

The Binding of CSL Proteins to Either Co-Activators or Co-Repressors Protects from Proteasomal Degradation Induced by MAPK-Dependent Phosphorylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
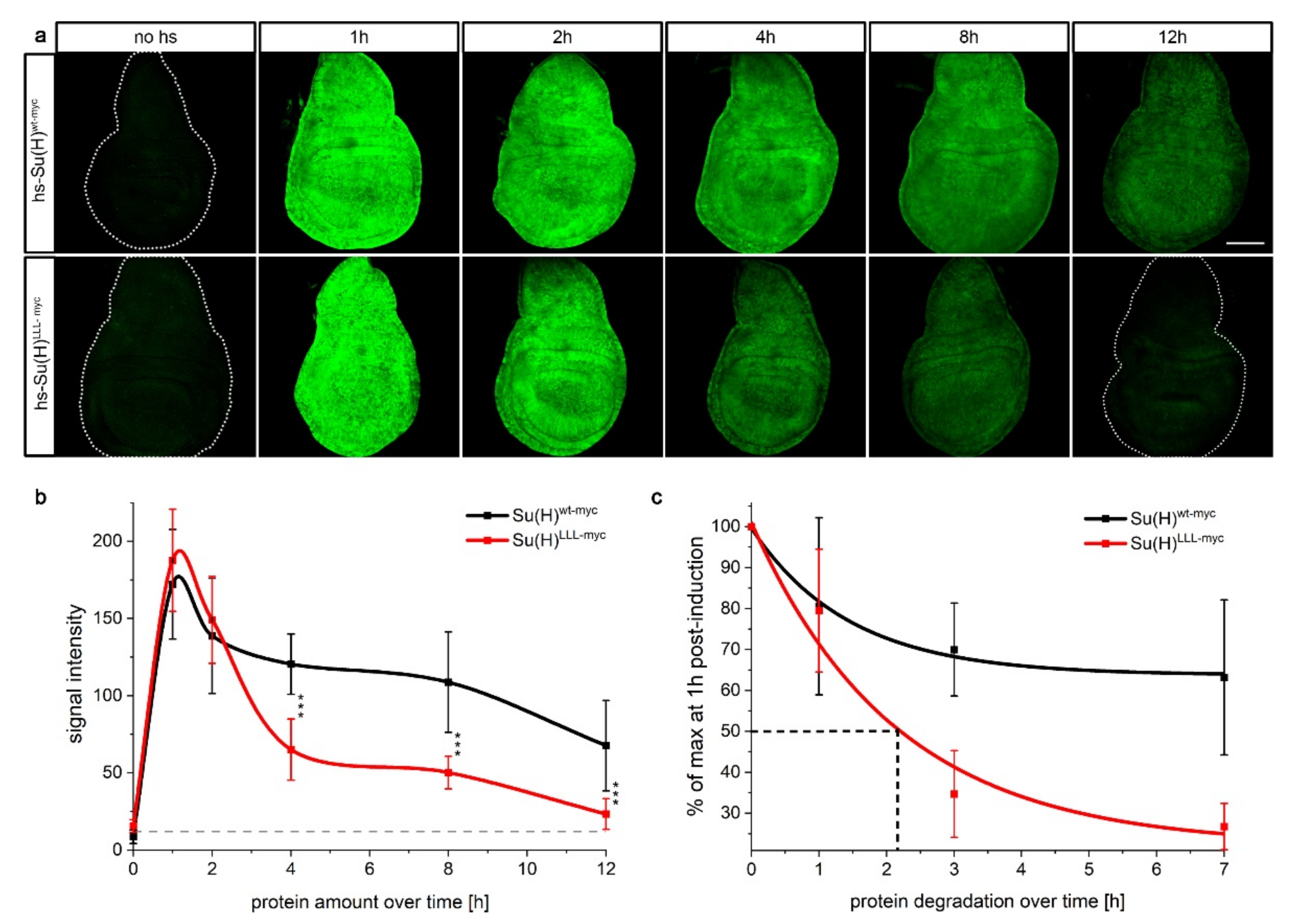
2.1. H Binding-Defective Su(H)LLL Protein Is Degraded More Rapidly Than Wildtype Su(H) Protein
2.2. Su(H) Protein Is Subjected to Proteasomal Degradation
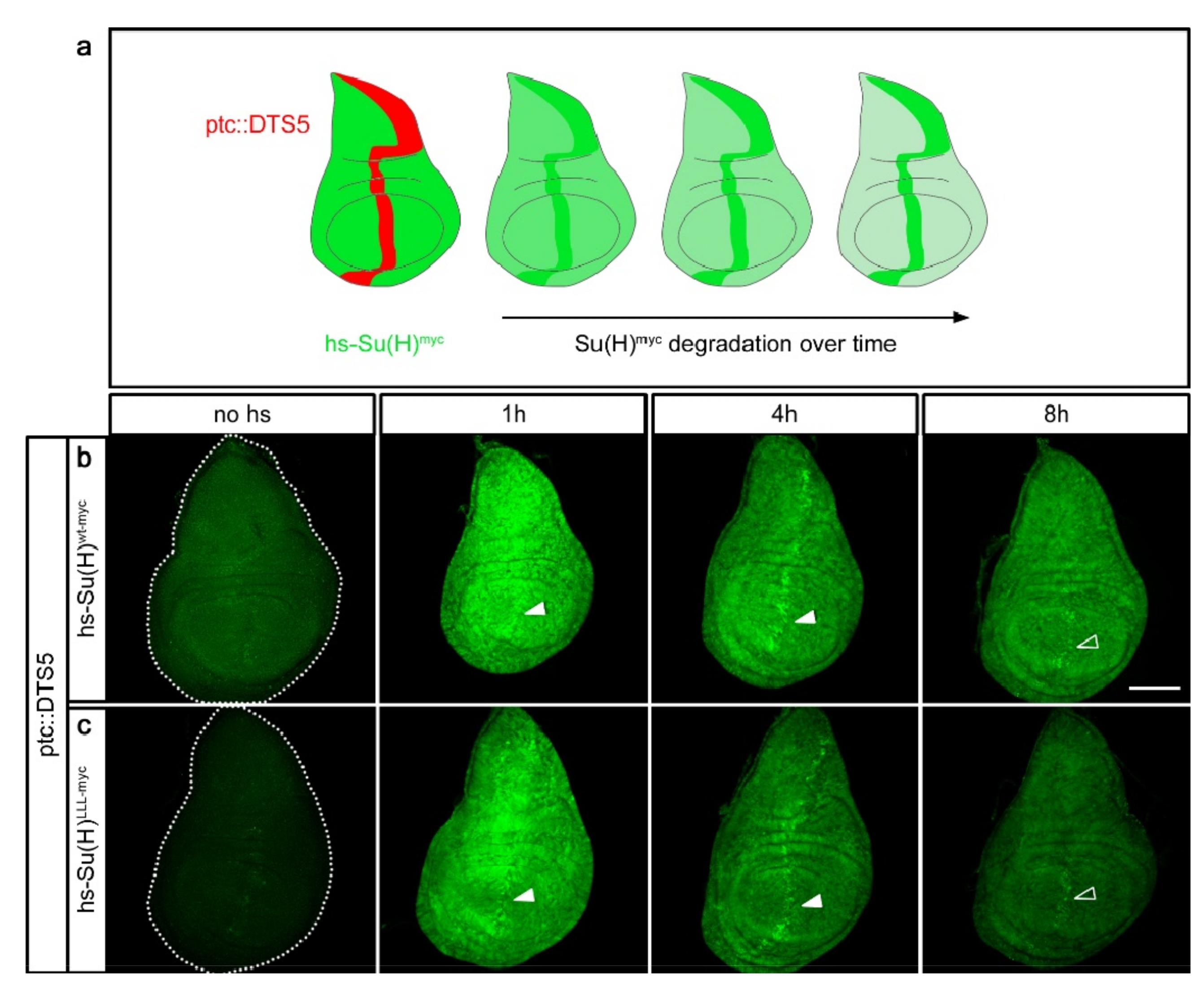
2.3. Su(H) Protein Levels Depend on the Presence of H
2.4. Su(H)–H Protein Interactions Are Short-Lived
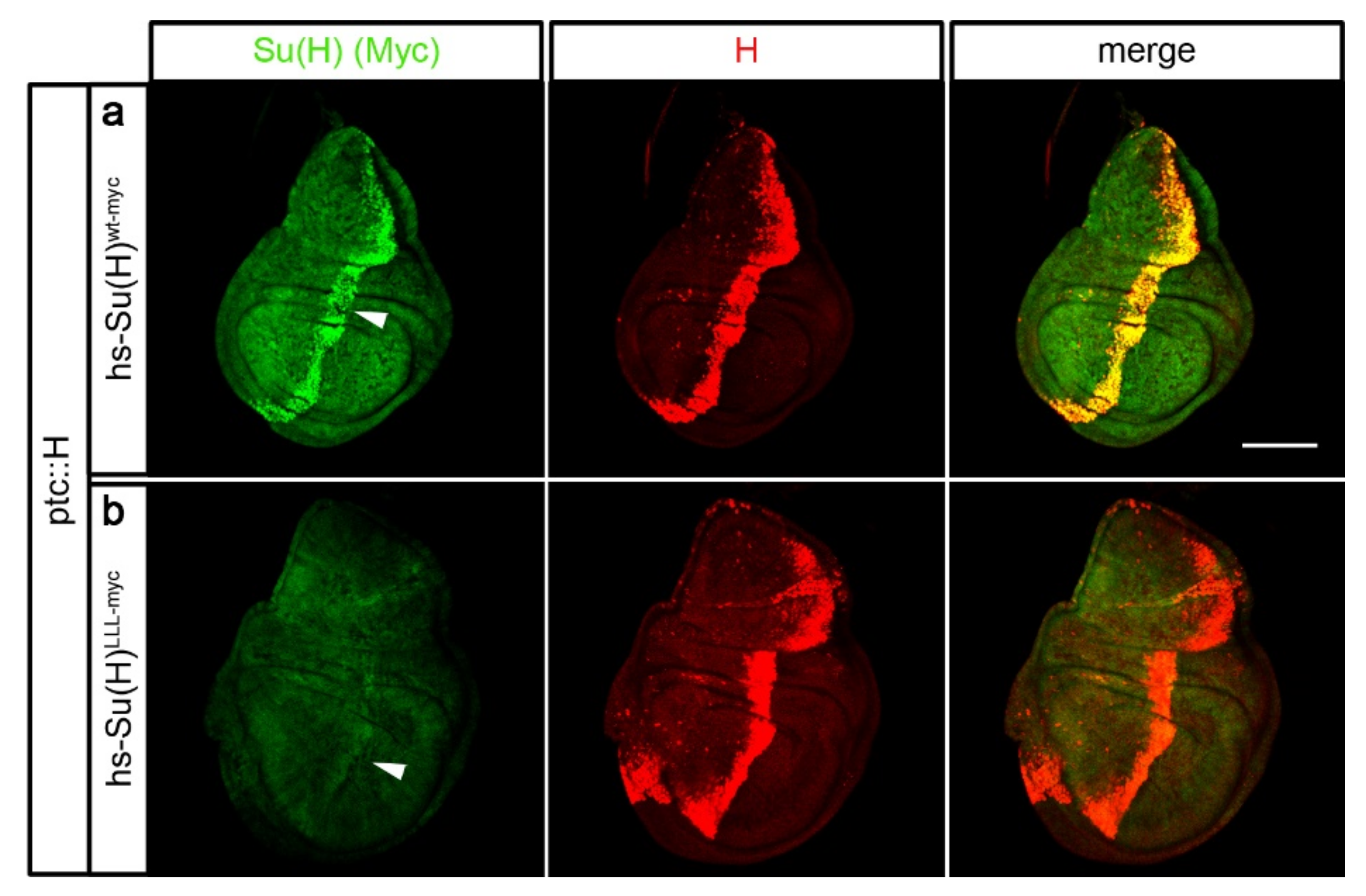
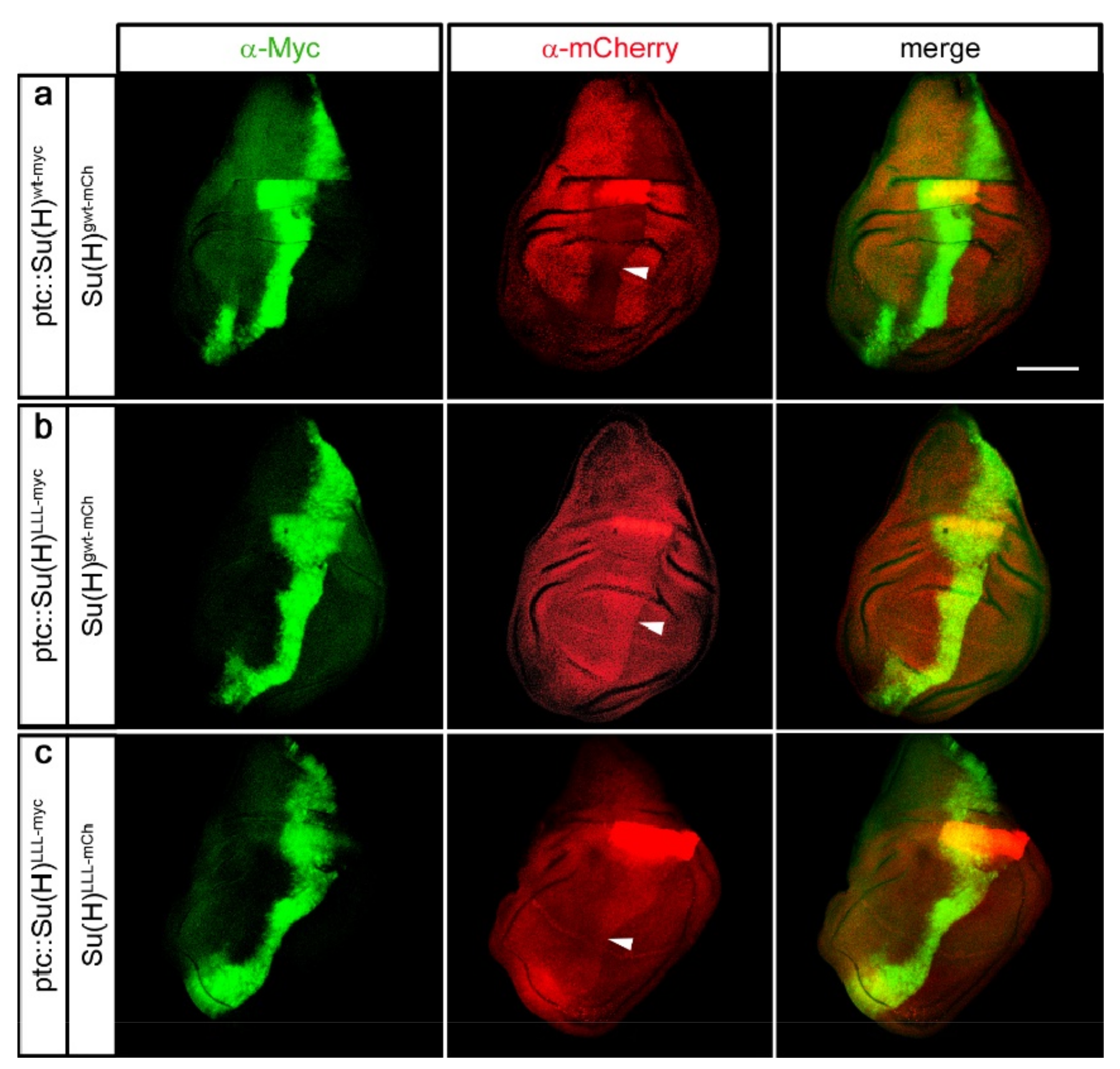
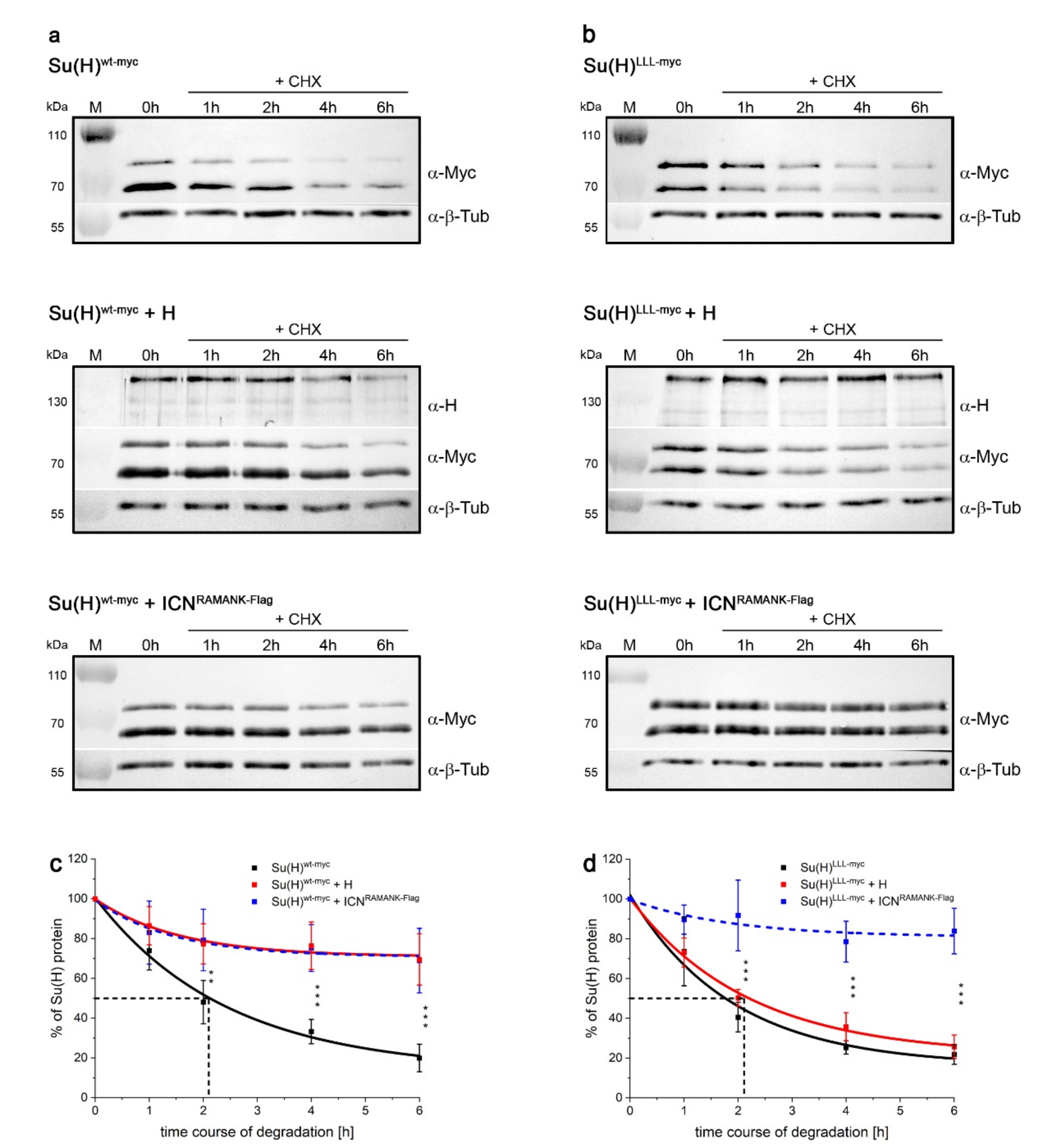
2.5. Protection of Su(H) by Co-Activator and Co-Repressor Alike
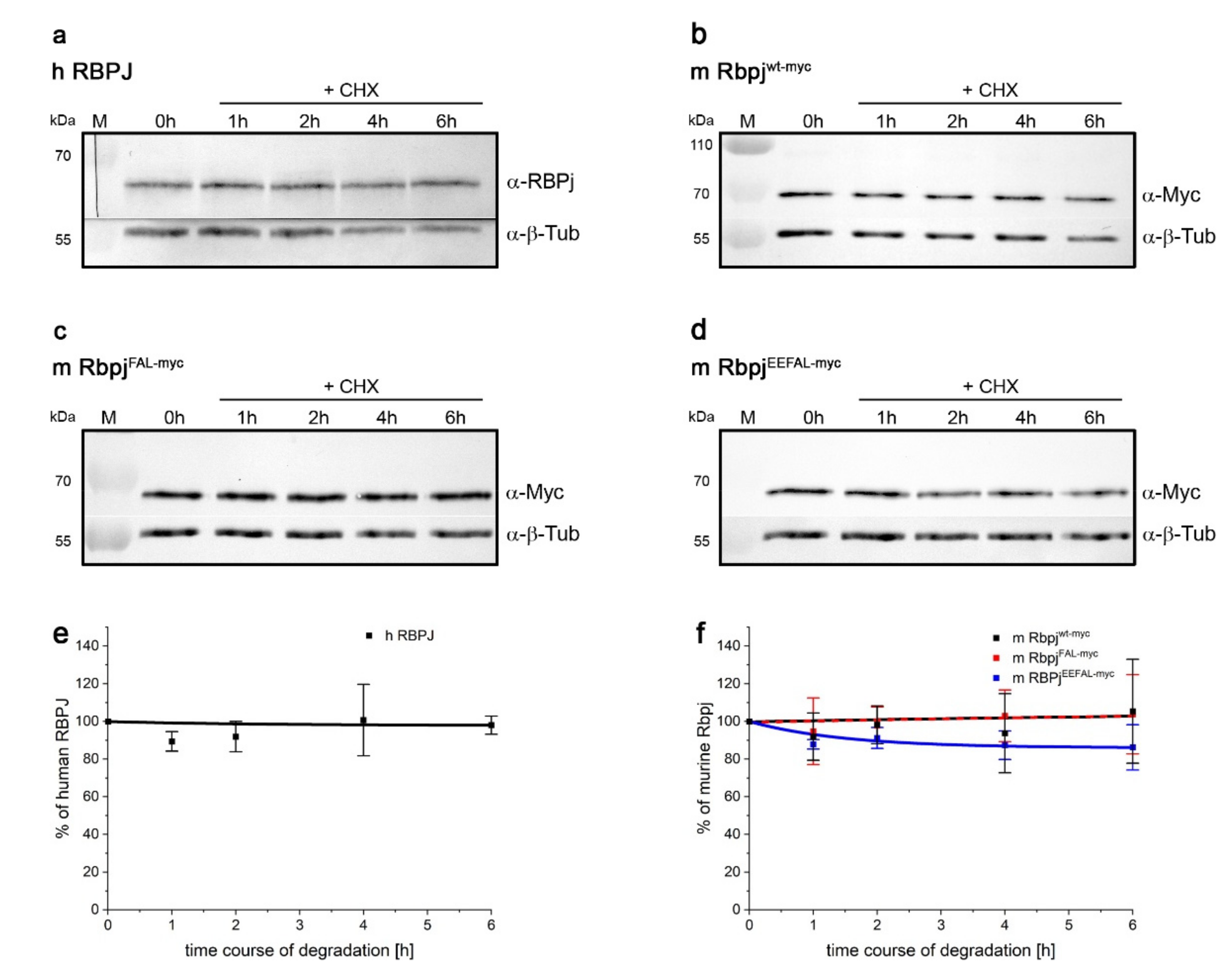
2.6. RBPJ Might Be Stabilised by Cofactors as well
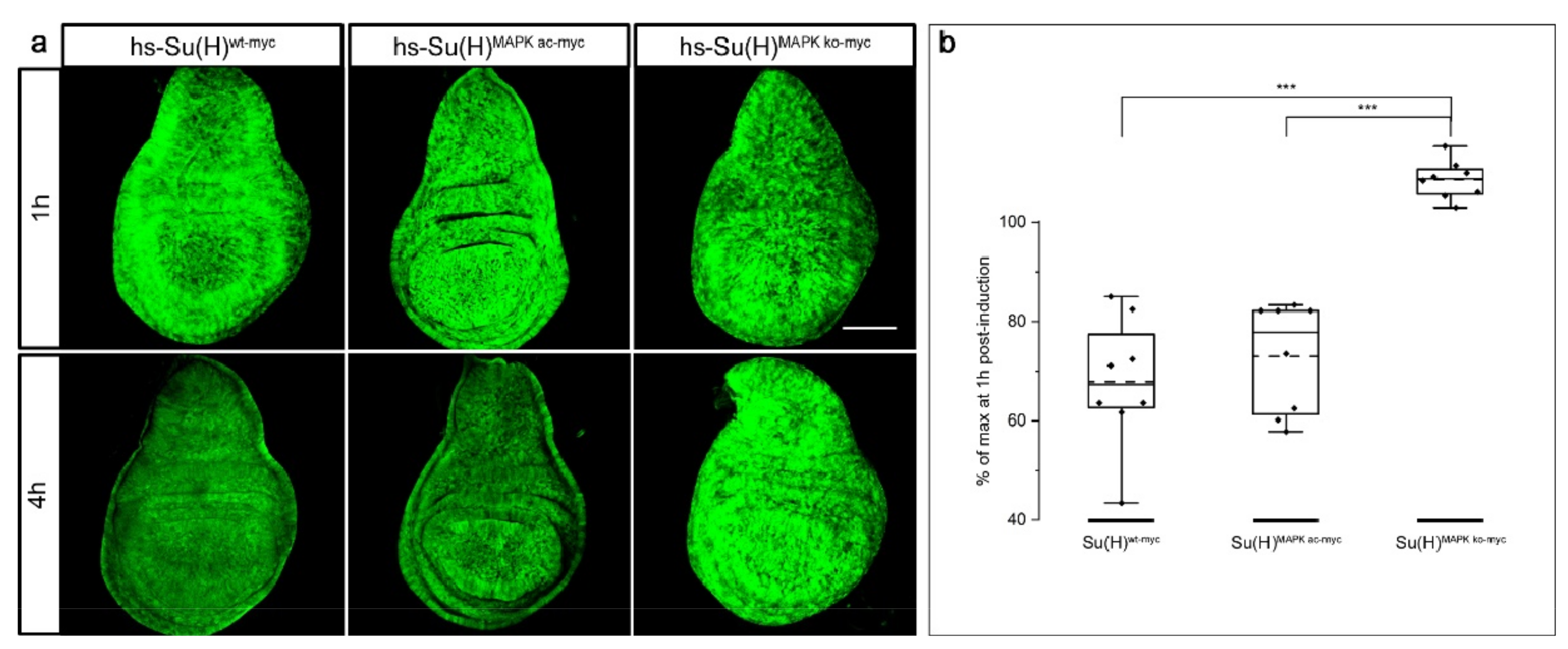
2.7. Stability of Su(H) Is Regulated by MAPK-Dependent Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Generation of Su(H) and RBPJ Constructs
4.2. Fly Work
4.2.1. Husbandry and Transgenic Lines
4.2.2. Heat Shock Regimen
4.2.3. DTS5 Experiment
4.2.4. Immunocytochemistry of Fly Tissue
4.3. Cell Culture Experiments
4.4. Yeast Two-Hybrid Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126 Pt 10, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The canonical Notch signaling pathway: Structural and biochemical insights into shape, sugar, and force. Dev. Cell. 2017, 41, 28–241. [Google Scholar] [CrossRef]
- Adams, J.M.; Jafar-Nejad, H. The Roles of Notch Signaling in Liver Development and Disease. Biomolecules 2019, 9, 608. [Google Scholar] [CrossRef]
- Louvi, A.; Artavanis-Tsakonas, S. Notch and disease: A growing field. Sem. Cell Dev. Biol. 2012, 23, 473–480. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch Signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for Notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef]
- Ho, D.M.; Artavanis-Tsakonas, S. The Notch-mediated proliferation circuitry. Curr. Top. Dev. Biol. 2016, 116, 17–33. [Google Scholar] [CrossRef]
- Falo-Sanjuan, J.; Bray, S.J. Decoding the Notch signal. Dev. Growth Differ. 2020, 62, 4–14. [Google Scholar] [CrossRef]
- Baron, M.; Aslam, H.; Flasza, M.; Fostier, M.; Higgs, J.E.; Mazaleyrat, S.L.; Wilkin, M.B. Multiple levels of Notch signal regulation (review). Mol. Membr. Biol. 2002, 19, 27–38. [Google Scholar] [CrossRef]
- Wang, H.; Zang, C.; Liu, X.S.; Aster, J.C. The role of Notch receptors in transcriptional regulation. J. Cell Physiol. 2015, 230, 982–988. [Google Scholar] [CrossRef]
