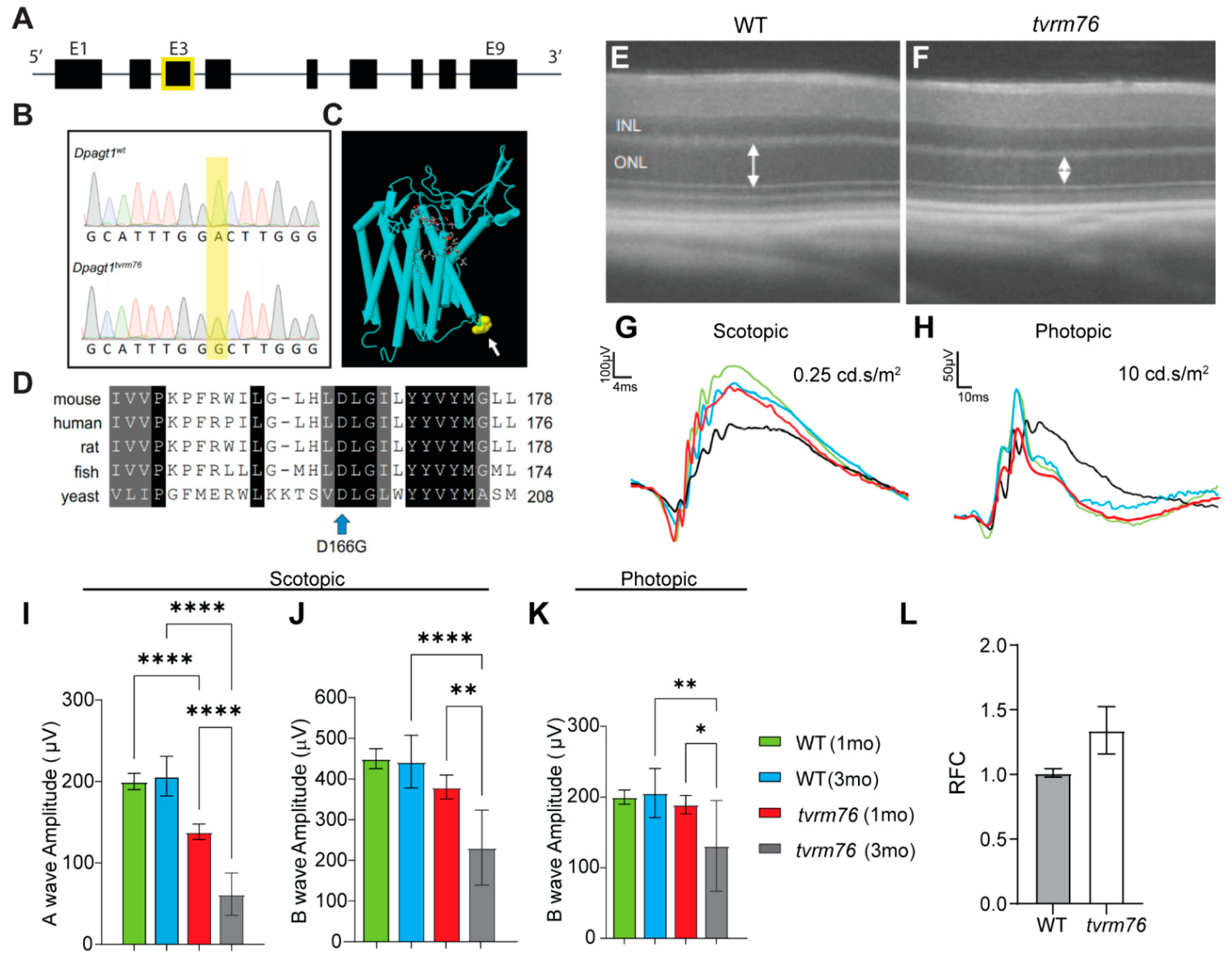{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
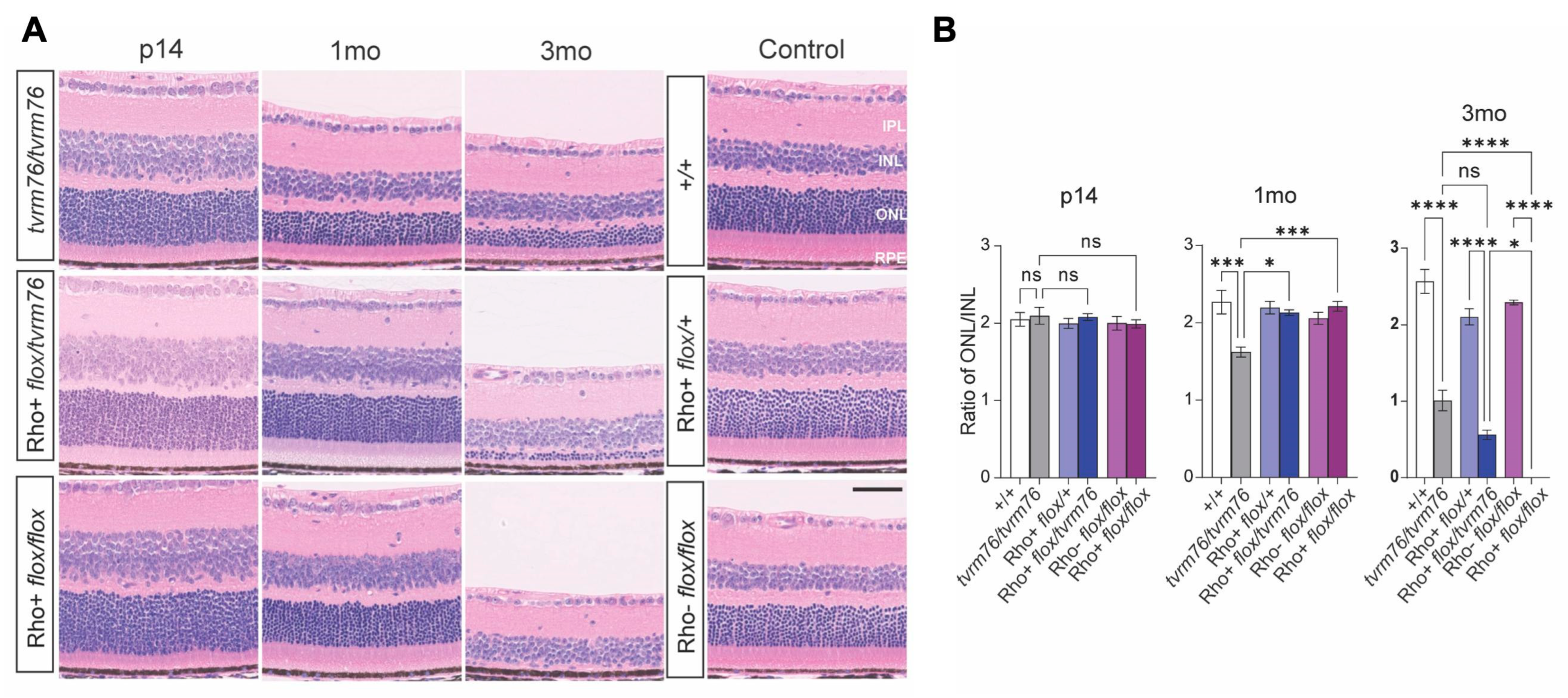
2.1. Homozygosity of the Dpagt1tvrm76 Allele Causes Early Onset Retinal Degeneration
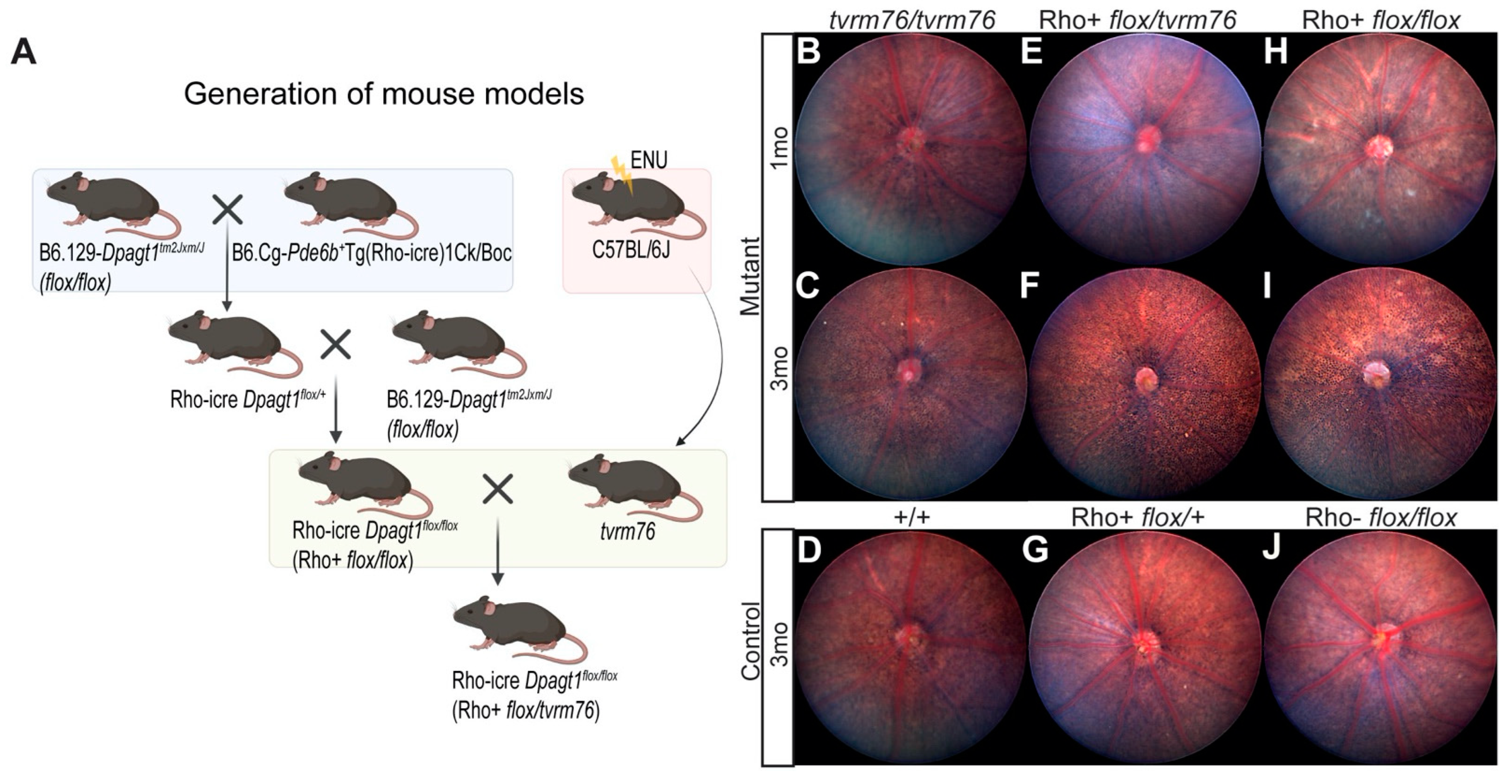
2.2. Rod Photoreceptor Specific Loss of DPAGT1 Function Causes Early Onset Retinal Degeneration
2.3. A Complementation Test Shows That the Dpagt1tvrm76 Mutation Causes the Retinal Degeneration
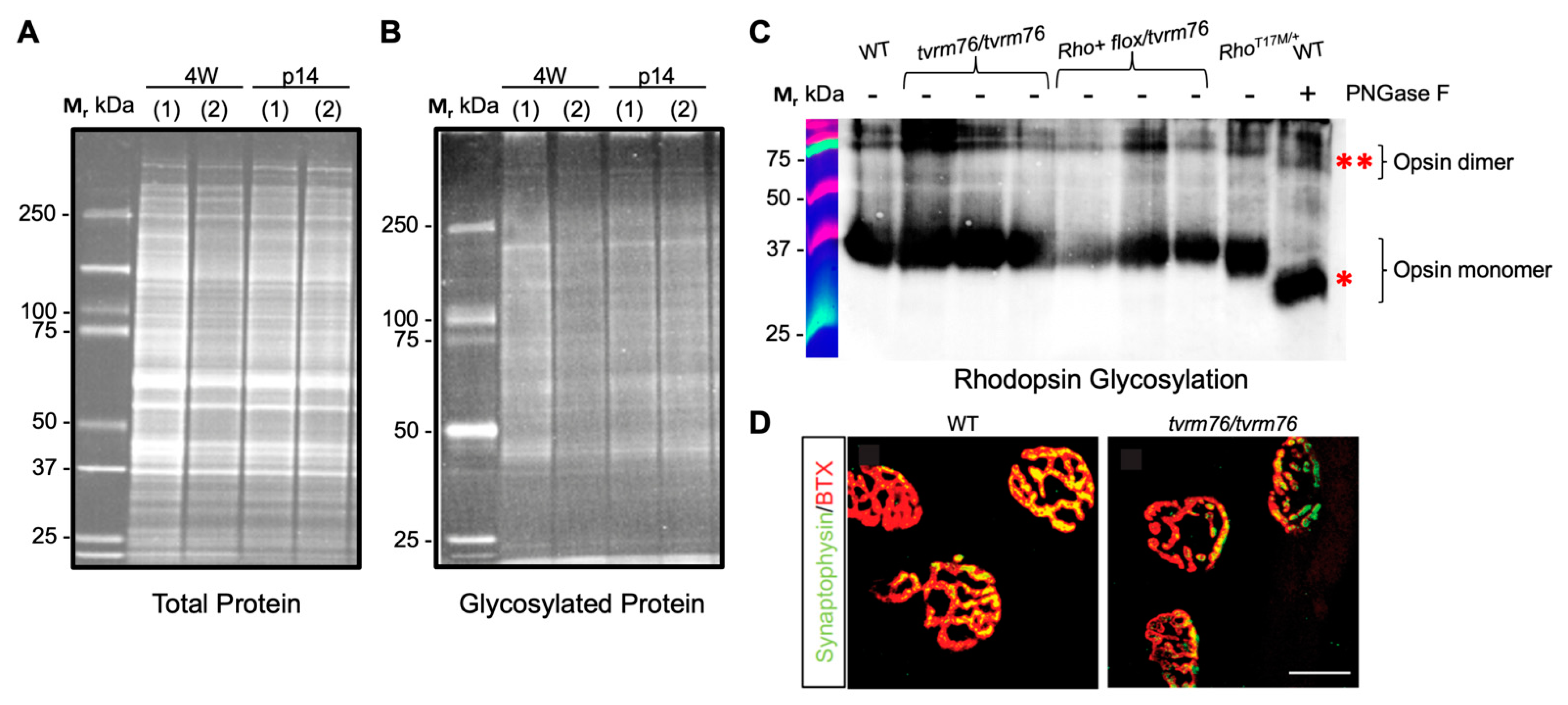
2.4. The Dpagt1tvrm76 Variant Is Not Associated with Aberrant Protein Glycosylation at an Early Age
2.5. Homozgyous Dpagt1tvrm76 Mice Do Not Recapitulate Early Muscular Abnormalities Observed in Patients with DPAGT1 Mutations
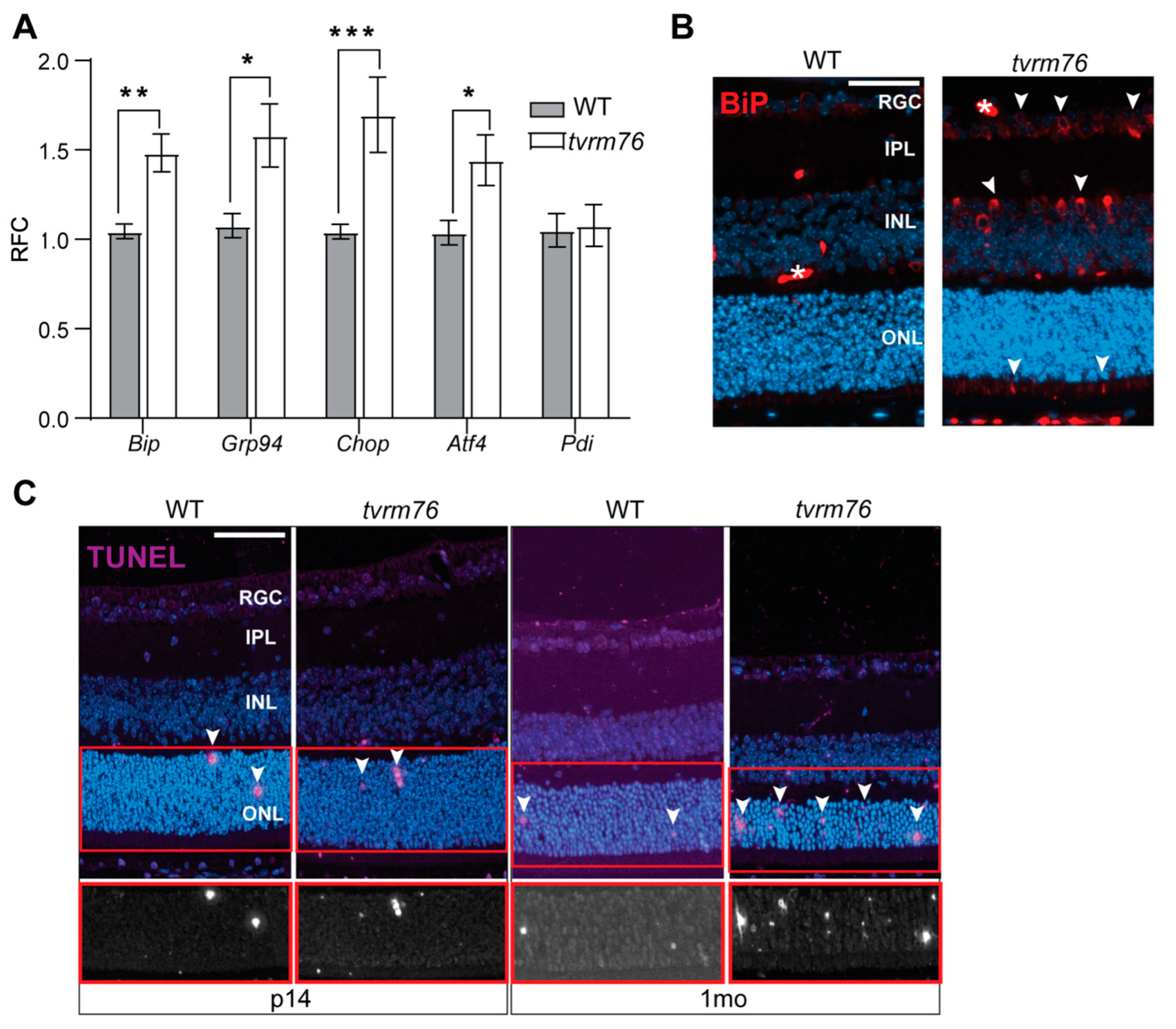
2.6. Homozygous Dpagt1tvrm76 Eyes Show Signs of ER Stress
2.7. Increased Number of Apoptotic Cells Are Detected in Dpagt1tvrm76 Retinas
3. Discussion
4. Materials and Methods
4.1. Mice, Mutagenesis, and Husbandry
4.2. Identification of the Molecular Basis of tvrm76
4.3. Phenotyping
4.4. Histology
4.5. Electron Microscopy
4.6. RNA Extraction and Analysis
4.7. Immunofluorescence
4.8. Antibodies
4.9. Western Blot
4.10. Image Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, B.G.; Freeze, H.H. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet. 2018, 34, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Francisco, R.; Brasil, S.; Marques-da-Silva, D.; Rijoff, T.; Pascoal, C.; Jaeken, J.; Videira, P.A.; Dos Reis Ferreira, V. Stakeholders’ views on drug development: The congenital disorders of glycosylation community perspective. Orphanet. J. Rare Dis. 2022, 17, 303. [Google Scholar] [CrossRef] [PubMed]
- Francisco, R.; Marques-da-Silva, D.; Brasil, S.; Pascoal, C.; Dos Reis Ferreira, V.; Morava, E.; Jaeken, J. The challenge of CDG diagnosis. Mol. Genet. Metab. 2019, 126, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H.; Jaeken, J.; Matthijs, G. CDG or not CDG. J. Inherit. Metab. Dis. 2022, 45, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Peanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Perez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Lefeber, D.J.; Freeze, H.H.; Steet, R.; Kinoshita, T. Congenital Disorders of Glycosylation. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.J., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 599–614. Available online: https://www.ncbi.nlm.nih.gov/books/NBK579918/ (accessed on 3 October 2022).
