
Midazolam Ameliorates Hyperglycemia-Induced Glomerular Endothelial Dysfunction by Inhibiting Transglutaminase 2 in Diabetes

 and
and
Abstract
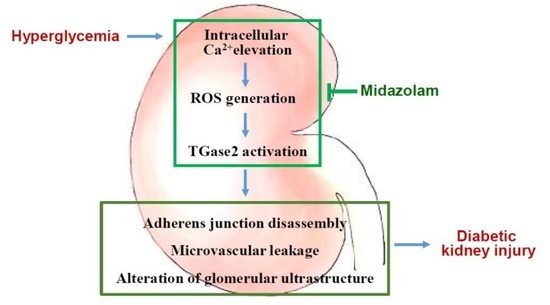
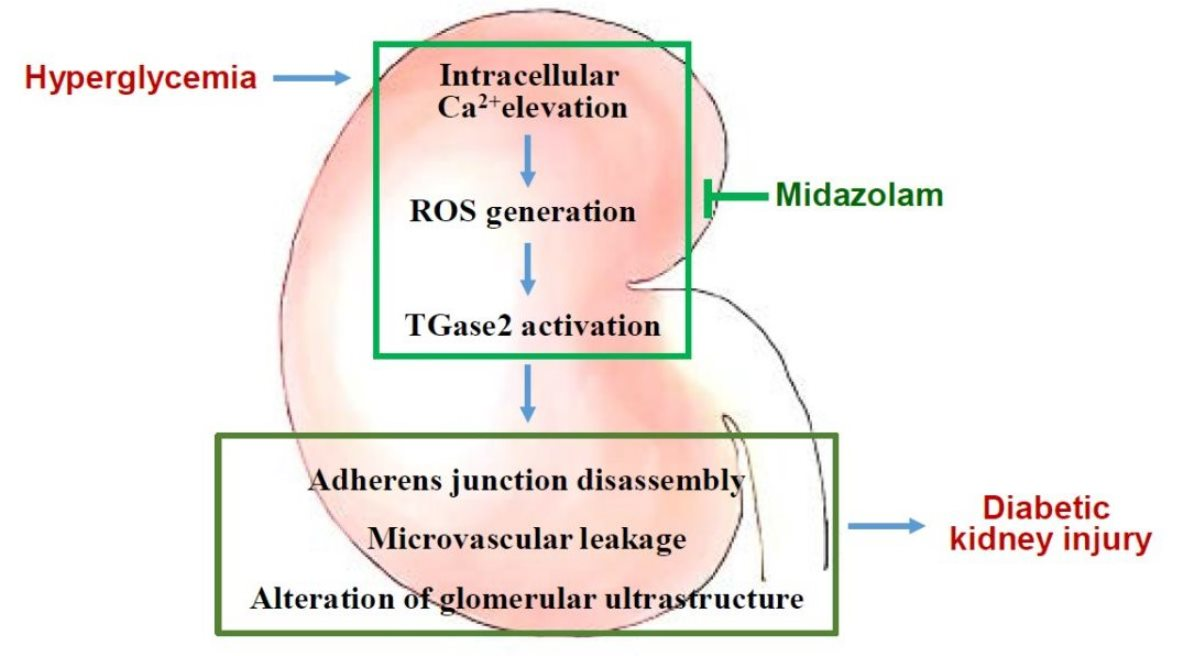
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
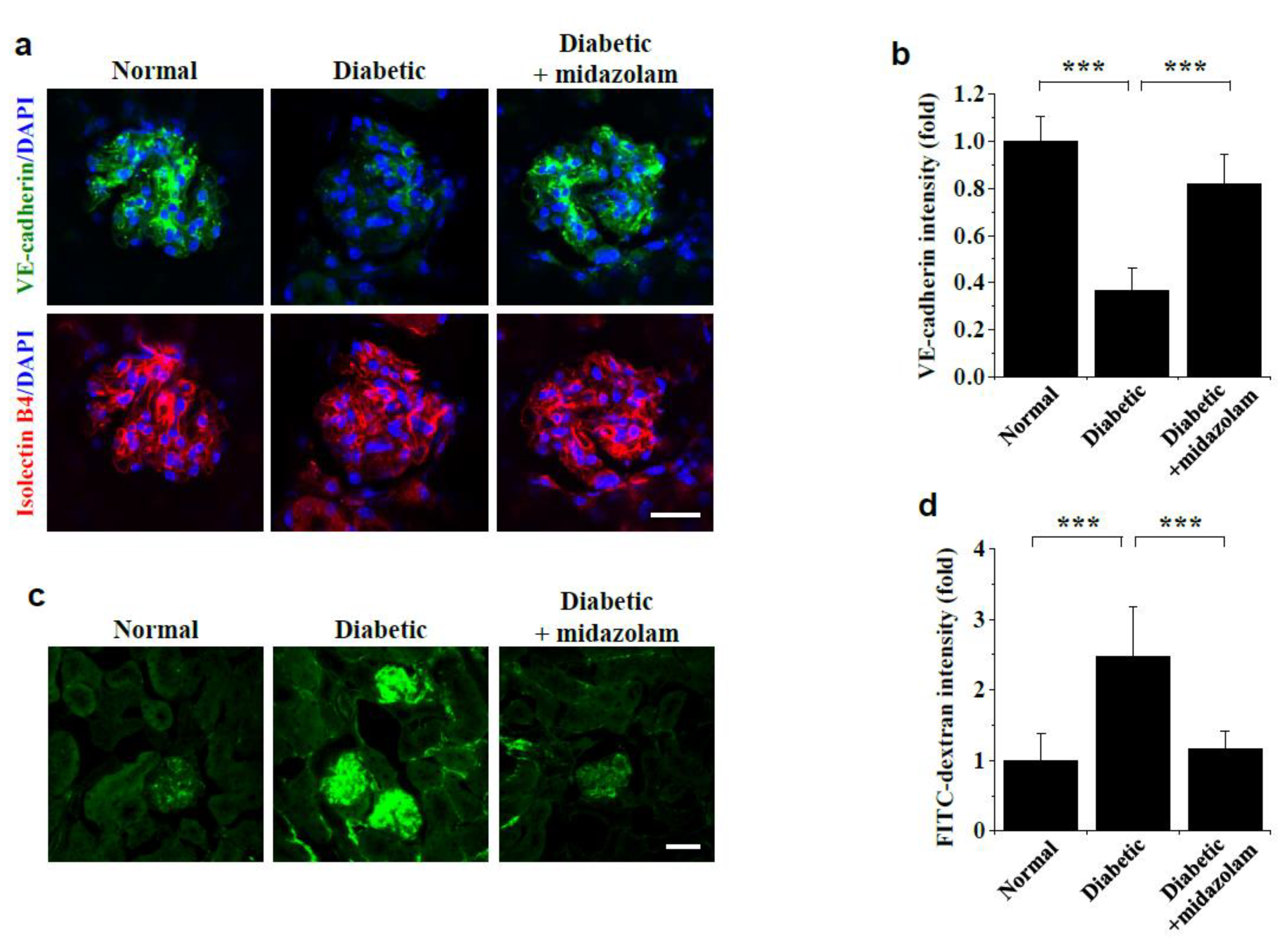
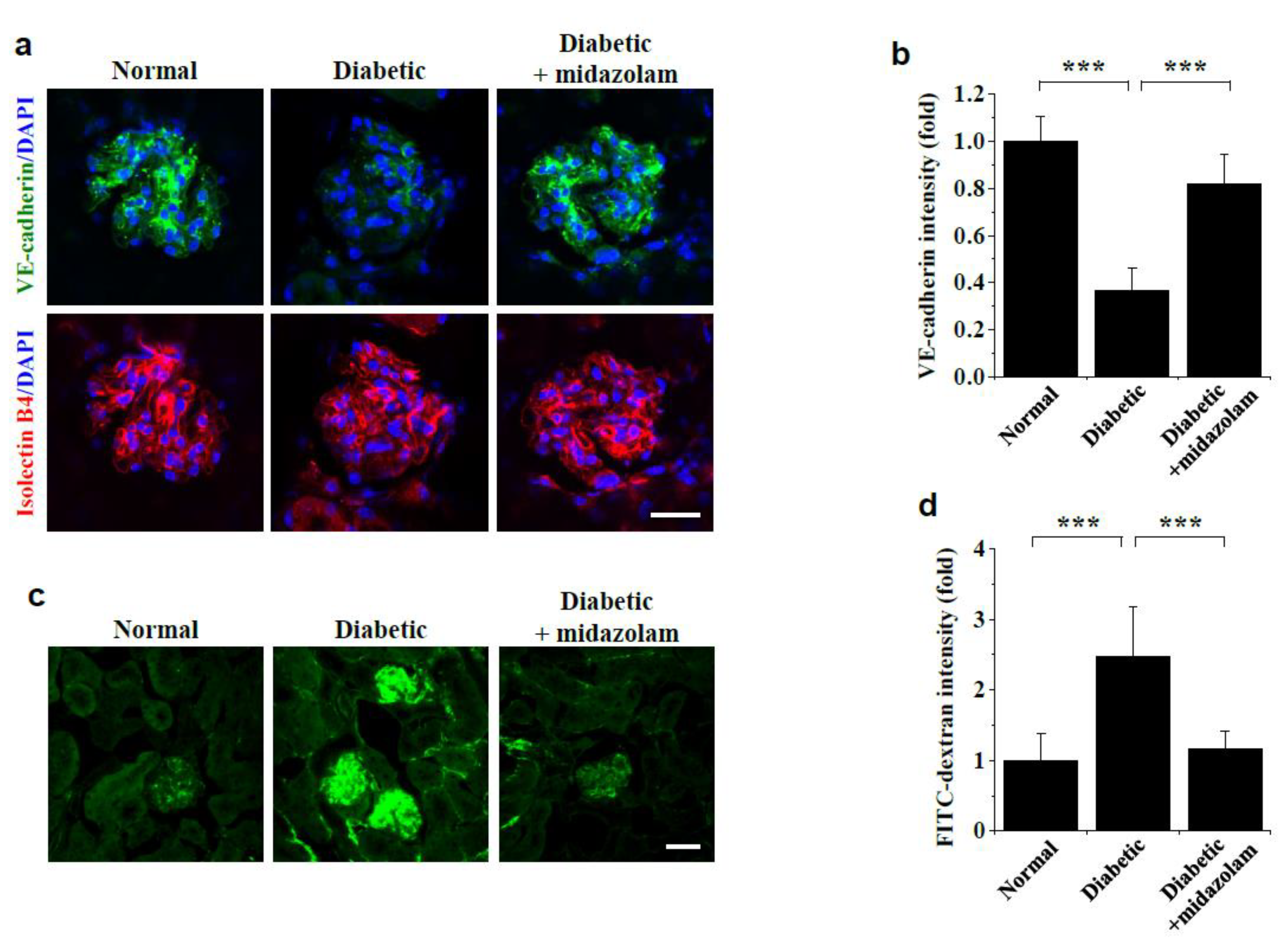
2.1. Midazolam Inhibits Hyperglycemia-Induced Adherens Junction Disruption and Vascular Leakage in Kidneys of Diabetic Mice