- Kovall, R.A.; Blacklow, S.C. Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr. Top. Dev. Biol. 2010, 92, 31–71. [Google Scholar] [CrossRef]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef]
- Oswald, F.; Kovall, R.A. CSL-Associated Corepressor and Coactivator Complexes. Adv. Exp. Med. Biol. 2018, 1066, 279–295. [Google Scholar] [CrossRef]
- Bray, S.; Furriols, M. Notch pathway: Making sense of Suppressor of Hairless. Curr. Biol. 2001, 11, R217–R221. [Google Scholar] [CrossRef]
- Maier, D. Hairless, the ignored antagonist of the Notch signalling pathway. Hereditas 2006, 143, 212–221. [Google Scholar] [CrossRef]
- Giaimo, B.D.; Gagliani, E.K.; Kovall, R.A.; Borggrefe, T. Transcription Factor RBPJ as a Molecular Switch in Regulating the Notch Response. Adv. Exp. Med. Biol. 2021, 1287, 9–30. [Google Scholar] [CrossRef]
- Johnson, S.E.; Ilagan, M.X.; Kopan, R.; Barrick, D. Thermodynamic analysis of the CSL x Notch interaction: Distribution of binding energy of the Notch RAM region to the CSL beta-trefoil domain and the mode of competition with the viral transactivator EBNA2. J. Biol. Chem. 2010, 285, 6681–6692. [Google Scholar] [CrossRef]
- VanderWielen, B.D.; Yuan, Z.; Friedmann, D.R.; Kovall, R.A. Transcriptional repression in the Notch pathway: Thermodynamic characterization of CSL-MINT (Msx2-interacting nuclear target protein) complexes. J. Biol. Chem. 2011, 286, 14892–14902. [Google Scholar] [CrossRef]
- Yuan, Z.; VanderWielen, B.D.; Giaimo, B.D.; Pan, L.; Collins, C.E.; Turkiewicz, A.; Hein, K.; Oswald, F.; Borggrefe, T.; Kovall, R.A. Structural and functional studies of the RBPJ-SHARP complex reveal a conserved corepressor binding site. Cell Rep. 2019, 26, 845–854.e6. [Google Scholar] [CrossRef]
- Maier, D.; Kurth, P.; Schulz, A.; Russell, A.; Yuan, Z.; Gruber, K.; Kovall, R.A.; Preiss, A. Structural and functional analysis of the repressor complex in the Notch signaling pathway of Drosophila melanogaster. Mol. Cell. Biol. 2011, 22, 3242–3252. [Google Scholar] [CrossRef]
- Yuan, Z.; Praxenthaler, H.; Tabaja, N.; Torella, R.; Preiss, A.; Maier, D.; Kovall, R.A. Structure and function of the Su(H)-Hairless repressor complex, the major antagonist of Notch signalling in Drosophila melanogaster. PLoS Biol. 2016, 14, e1002509. [Google Scholar] [CrossRef]
- Friedmann, D.R.; Wilson, J.J.; Kovall, R.A. RAM-induced allostery facilitates assembly of a Notch pathway active transcription complex. J. Biol. Chem. 2008, 283, 14781–14791. [Google Scholar] [CrossRef]
- Friedmann, D.R.; Kovall, R.A. Thermodynamic and structural insights into CSL-DNA complexes. Protein Sci. 2010, 19, 34–46. [Google Scholar] [CrossRef]