- Park, J.H.; Marquardt, T. Treatment Options in Congenital Disorders of Glycosylation. Front. Genet. 2021, 12, 735348. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.W.; Johnsen, C.; Morava, E. Nutrition interventions in congenital disorders of glycosylation. Trends. Mol. Med. 2022, 28, 463–481. [Google Scholar] [CrossRef]
- Janssen, M.C.; de Kleine, R.H.; van den Berg, A.P.; Heijdra, Y.; van Scherpenzeel, M.; Lefeber, D.J.; Morava, E. Successful liver transplantation and long-term follow-up in a patient with MPI-CDG. Pediatrics 2014, 134, e279–e283. [Google Scholar] [CrossRef]
- Chang, I.J.; He, M.; Lam, C.T. Congenital disorders of glycosylation. Ann. Transl. Med. 2018, 6, 477–490. [Google Scholar] [CrossRef]
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef]
- Wilson, M.P.; Matthijs, G. The evolving genetic landscape of congenital disorders of glycosylation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129976. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Lefeber, D.; Matthijs, G. Clinical utility gene card for: DPAGT1 defective congenital disorder of glycosylation. Eur. J. Hum. Genet. 2015, 23, 1749–1752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Selcen, D.; Shen, X.M.; Brengman, J.; Li, Y.; Stans, A.A.; Wieben, E.; Engel, A.G. DPAGT1 myasthenia and myopathy: Genetic, phenotypic, and expression studies. Neurology 2014, 82, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.Y.; Wang, H.; Pike, A.C.W.; Cochrane, S.A.; Hamedzadeh, S.; Wyszynski, F.J.; Bushell, S.R.; Royer, S.F.; Widdick, D.A.; Sajid, A.; et al. Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design. Cell 2018, 175, 1045–1058. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell. Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Paprocka, J.; Jezela-Stanek, A.; Tylki-Szymanska, A.; Grunewald, S. Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci. 2021, 11, 88. [Google Scholar] [CrossRef]