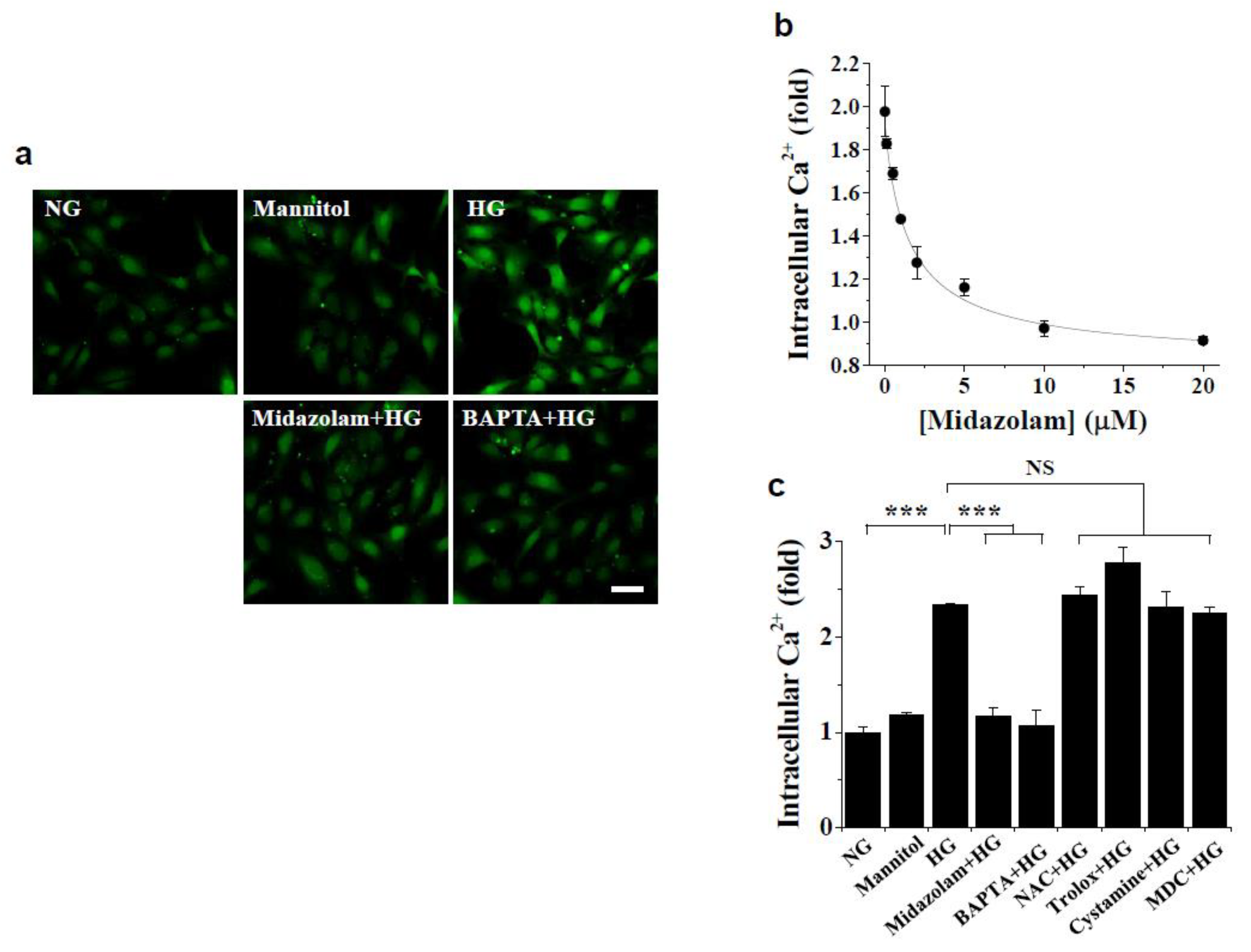
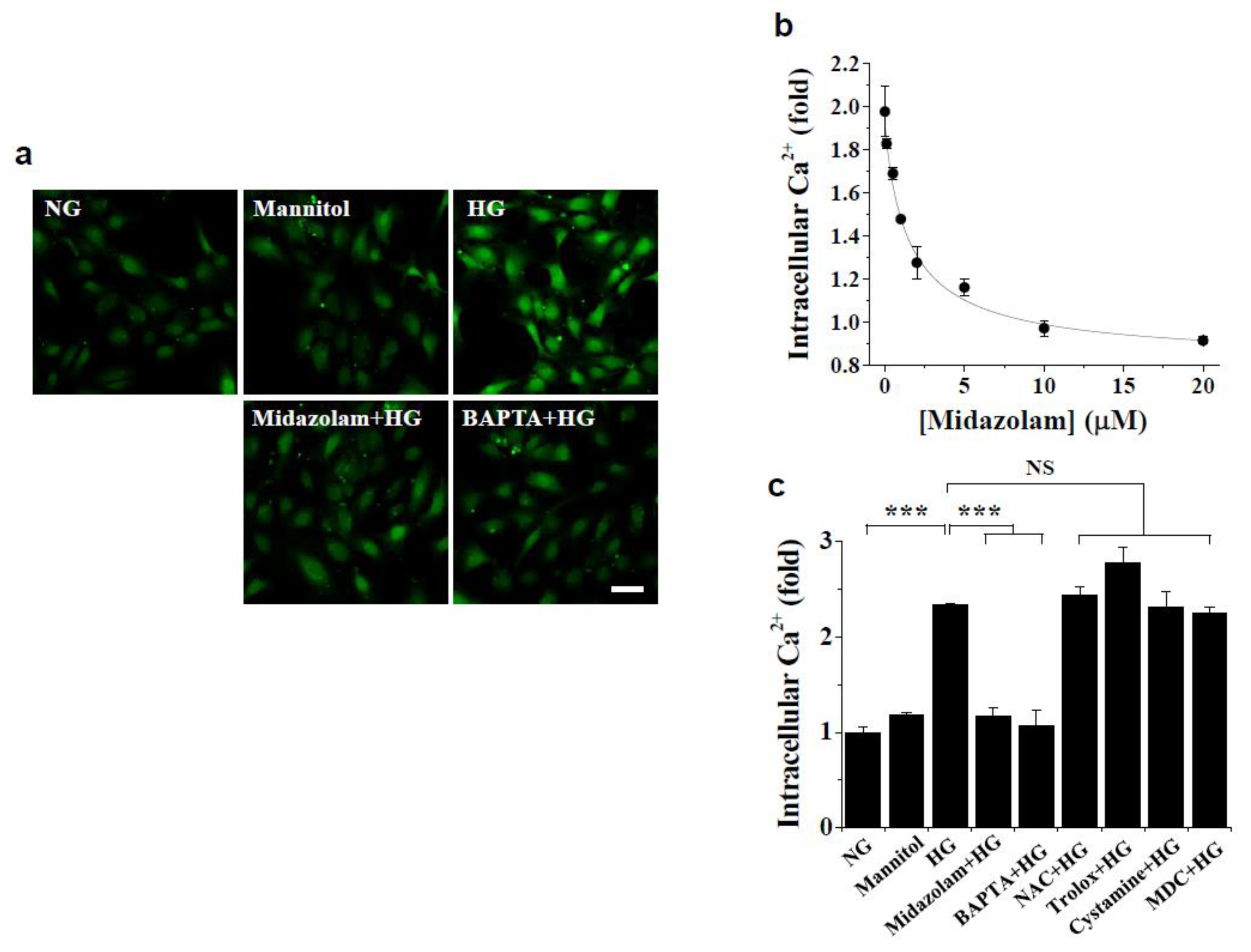
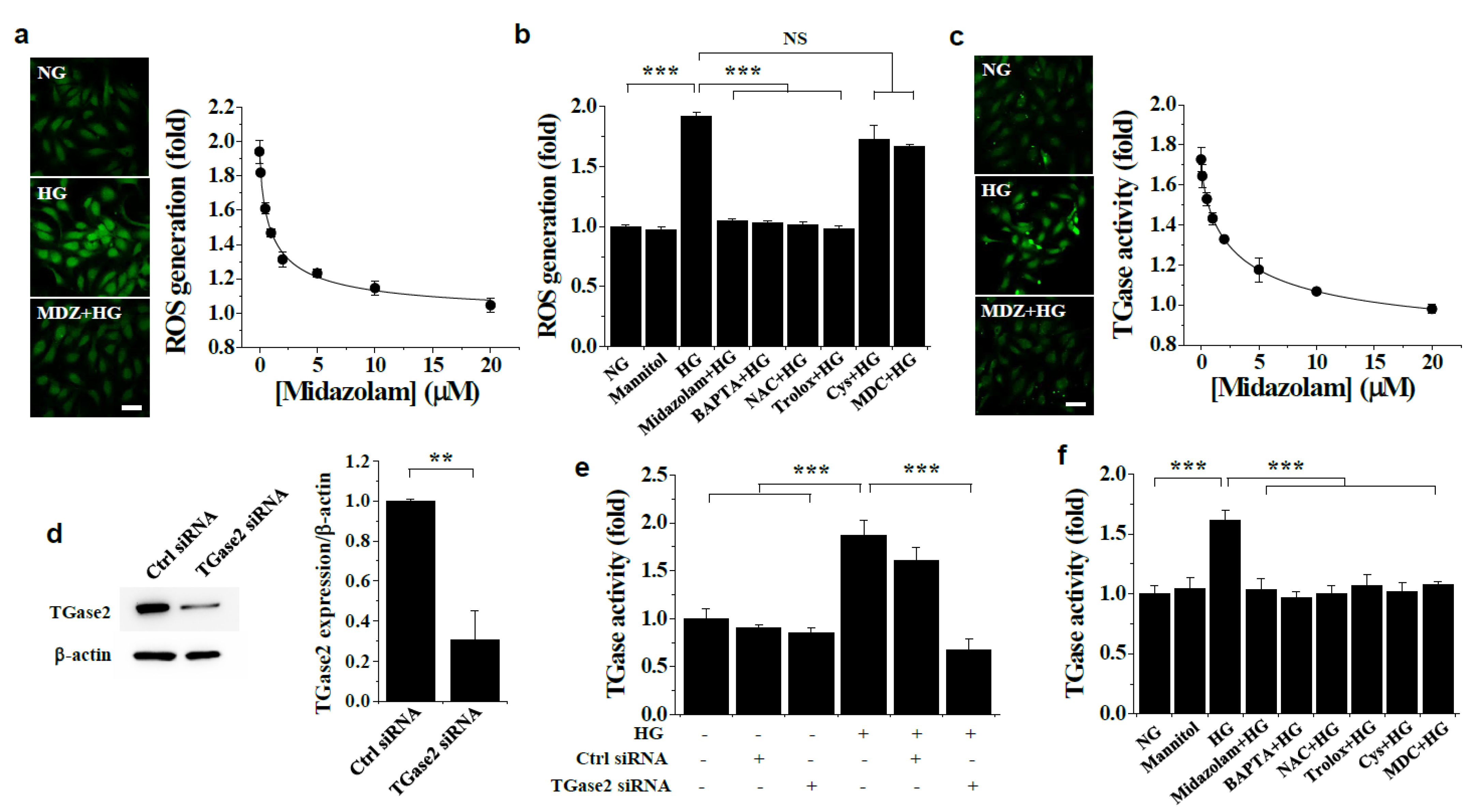
2.2. Midazolam Inhibits High Glucose-Induced Elevation of Intracellular Ca2+ and Subsequent ROS Generation and TGase2 Activation in HGECs
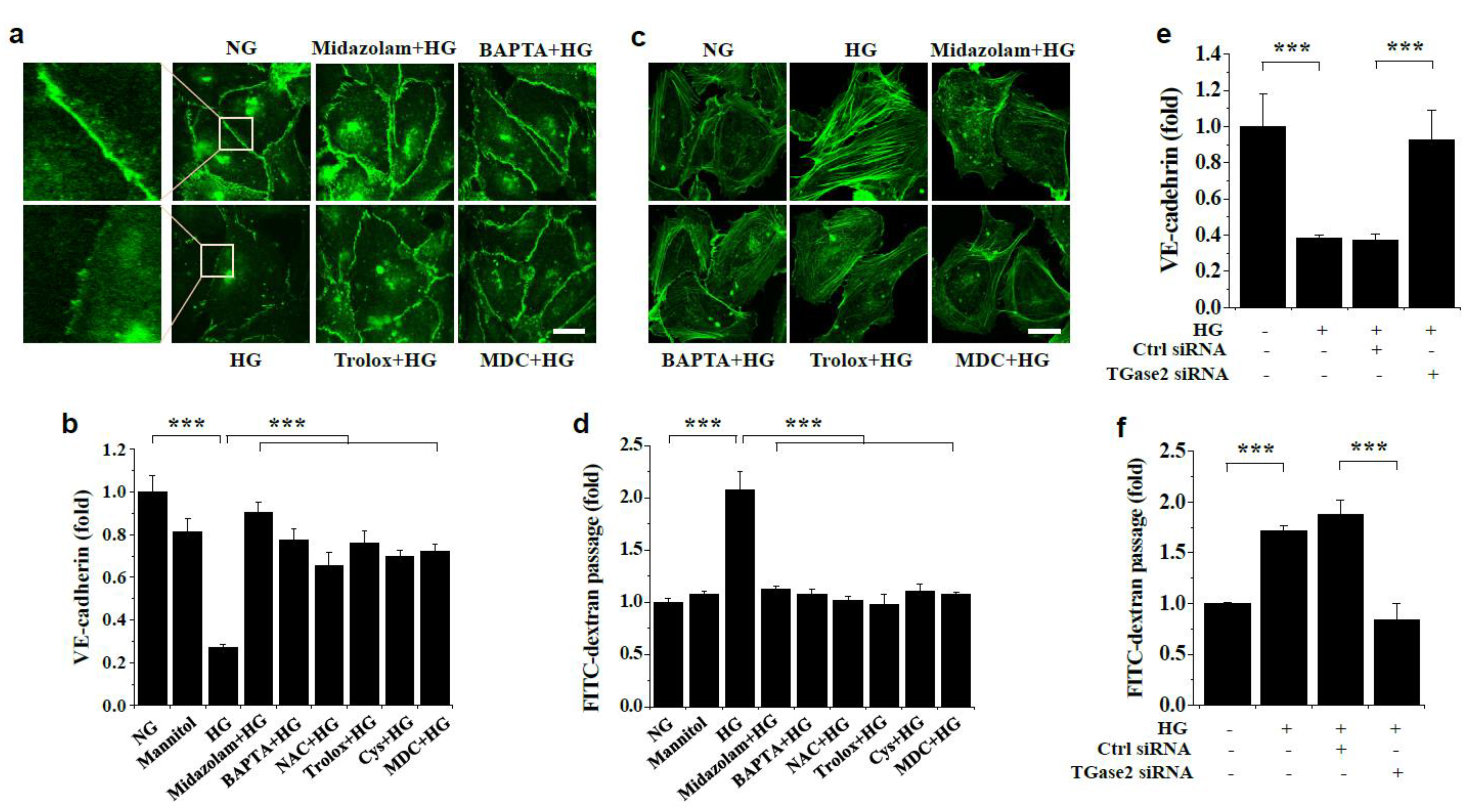
2.3. Midazolam Inhibits High Glucose-Induced Adherens Junction Disruption, Stress Fiber Formation, and Endothelial Cell Permeability in HGECs