- Torella, R.; Li, J.; Kinrade, E.; Cerda-Moya, G.; Contreras, A.N.; Foy, R.; Stojnic, R.; Glen, R.C.; Kovall, R.A.; Adryan, B.; et al. A combination of computational and experimental approaches identifies DNA sequence constraints associated with target site binding specificity of the transcription factor CSL. Nucleic Acids Res. 2014, 42, 10550–10563. [Google Scholar] [CrossRef]
- Zhou, S.; Hayward, S.D. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol. Cell Biol. 2001, 21, 6222–6232. [Google Scholar] [CrossRef]
- Wolf, D.; Smylla, T.K.; Reichmuth, J.; Hoffmeister, P.; Kober, L.; Zimmermann, M.; Turkiewicz, A.; Borggrefe, T.; Nagel, A.C.; Oswald, F.; et al. Nucleo-cytoplasmic shuttling of Drosophila Hairless/Su(H) heterodimer as a means of regulating Notch dependent transcription. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1520–1532. [Google Scholar] [CrossRef]
- Wolf, D.B.; Maier, D.; Nagel, A.C. Nucleo-cytoplasmic shuttling of murine RBPJ by Hairless protein matches that of Su(H) protein in the model system Drosophila melanogaster. Hereditas 2021, 158, 11. [Google Scholar] [CrossRef]
- Krejcí, A.; Bray, S. Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev. 2007, 21, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Mourikis, P.; Barteis, S.J.; Brinkman, A.B.; Tajbakhsh, S.; Stunnenberg, H.G. Dynamic binding of RBPJ is determined by Notch signaling Status. Genes Dev. 2013, 27, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zang, C.; Taing, L.; Arnett, K.L.; Wong, Y.J.; Pear, W.S.; Blacklow, S.C.; Liu, X.S.; Aster, J.C. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. USA 2014, 111, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lamarca, M.J.; Falo-Sanjuan, J.; Stojnic, R.; Abdul Rehman, S.; Muresan, L.; Jones, M.L.; Pillidge, Z.; Cerda-Moya, G.; Yuan, Z.; Baloul, S.; et al. Activation of the Notch signaling pathway in vivo elicits changes in CSL nuclear dynamics. Dev. Cell 2018, 44, 611–623.e7. [Google Scholar] [CrossRef]
- Falo-Sanjuan, J.; Lammers, N.C.; Garcia, H.G.; Bray, S.J. Enhancer Priming Enables Fast and Sustained Transcriptional Responses to Notch Signaling. Dev. Cell. 2019, 50, 411–425.e8. [Google Scholar] [CrossRef]
- Lee, C.; Shin, H.; Kimble, J. Dynamics of Notch-Dependent Transcriptional Bursting in Its Native Context. Dev. Cell. 2019, 50, 426–435.e4. [Google Scholar] [CrossRef]
- Hall, D.P.; Kovall, R.A. Structurally conserved binding motifs of transcriptional regulators to Notch nuclear effector CSL. Exp. Biol. Med. 2019, 244, 1520–1529. [Google Scholar] [CrossRef]