- Belaya, K.; Finlayson, S.; Slater, C.R.; Cossins, J.; Liu, W.W.; Maxwell, S.; McGowan, S.J.; Maslau, S.; Twigg, S.R.; Walls, T.J.; et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am. J. Hum. Genet. 2012, 91, 193–201. [Google Scholar] [CrossRef]
- Belaya, K.; Finlayson, S.; Cossins, J.; Liu, W.W.; Maxwell, S.; Palace, J.; Beeson, D. Identification of DPAGT1 as a new gene in which mutations cause a congenital myasthenic syndrome. Ann. N. Y. Acad. Sci. 2012, 1275, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Vega, A.I.; Martin-Higueras, C.; Medrano, C.; Gamez, A.; Desviat, L.R.; Ugarte, M.; Perez-Cerda, C.; Perez, B. DPAGT1-CDG: Functional analysis of disease-causing pathogenic mutations and role of endoplasmic reticulum stress. PLoS ONE 2017, 12, e0179456. [Google Scholar] [CrossRef]
- Messenger, W.B.; Yang, P.; Pennesi, M.E. Ophthalmic findings in an infant with phosphomannomutase deficiency. Doc. Ophthalmol. 2014, 128, 149–153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thompson, D.A.; Lyons, R.J.; Russell-Eggitt, I.; Liasis, A.; Jagle, H.; Grunewald, S. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J. Inherit. Metab. Dis. 2013, 36, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, A.; Keenan, R.W.; Elbein, A.D. Mechanism of action of tunicamycin on the UDP-GlcNAc: Dolichyl-phosphate GlcNAc-1-phosphate transferase. Biochemistry 1979, 18, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J.; Basinger, S.F. Tunicamycin blocks the incorporation of opsin into retinal rod outer segment membranes. Proc. Natl. Acad. Sci. USA 1985, 82, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J.; Rapp, L.M.; Hollyfield, J.G. Photoreceptor-specific degeneration caused by tunicamycin. Nature 1984, 311, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.W.; Vijay, I.K.; Marth, J.D. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology 1999, 9, 1263–1271. [Google Scholar] [CrossRef]
- Krebs, M.P.; Collin, G.B.; Hicks, W.L.; Yu, M.; Charette, J.R.; Shi, L.Y.; Wang, J.; Naggert, J.K.; Peachey, N.S.; Nishina, P.M. Mouse models of human ocular disease for translational research. PLoS ONE 2017, 12, e0183837. [Google Scholar] [CrossRef]