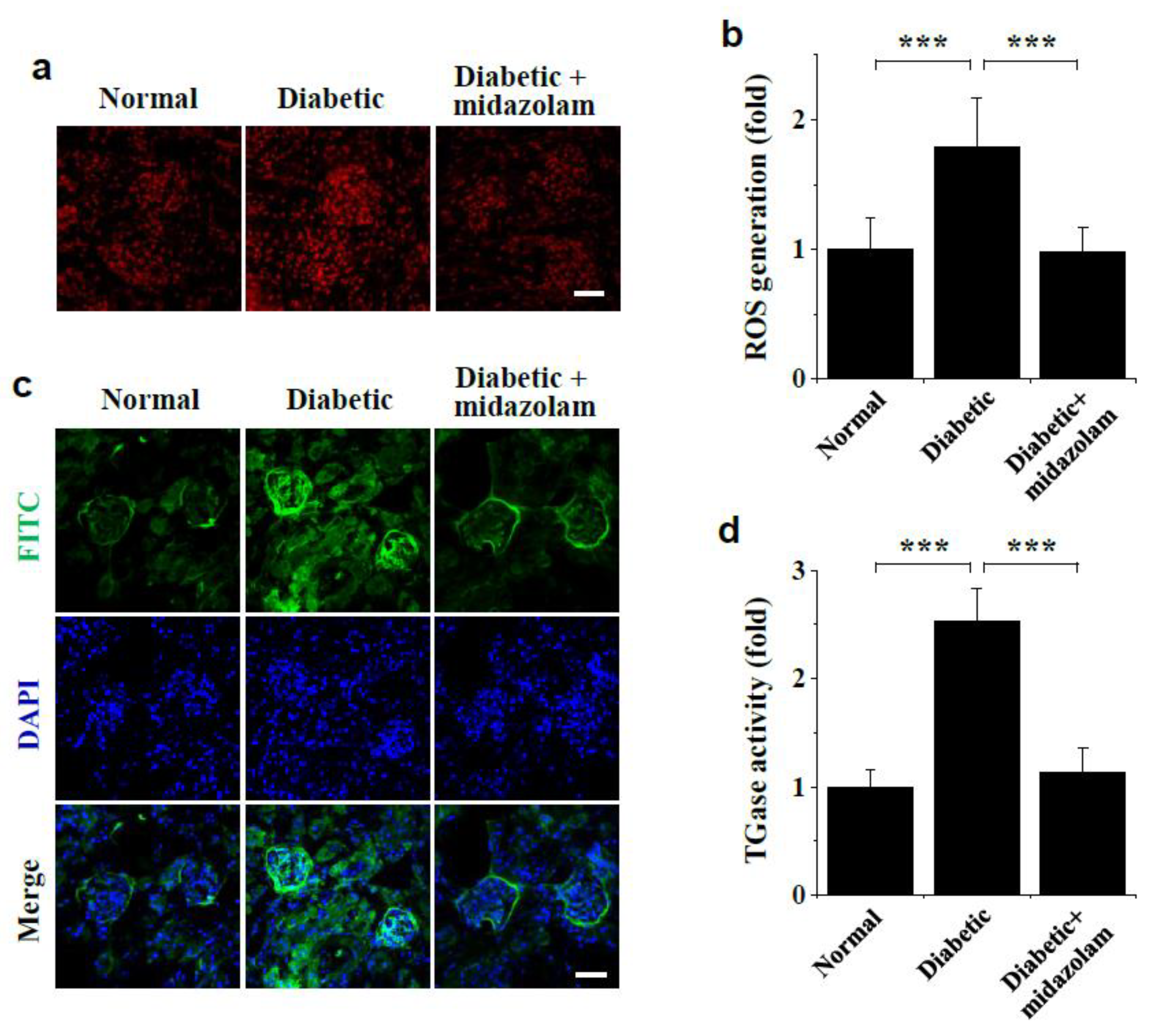
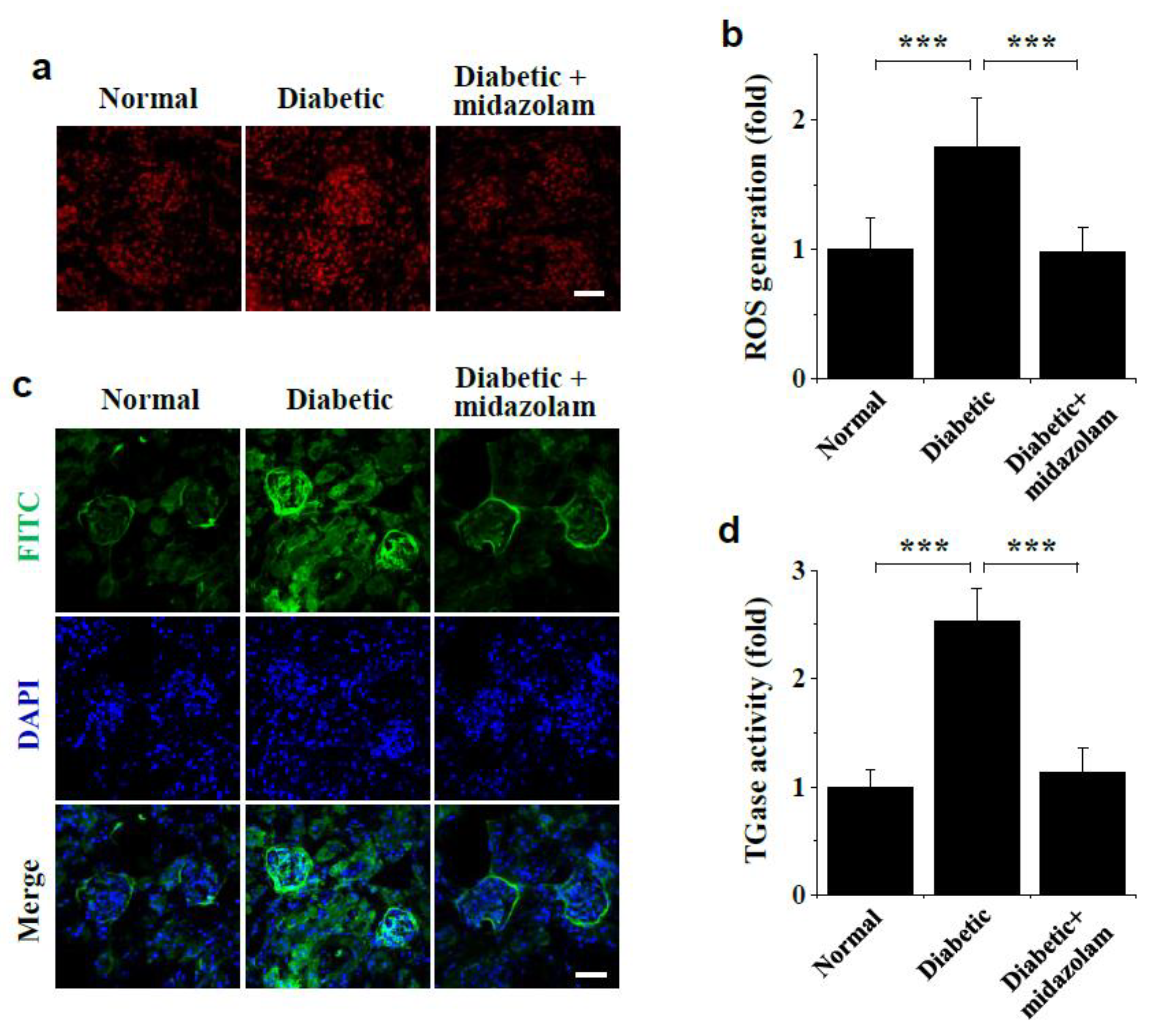
2.4. Midazolam Inhibits Hyperglycemia-Induced ROS Generation and TGase Activation in Kidneys of Diabetic Mice
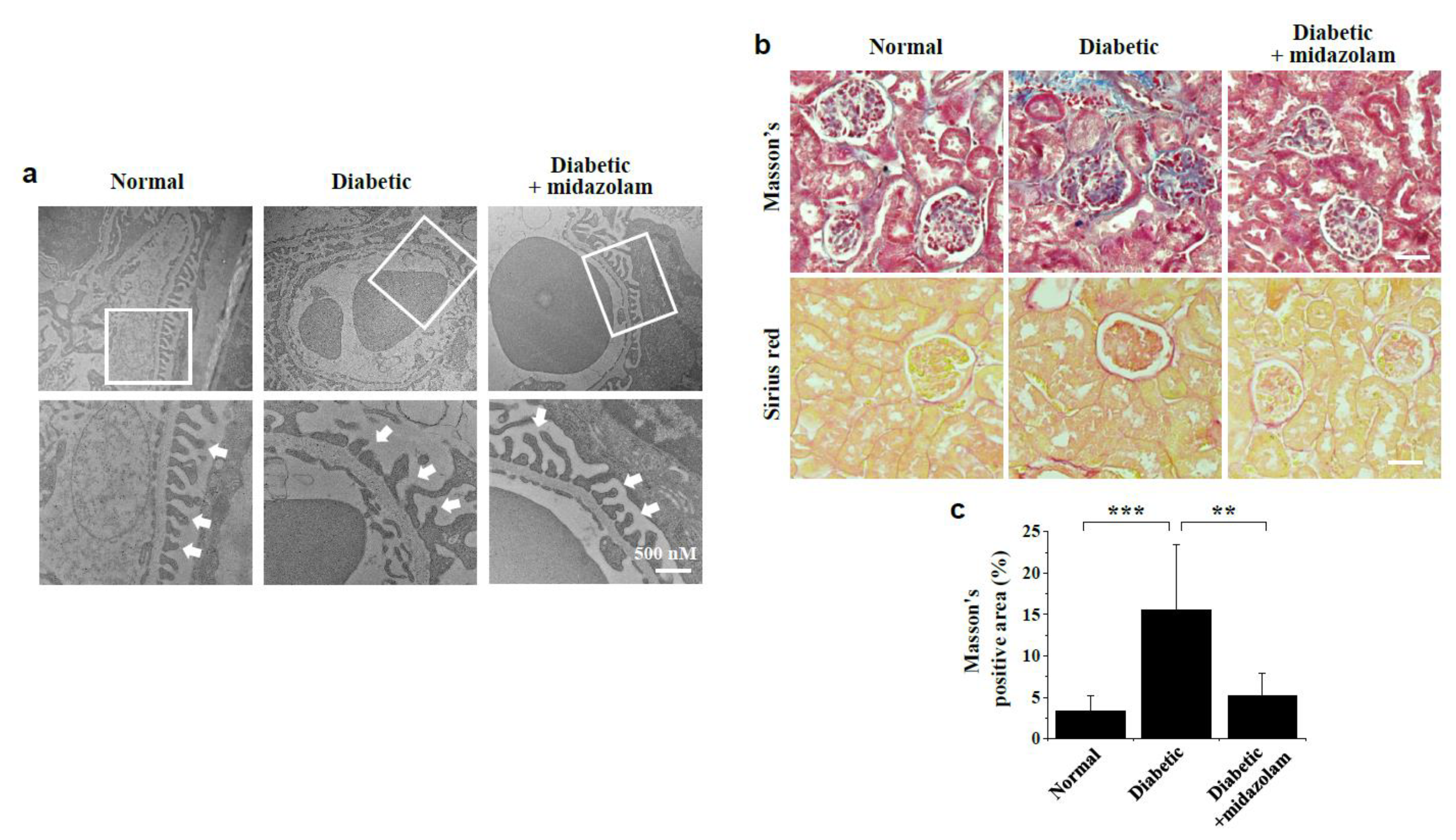
2.5. Midazolam Inhibits Hyperglycemia-Induced Pathological Alterations in Glomerular Ultrastructure and Renal Fibrosis in Kidneys of Diabetic Mice
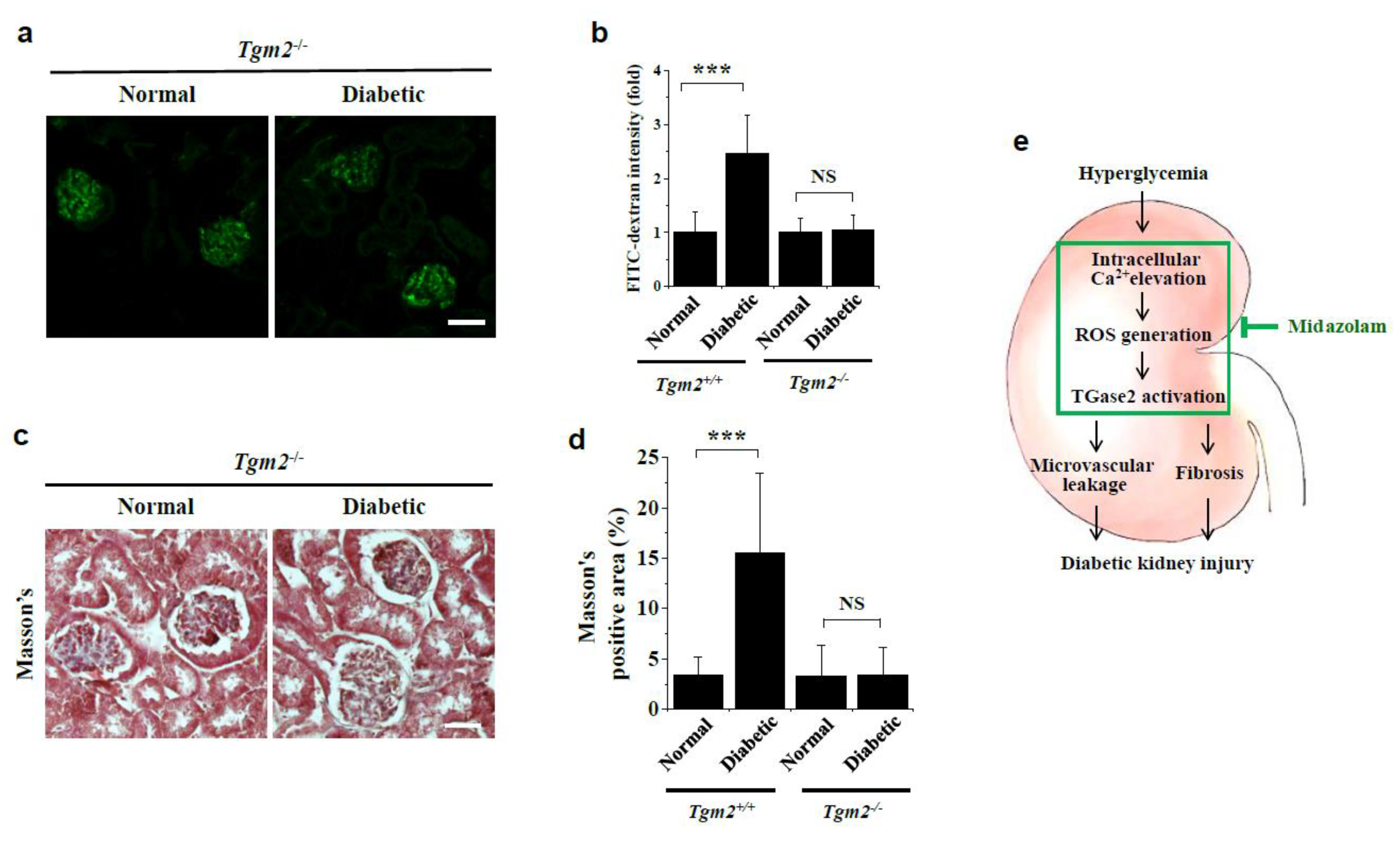