- Ohnuki, H.; Inoue, H.; Takemori, N.; Nakayama, H.; Sakaue, T.; Fukuda, S.; Miwa, D.; Nishiwaki, E.; Hatano, M.; Tokuhisa, T.; et al. BAZF, a novel component of cullin3-based E3 ligase complex, mediates VEGFR and Notch cross-signaling in angiogenesis. Blood 2012, 119, 2688–2698. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, M.Y.; Ann, E.J.; Mo, J.S.; Yoon, J.H.; Park, H.S. Presenilin-2 regulates the degradation of RBP-Jk protein through p38 mitogen-activated protein kinase. J. Cell Sci. 2012, 125, 1296–1308. [Google Scholar] [CrossRef][Green Version]
- Deshmukh, R.S.; Sharma, S.; Das, S. Cyclin F-Dependent Degradation of RBPJ Inhibits IDH1R132H-Mediated Tumorigenesis. Cancer Res. 2018, 78, 6386–6398. [Google Scholar] [CrossRef]
- Lecourtois, M.; Schweisguth, F. The neurogenic Suppressor of Hairless DNA-binding protein mediates the transcriptional activation of the Enhancer of split complex genes triggered by Notch signaling. Genes Dev. 1995, 9, 2598–2608. [Google Scholar] [CrossRef]
- Eastman, D.S.; Slee, R.; Skoufos, E.; Bangalore, L.; Bray, S.; Delidakis, C. Synergy between Suppressor of Hairless and Notch in regulation of Enhancer of split m gamma and m delta expression. Mol. Cell. Biol. 1997, 17, 5620–5628. [Google Scholar] [CrossRef]
- Wesley, C.S.; Mok, L.P. Regulation of Notch signaling by a novel mechanism involving Suppressor of Hairless stability and carboxyl terminus-truncated Notch. Mol. Cell Biol. 2003, 23, 5581–5593. [Google Scholar] [CrossRef]
- Praxenthaler, H.; Nagel, A.C.; Schulz, A.; Zimmermann, M.; Meier, M.; Schmid, H.; Preiss, A.; Maier, D. Hairless-binding deficient Suppressor of Hairless alleles reveal Su(H) protein levels are dependent on complex formation with Hairless. PLoS Genet. 2017, 13, e1006774. [Google Scholar] [CrossRef]
- Schedl, P.; Artavanis-Tsakonas, S.; Steward, R.; Gehring, W.J.; Mirault, M.E.; Goldschmidt-Clermont, M.; Moran, L.; Tissières, A. Two hybrid plasmids with D. melanogaster DNA sequences complementary to mRNA coding for the major heat shock protein. Cell 1978, 14, 921–929. [Google Scholar] [CrossRef]
- Török, I.; Karch, F. Nucleotide sequences of heat shock activated genes in Drosophila melanogaster. I. Sequences in the regions of the 5′ and 3′ ends of the hsp 70 gene in the hybrid plasmid 56H8. Nucleic Acids Res. 1980, 8, 3105–3123. [Google Scholar] [CrossRef]
- Pelham, H.R. A regulatory upstream promoter element in the Drosophila hsp 70 heat-shock gene. Cell 1982, 30, 517–528. [Google Scholar] [CrossRef]
- Pirrotta, V. Vectors for P-mediated transformation in Drosophila. Biotechnology 1988, 10, 437–456. [Google Scholar] [CrossRef]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An optimized transgenesis system for Drosophila using germ-line-specific PhiC31 integrases. Proc. Natl. Acad. Sci. USA 2007, 104, 3312–3317. [Google Scholar] [CrossRef]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell. Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef]