- Won, J.; Shi, L.Y.; Hicks, W.; Wang, J.; Hurd, R.; Naggert, J.K.; Chang, B.; Nishina, P.M. Mouse model resources for vision research. J. Ophthalmol. 2011, 2011, 391384. [Google Scholar] [CrossRef]
- Won, J.; Gifford, E.; Smith, R.S.; Yi, H.; Ferreira, P.A.; Hicks, W.L.; Li, T.; Naggert, J.K.; Nishina, P.M. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum. Mol. Genet. 2009, 18, 4329–4339. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vis. Res 2002, 42, 517–525. [Google Scholar] [CrossRef]
- Salom, D.; Jin, H.; Gerken, T.A.; Yu, C.; Huang, L.; Palczewski, K. Human red and green cone opsins are O-glycosylated at an N-terminal Ser/Thr–rich domain conserved in vertebrates. J. Biol. Chem. 2019, 294, 8123–8133. [Google Scholar] [CrossRef]
- Murray, A.R.; Vuong, L.; Brobst, D.; Fliesler, S.J.; Peachey, N.S.; Gorbatyuk, M.S.; Naash, M.I.; Al-Ubaidi, M.R. Glycosylation of rhodopsin is necessary for its stability and incorporation into photoreceptor outer segment discs. Hum. Mol. Genet. 2015, 24, 2709–2723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berson, E.L.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Sandberg, M.A. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3027–3036. [Google Scholar]
- Nashine, S.; Bhootada, Y.; Lewin, A.S.; Gorbatyuk, M. Ablation of C/EBP homologous protein does not protect T17M RHO mice from retinal degeneration. PLoS ONE 2013, 8, e63205. [Google Scholar] [CrossRef] [PubMed]
- Michele, D.E.; Barresi, R.; Kanagawa, M.; Saito, F.; Cohn, R.D.; Satz, J.S.; Dollar, J.; Nishino, I.; Kelley, R.I.; Somer, H. Post-translational disruption of dystroglycan–ligand interactions in congenital muscular dystrophies. Nature 2002, 418, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Shinde, V.M.; Starr, C.R.; Kruglov, A.A.; Boitet, E.R.; Kotla, P.; Zolotukhin, S.; Gross, A.K.; Gorbatyuk, M.S. An activated unfolded protein response promotes retinal degeneration and triggers an inflammatory response in the mouse retina. Cell Death Dis. 2014, 5, e1578. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, H.; Jia, Y.; Pan, H.; Ding, H. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother. Pharmacol. 2017, 79, 1031–1041. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Li, H.; You, L.; Tian, Y.; Guo, J.; Fang, X.; Zhou, C.; Shi, L.; Su, Y.Q. DPAGT1-Mediated Protein N-Glycosylation Is Indispensable for Oocyte and Follicle Development in Mice. Adv. Sci. 2020, 7, 2000531. [Google Scholar] [CrossRef]
- Lehle, L.; Strahl, S.; Tanner, W. Protein glycosylation, conserved from yeast to man: A model organism helps elucidate congenital human diseases. Angew. Chem. Int. Ed. 2006, 45, 6802–6818. [Google Scholar] [CrossRef]
- Ramachandra Rao, S.; Skelton, L.A.; Wu, F.; Onysk, A.; Spolnik, G.; Danikiewicz, W.; Butler, M.C.; Stacks, D.A.; Surmacz, L.; Mu, X.; et al. Retinal Degeneration Caused by Rod-Specific Dhdds Ablation Occurs without Concomitant Inhibition of Protein N-Glycosylation. iScience 2020, 23, 101198. [Google Scholar] [CrossRef] [PubMed]
- Nicole, S.; Azuma, Y.; Bauche, S.; Eymard, B.; Lochmuller, H.; Slater, C. Congenital Myasthenic Syndromes or Inherited Disorders of Neuromuscular Transmission: Recent Discoveries and Open Questions. J. Neuromuscul. Dis. 2017, 4, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Ng, B.G.; Underhill, H.R.; Palm, L.; Bengtson, P.; Rozet, J.M.; Gerber, S.; Munnich, A.; Zanlonghi, X.; Stevens, C.A.; Kircher, M.; et al. DPAGT1 Deficiency with Encephalopathy (DPAGT1-CDG): Clinical and Genetic Description of 11 New Patients. JIMD Rep. 2019, 44, 85–92. [Google Scholar]
- Engel, A.G. Genetic basis and phenotypic features of congenital myasthenic syndromes. Handb. Clin. Neurol. 2018, 148, 565–589. [Google Scholar]
- Issop, Y.; Hathazi, D.; Khan, M.M.; Rudolf, R.; Weis, J.; Spendiff, S.; Slater, C.R.; Roos, A.; Lochmüller, H. GFPT1 deficiency in muscle leads to myasthenia and myopathy in mice. Hum. Mol. Genet. 2018, 27, 3218–3232. [Google Scholar] [CrossRef]
- Gidaro, T.; Vandenbrande, L.; Malfatti, E.; Labasse, C.; Carlier, P.; Romero, N.; Servais, L.; Böhm, J. New homozygous mutation in DPAGT1 gene leading to LG-CMS with tubular aggregates. Neuromuscul. Disord. 2018, 28, S51. [Google Scholar] [CrossRef]