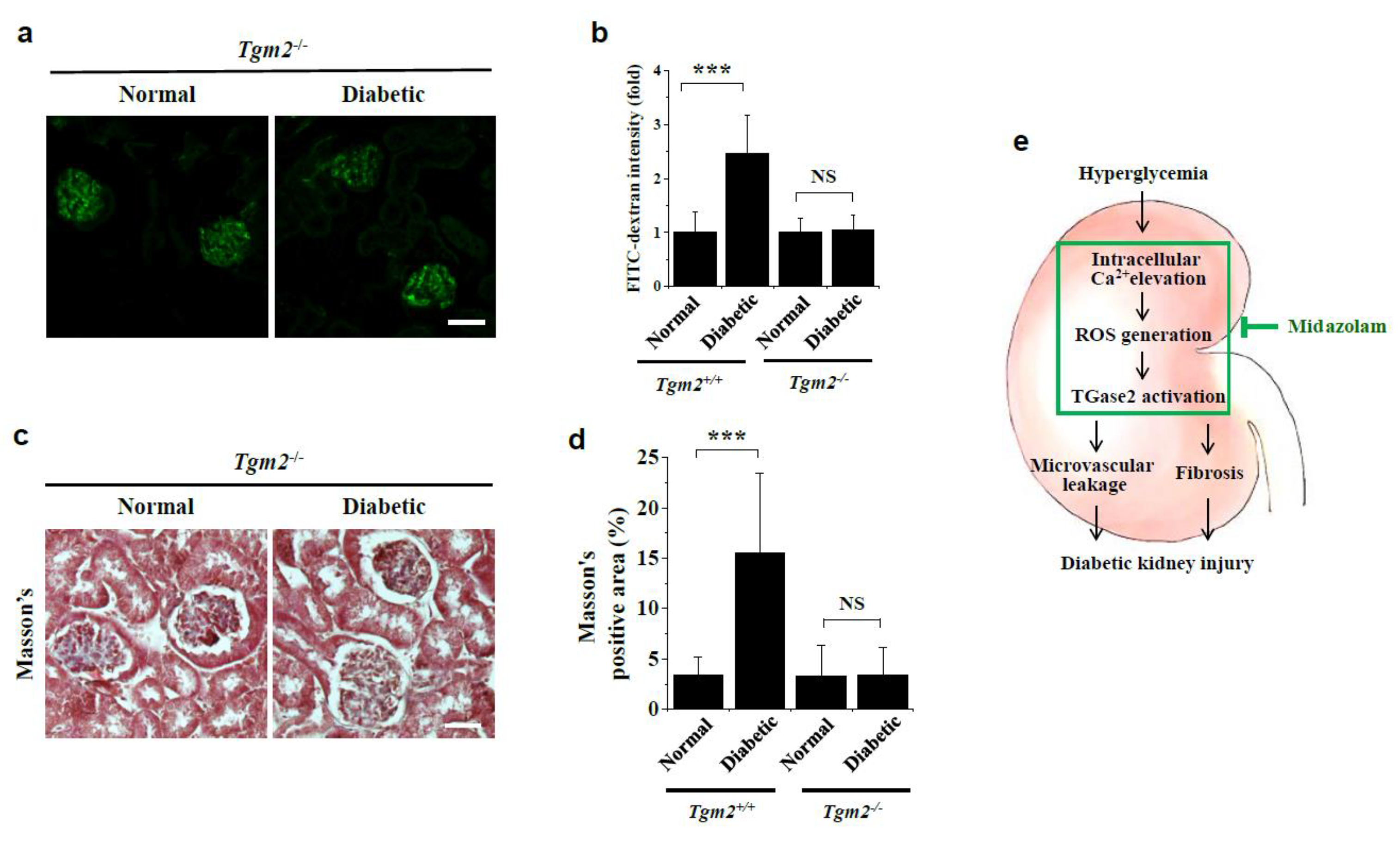
2.6. Vascular Leakage and Renal Fibrosis Are Not Observed in Kidneys of Diabetic Tgm2−/− Mice
3. Discussion
4. Materials and Methods
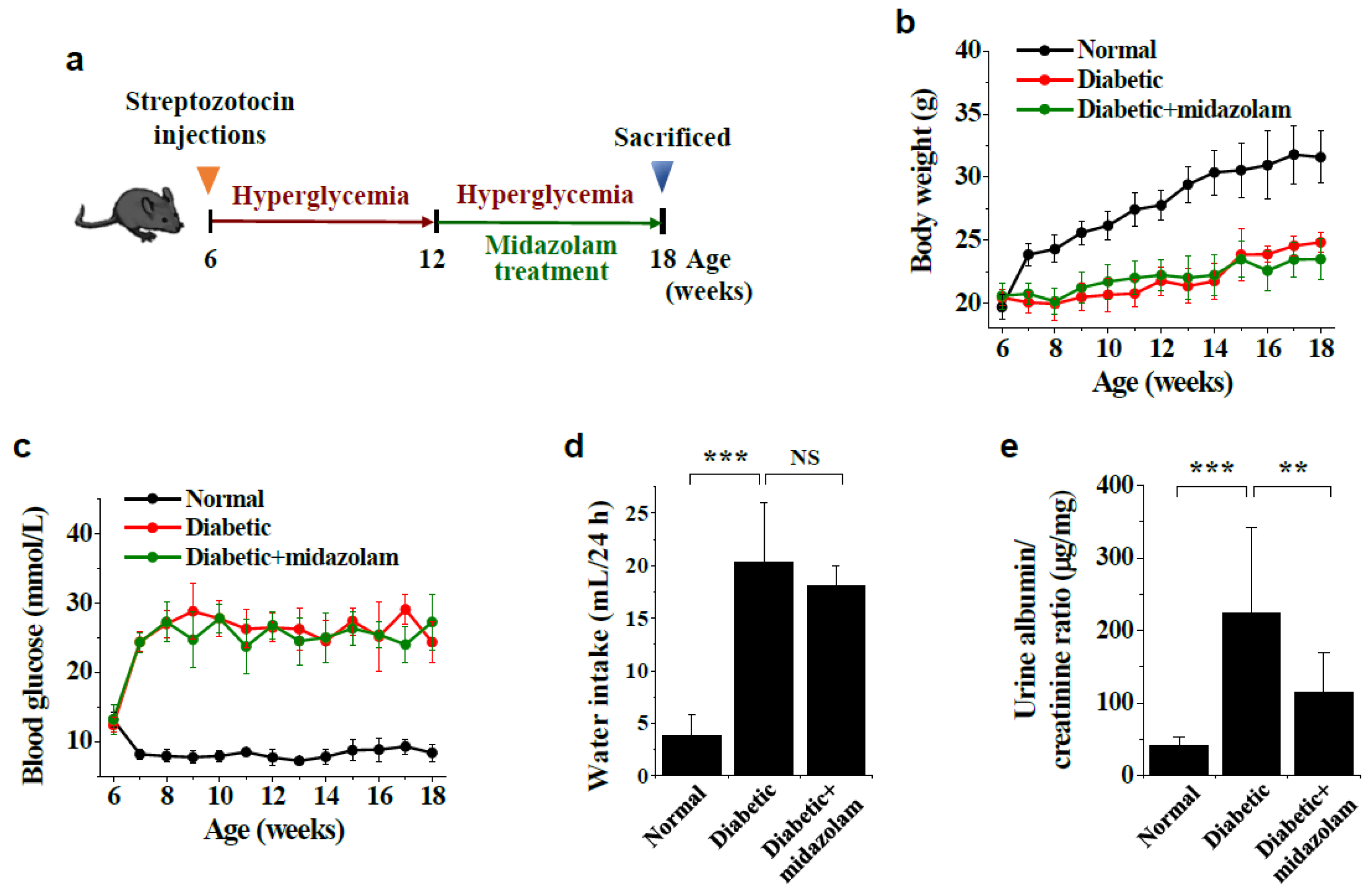
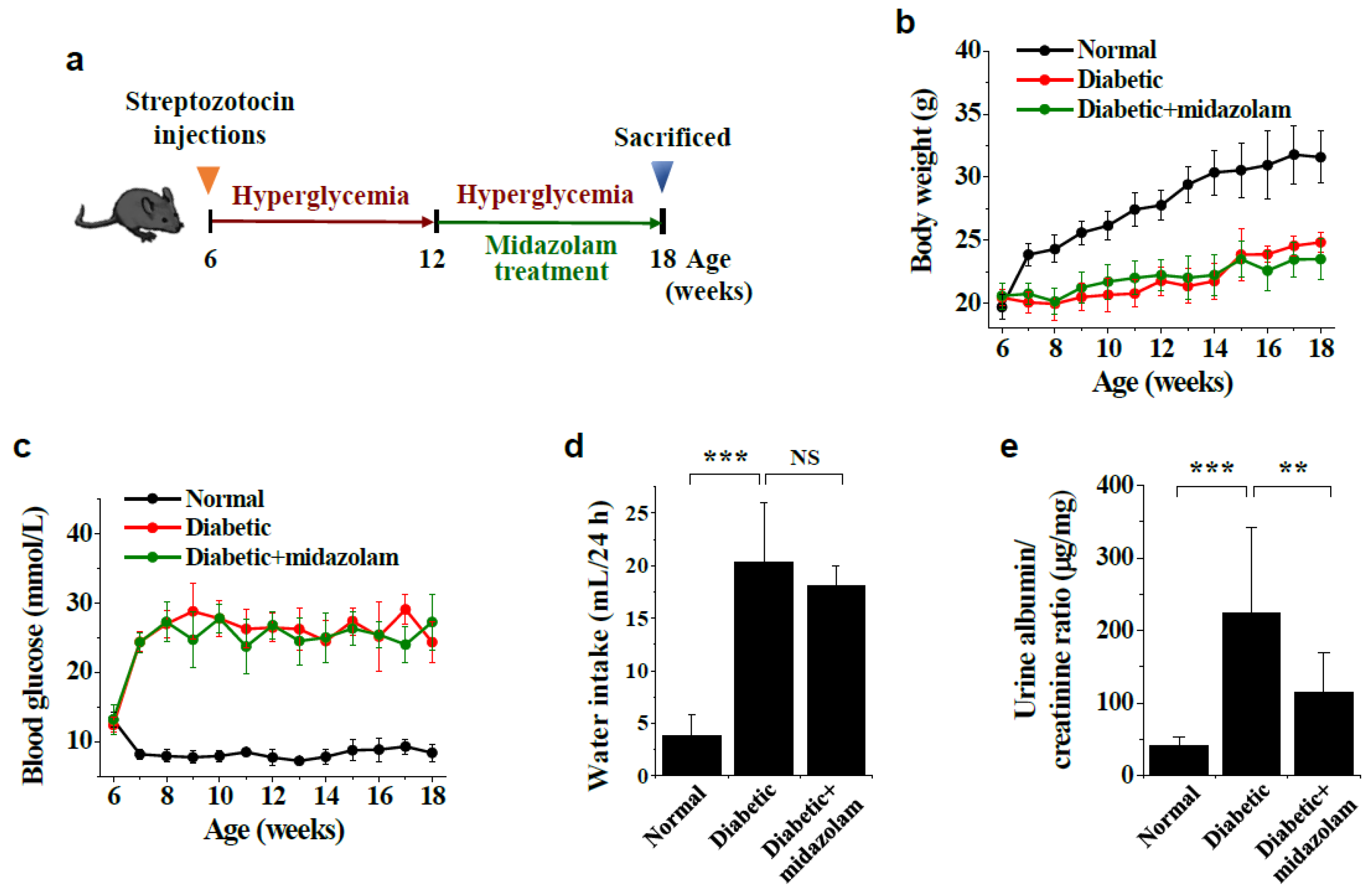
4.1. Diabetic Mouse Models and Midazolam Treatment
4.2. Measurement of Vascular Leakage in Mouse Kidneys
4.3. Visualization of VE-Cadherin in Mouse Kidneys
4.4. Measurement of ROS Generation in Mouse Kidneys