- Schweisguth, F. Dominant-negative mutation in the beta2 and beta6 proteasome subunit genes affect alternative cell fate decisions in the Drosophila sense organ lineage. Proc. Natl. Acad. Sci USA 1999, 96, 11382–11386. [Google Scholar] [CrossRef]
- Saville, K.J.; Belote, J.M. Identification of an essential gene, l(3)73Ai, with a dominant temperature-sensitive lethal allele, encoding a Drosophila proteasome subunit. Proc. Natl. Acad. Sci USA 1993, 90, 8842–8846. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef]
- Speicher, S.A.; Thomas, U.; Hinz, U.; Knust, E. The Serrate locus of Drosophila and its role in morphogenesis of the wing imaginal discs: Control of cell proliferation. Development 1994, 120, 535–544. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, Analysis and Prediction of Protein Ubiquitination Sites. Proteins 2011, 78, 365–380. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Schölz, C.; Kelstrup, C.D.; Young, C.; Nielsen, M.L.; Olsen, J.V.; Brakebusch, C.; Choudhary, C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell. Proteom. 2012, 11, 1578–1585. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Wilson, J.J.; Kovall, R.A. Crystal structure of the CSL-Notch-Mastermind Ternary complex bound to DNA. Cell 2006, 124, 985–996. [Google Scholar] [CrossRef]
- Maier, D. The evolution of transcriptional repressors in the Notch signaling pathway: A computational analysis. Hereditas 2019, 156, 5. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lee, B.H.; King, R.W.; Finley, D.; Kirschner, M.W. Substrate degradation by the proteasome: A single-molecule kinetic analysis. Science 2015, 348, 1250834. [Google Scholar] [CrossRef] [PubMed]
- Praxenthaler, H.; Smylla, T.K.; Nagel, A.C.; Preiss, A.; Maier, D. Generation of new Hairless alleles by genomic engineering at the Hairless locus in Drosophila melanogaster. PLoS ONE 2015, 10, e0140007. [Google Scholar] [CrossRef] [PubMed]
- Gahr, B.M.; Brändle, F.; Zimmermann, M.; Nagel, A.C. An RBPJ-Drosophila model reveals dependence of RBPJ protein stability on the formation of transcription-regulator complexes. Cells 2019, 8, 1252. [Google Scholar] [CrossRef]
- Nam, Y.; Sliz, P.; Song, L.; Aster, J.C.; Blacklow, S.C. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 2006, 124, 973–983. [Google Scholar] [CrossRef]