- Nicolau, S.; Kao, J.C.; Liewluck, T. Trouble at the junction: When myopathy and myasthenia overlap. Muscle Nerve 2019, 60, 648–657. [Google Scholar] [CrossRef]
- Bogdanova-Mihaylova, P.; Murphy, R.; Alexander, M.; McHugh, J.; Foley, A.R.; Brett, F.; Murphy, S. Congenital myasthenic syndrome due to DPAGT1 mutations mimicking congenital myopathy in an Irish family. Eur. J. Neurol. 2018, 25, e22–e23. [Google Scholar] [CrossRef]
- Kanaki, N.; Matsuda, A.; Dejima, K.; Murata, D.; Nomura, K.H.; Ohkura, T.; Gengyo-Ando, K.; Yoshina, S.; Mitani, S.; Nomura, K. UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase is indispensable for oogenesis, oocyte-to-embryo transition, and larval development of the nematode Caenorhabditis elegans. Glycobiology 2019, 29, 163–178. [Google Scholar] [CrossRef]
- Finlayson, S.; Palace, J.; Belaya, K.; Walls, T.J.; Norwood, F.; Burke, G.; Holton, J.L.; Pascual-Pascual, S.I.; Cossins, J.; Beeson, D. Clinical features of congenital myasthenic syndrome due to mutations in DPAGT1. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Basiri, K.; Belaya, K.; Liu, W.W.; Maxwell, S.; Sedghi, M.; Beeson, D. Clinical features in a large Iranian family with a limb-girdle congenital myasthenic syndrome due to a mutation in DPAGT1. Neuromuscul. Disord. 2013, 23, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Estephan, E.P.; Zambon, A.A.; Thompson, R.; Polavarapu, K.; Jomaa, D.; Töpf, A.; Helito, P.V.; Heise, C.O.; Moreno, C.A.; Silva, A.M. Congenital myasthenic syndrome: Correlation between clinical features and molecular diagnosis. Eur. J. Neurol. 2022, 29, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cimmaruta, C.; Monticelli, M.; Riccio, G.; Hay Mele, B.; Cubellis, M.V.; Andreotti, G. The analysis of variants in the general population reveals that PMM2 is extremely tolerant to missense mutations and that diagnosis of PMM2-CDG can benefit from the identification of modifiers. Int. J. Mol. Sci. 2018, 19, 2218. [Google Scholar] [CrossRef]
- Eichler, J. Protein glycosylation. Curr. Biol. 2019, 29, R229–R231. [Google Scholar] [CrossRef]