- Collins, K.J.; Yuan, Z.; Kovall, R.A. Structure and function of the CSL-KyoT2 corepressor complex: A negative regulator of Notch signaling. Structure 2014, 22, 70–81. [Google Scholar] [CrossRef]
- Tabaja, N.; Yuan, Z.; Oswald, F.; Kovall, R.A. Structure-function analysis of RBP-J-interacting and tubulin-associated (RITA) reveals regions critical for repression of Notch target genes. J. Biol. Chem. 2017, 292, 10549–10563. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 14 September 2022).
- Hsieh, J.J.; Hayward, S.D. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science 1995, 268, 560–563. [Google Scholar] [CrossRef]
- Fuchs, K.P.; Bommer, G.; Dumont, E.; Christoph, B.; Vidal, M.; Kremmer, E.; Kempkes, B. Mutational analysis of the J recombination signal sequence binding protein (RBP-J)/Epstein-Barr virus nuclear antigen 2 (EBNA2) and RBPJ/Notch interaction. Eur. J. Biochem. 2001, 268, 4639–4646. [Google Scholar] [CrossRef]
- Ella, H.; Reiss, Y.; Ravid, T. The hunt for degrons of the 26S Proteasome. Biomolecules 2019, 9, 230. [Google Scholar] [CrossRef]
- Auer, J.S.; Nagel, A.C.; Schulz, A.; Wahl, V.; Preiss, A. MAPK-dependent phosphorylation modulates the activity of Suppressor of Hairless in Drosophila. Cell. Signal. 2015, 27, 115–124. [Google Scholar] [CrossRef]
- Auer, J.S.; Nagel, A.C.; Schulz, A.; Wahl, V.; Preiss, A. Local overexpression of Su(H)-MAPK variants affects Notch target gene expression and adult phenotypes in Drosophila. Data Brief 2015, 5, 852–863. [Google Scholar] [CrossRef][Green Version]
- Antfolk, D.; Antila, C.; Kemppainen, K.; Landor, S.K.; Sahlgren, C. Decoding the PTM-switchboard of Notch. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 118507. [Google Scholar] [CrossRef]
- Nagel, A.C.; Auer, J.S.; Schulz, A.; Pfannstiel, J.; Yuan, Z.; Collins, C.E.; Kovall, R.A.; Preiss, A. Phosphorylation of Suppressor of Hairless impedes its DNA-binding activity. Sci. Rep. 2017, 7, 11820. [Google Scholar] [CrossRef]
- Frankenreiter, L.; Gahr, B.M.; Schmid, H.; Zimmermann, M.; Deichsel, S.; Hoffmeister, P.; Turkiewicz, A.; Borggrefe, T.; Oswald, F.; Nagel, A.C. Phospho-site mutations in transcription factor Suppressor of Hairless impact Notch signaling activity during hematopoiesis in Drosophila. Front. Cell Dev. Biol. 2021, 9, 658820. [Google Scholar] [CrossRef]
- Sundaram, M.V. The love-hate relationship between Ras and Notch. Genes Dev. 2005, 19, 1825–1839. [Google Scholar] [CrossRef]
- Hurlbut, G.D.; Kankel, M.W.; Lake, R.J.; Artavanis-Tsakonas, S. Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 2007, 19, 166–175. [Google Scholar] [CrossRef]
- Baker, A.T.; Zlobin, A.; Osipo, C. Notch-EGFR/HER2 Bidirectional Crosstalk in Breast Cancer. Front. Oncol. 2014, 4, 360. [Google Scholar] [CrossRef]
- Tang, H.; Li, X.; Jiang, L.; Liu, Z.; Chen, L.; Chen, J.; Deng, M.; Zhou, F.; Zheng, X.; Liu, Z. RITA1 drives the growth of bladder cancer cells by recruiting TRIM25 to facilitate the proteasomal degradation of RBPJ. Cancer Sci. 2022, 113, 3071–3084. [Google Scholar] [CrossRef]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef]
- Zhou, S.; Peng, J.; Xiao, L.; Zhou, C.; Fang, Y.; Ou, Q.; Qin, J.; Liu, M.; Pan, Z.; Hou, Z. TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell Death Dis. 2021, 12, 463. [Google Scholar] [CrossRef]
- Lai, E.C. Protein degradation: Four E3s for the Notch pathway. Curr. Biol. 2002, 12, R74–R78. [Google Scholar] [CrossRef]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell. 2004, 16, 509–520. [Google Scholar] [CrossRef]
- Muratani, M.; Tansey, W.P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell. Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef]
- Ezhkova, E.; Tansey, W.P. Proteasomal ATPases link ubiquitylation of histone H2B to methylation of histone H3. Mol. Cell. 2004, 13, 435–442. [Google Scholar] [CrossRef]
- Müller, D.; Nagel, A.C.; Maier, D.; Preiss, A. A molecular link between Hairless and Pros26.4, a member of the AAA-ATPase subunits of the proteasome 19S regulatory particle in Drosophila. J. Cell Sci. 2006, 119 Pt 2, 250–258. [Google Scholar] [CrossRef][Green Version]
- Bray, S.; Musisi, H.; Bienz, M. Bre1 is required for Notch signaling and histone modification. Dev. Cell 2005, 8, 279–286. [Google Scholar] [CrossRef]
- Maier, D.; Marquart, J.; Thompson-Fontaine, A.; Beck, I.; Wurmbach, E.; Preiss, A. In vivo structure-function analysis of Drosophila Hairless. Mech. Dev. 1997, 67, 97–106. [Google Scholar] [CrossRef]
- Broadus, M.R.; Chen, T.W.; Neitzel, L.R.; Ng, V.H.; Jodoin, J.N.; Lee, L.A.; Salic, A.; Robbins, D.J.; Capobianco, A.J.; Patton, J.G.; et al. Identification of a Paralog-Specific Notch1 Intracellular Domain Degron. Cell Rep. 2016, 15, 1920–1929. [Google Scholar] [CrossRef]
- Bunch, T.A.; Grinblat, Y.; Goldstein, L.S. Characterization and use of the Drosophila metallothionein promoter in cultured Drosophila melanogaster cells. Nucleic Acids Res. 1988, 16, 1043–1061. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.C.; Krejci, A.; Tenin, G.; Bravo-Patiño, A.; Bray, S.; Maier, D.; Preiss, A. Hairless-mediated repression of Notch target genes requires the combined activity of Groucho and CtBP co-repressors. Mol. Cell. Biol. 2005, 25, 10433–10441. [Google Scholar] [CrossRef] [PubMed]