- Van Landuyt, L.; Lonigro, C.; Meuris, L.; Callewaert, N. Customized protein glycosylation to improve biopharmaceutical function and targeting. Curr. Opin. Biotechnol. 2019, 60, 17–28. [Google Scholar] [CrossRef]
- Esmail, S.; Manolson, M.F. Advances in understanding N-glycosylation structure, function, and regulation in health and disease. Eur. J. Cell Biol. 2021, 100, 151186. [Google Scholar] [CrossRef]
- Qi, Z.; Chen, L. Endoplasmic reticulum stress and autophagy. Autophagy Biol. Dis. 2019, 1206, 167–177. [Google Scholar]
- Choy, K.W.; Murugan, D.; Mustafa, M.R. Natural products targeting ER stress pathway for the treatment of cardiovascular diseases. Pharmacol. Res. 2018, 132, 119–129. [Google Scholar] [CrossRef]
- Medinas, D.B.; Rozas, P.; Traub, F.M.; Woehlbier, U.; Brown, R.H.; Bosco, D.A.; Hetz, C. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 8209–8214. [Google Scholar] [CrossRef]
- Lebedeva, I.V.; Wagner, M.V.; Sahdeo, S.; Lu, Y.-F.; Anyanwu-Ofili, A.; Matthew, B. Harms, M.B.; Wadia, J.S.; Rajagopal, G.; Boland, M.J.; et al. Precision genetic cellular models identify therapies protective against ER stress. Cell Death Dis. 2021, 12, 770–781. [Google Scholar] [CrossRef]
- Fairfield, H.; Gilbert, G.J.; Barter, M.; Corrigan, R.R.; Curtain, M.; Ding, Y.; D’Ascenzo, M.; Gerhardt, D.J.; He, C.; Huang, W.; et al. Mutation discovery in mice by whole exome sequencing. Genome Biol. 2011, 12, R86. [Google Scholar] [CrossRef] [PubMed]
- Hawes, N.L.; Chang, B.; Hageman, G.S.; Nusinowitz, S.; Nishina, P.M.; Schneider, B.S.; Smith, R.S.; Roderick, T.H.; Davisson, M.T.; Heckenlively, J.R. Retinal degeneration 6 (rd6): A new mouse model for human retinitis punctata albescens. Invest. Ophthalmol. Vis. Sci. 2000, 41, 3149–3157. [Google Scholar] [PubMed]
- Krebs, M.P.; Xiao, M.; Sheppard, K.; Hicks, W.; Nishina, P.M. Bright-Field Imaging and Optical Coherence Tomography of the Mouse Posterior Eye. Methods Mol. Biol. 2016, 1438, 395–415. [Google Scholar] [PubMed]