- Oswald, F.; Liptay, S.; Adler, G.; Schmid, R.M. NF-kappaB2 is a putative target gene of activated Notch-1 via RBP-Jkappa. Mol. Cell. Biol. 1998, 18, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Kugler, S.J.; Preiss, A.; Maier, D.; Nagel, A.C. Genetic modifier screens on Hairless gain-of-function phenotypes reveal genes involved in cell differentiation, cell growth and apoptosis in Drosophila melanogaster. Genetics 2005, 171, 1137–1152. [Google Scholar] [CrossRef]
- Maier, D.; Praxenthaler, H.; Schulz, A.; Preiss, A. Gain of function Notch phenotypes associated with ectopic expression of the Su(H) C-terminal domain illustrate separability of Notch and Hairless-mediated activities. PLoS ONE 2013, 8, e81578. [Google Scholar] [CrossRef]
- Golemis, E.A.; Brent, R. Searching for interacting proteins with the two-hybrid system III. In The Yeast Two-Hybrid System; Bartel, P.L., Fields, S., Eds.; Oxford University Press: Oxford, UK, 1997; pp. 43–72. [Google Scholar]
- Brockmann, B.; Mastel, H.; Oswald, F.; Maier, D. Analysis of the interaction between human RITA and Drosophila Suppressor of Hairless. Hereditas 2014, 151, 209–219. [Google Scholar] [CrossRef]
- Matsuno, K.; Go, M.J.; Sun, X.; Eastman, D.S.; Artavanis-Tsakonas, S. Suppressor of Hairless-independent events in Notch signaling imply novel pathway elements. Development 1997, 124, 4265–4273. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fechner, J.; Ketelhut, M.; Maier, D.; Preiss, A.; Nagel, A.C. The Binding of CSL Proteins to Either Co-Activators or Co-Repressors Protects from Proteasomal Degradation Induced by MAPK-Dependent Phosphorylation. Int. J. Mol. Sci. 2022, 23, 12336. https://doi.org/10.3390/ijms232012336
Fechner J, Ketelhut M, Maier D, Preiss A, Nagel AC. The Binding of CSL Proteins to Either Co-Activators or Co-Repressors Protects from Proteasomal Degradation Induced by MAPK-Dependent Phosphorylation. International Journal of Molecular Sciences. 2022; 23(20):12336. https://doi.org/10.3390/ijms232012336
Chicago/Turabian StyleFechner, Johannes, Manuela Ketelhut, Dieter Maier, Anette Preiss, and Anja C. Nagel. 2022. "The Binding of CSL Proteins to Either Co-Activators or Co-Repressors Protects from Proteasomal Degradation Induced by MAPK-Dependent Phosphorylation" International Journal of Molecular Sciences 23, no. 20: 12336. https://doi.org/10.3390/ijms232012336
APA StyleFechner, J., Ketelhut, M., Maier, D., Preiss, A., & Nagel, A. C. (2022). The Binding of CSL Proteins to Either Co-Activators or Co-Repressors Protects from Proteasomal Degradation Induced by MAPK-Dependent Phosphorylation. International Journal of Molecular Sciences, 23(20), 12336. https://doi.org/10.3390/ijms232012336