- Kong, Y.; Zhao, L.; Charette, J.R.; Hicks, W.L.; Stone, L.; Nishina, P.M.; Naggert, J.K. An FRMD4B variant suppresses dysplastic photoreceptor lesions in models of enhanced S-cone syndrome and of Nrl deficiency. Hum. Mol. Genet. 2018, 27, 3340–3352. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, H.; Yamamoto, K.; Nozato, S.; Inagaki, T.; Tsuchimochi, H.; Shirai, M.; Yamamoto, R.; Imaizumi, Y.; Hongyo, K.; Yokoyama, S.; et al. Modified forelimb grip strength test detects aging-associated physiological decline in skeletal muscle function in male mice. Sci. Rep. 2017, 7, 42323. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.W.; Cox, G.A.; Seburn, K.L. Neuromuscular disease models and analysis. Methods Mol. Biol. 2010, 602, 347–393. [Google Scholar] [PubMed]
- Motley, W.W.; Seburn, K.L.; Nawaz, M.H.; Miers, K.E.; Cheng, J.; Antonellis, A.; Green, E.D.; Talbot, K.; Yang, X.L.; Fischbeck, K.H.; et al. Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 2011, 7, e1002399. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyde, L.F.; Kong, Y.; Zhao, L.; Rao, S.R.; Wang, J.; Stone, L.; Njaa, A.; Collin, G.B.; Krebs, M.P.; Chang, B.; et al. A Dpagt1 Missense Variant Causes Degenerative Retinopathy without Myasthenic Syndrome in Mice. Int. J. Mol. Sci. 2022, 23, 12005. https://doi.org/10.3390/ijms231912005
Hyde LF, Kong Y, Zhao L, Rao SR, Wang J, Stone L, Njaa A, Collin GB, Krebs MP, Chang B, et al. A Dpagt1 Missense Variant Causes Degenerative Retinopathy without Myasthenic Syndrome in Mice. International Journal of Molecular Sciences. 2022; 23(19):12005. https://doi.org/10.3390/ijms231912005
Chicago/Turabian StyleHyde, Lillian F., Yang Kong, Lihong Zhao, Sriganesh Ramachandra Rao, Jieping Wang, Lisa Stone, Andrew Njaa, Gayle B. Collin, Mark P. Krebs, Bo Chang, and et al. 2022. "A Dpagt1 Missense Variant Causes Degenerative Retinopathy without Myasthenic Syndrome in Mice" International Journal of Molecular Sciences 23, no. 19: 12005. https://doi.org/10.3390/ijms231912005
APA StyleHyde, L. F., Kong, Y., Zhao, L., Rao, S. R., Wang, J., Stone, L., Njaa, A., Collin, G. B., Krebs, M. P., Chang, B., Fliesler, S. J., Nishina, P. M., & Naggert, J. K. (2022). A Dpagt1 Missense Variant Causes Degenerative Retinopathy without Myasthenic Syndrome in Mice. International Journal of Molecular Sciences, 23(19), 12005. https://doi.org/10.3390/ijms231912005