4.5. Measurement of In Vivo TGase Activity in Mouse Kidneys
4.6. Renal Histopathological Analysis
4.7. Cell Culture
4.8. Measurement of Intracellular Ca2+ and ROS Levels
4.9. Measurement of In Situ TGase Activity
4.10. Visualization of VE-Cadherin and Actin Filaments in HGECs
4.11. In Vitro Glomerular Endothelial Cell Monolayer Permeability Assay
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Said, S.M.; Nasr, S.H. Silent diabetic nephropathy. Kidney Int. 2016, 90, 24–26. [Google Scholar] [CrossRef]
- Wang, G.; Ouyang, J.; Li, S.; Wang, H.; Lian, B.; Liu, Z.; Xie, L. The analysis of risk factors for diabetic nephropathy progression and the construction of a prognostic database for chronic kidney diseases. J. Transl. Med. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Stehouwer, C.D. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle with Widespread Consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef] [Green Version]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Prat-Duran, J.; Pinilla, E.; Nørregaard, R.; Simonsen, U.; Buus, N.H. Transglutaminase 2 as a novel target in chronic kidney disease—Methods, mechanisms and pharmacological inhibition. Pharmacol. Ther. 2021, 222, 107787. [Google Scholar] [CrossRef]
- Klessens, C.Q.; Woutman, T.D.; Veraar, K.A.; Zandbergen, M.; Valk, E.J.; Rotmans, J.; Wolterbeek, R.; Bruijn, J.A.; Bajema, I.M. An autopsy study suggests that diabetic nephropathy is underdiagnosed. Kidney Int. 2016, 90, 149–156. [Google Scholar] [CrossRef]
- Cai, A.; Chatziantoniou, C.; Calmont, A. Vascular Permeability: Regulation Pathways and Role in Kidney Diseases. Nephron 2021, 145, 297–310. [Google Scholar] [CrossRef]
- Cheng, H.; Harris, R.C. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Jung, S.H.; Kim, S.H.; Kim, M.S.; Lee, S.; Hwang, J.; Kim, S.Y.; Kim, Y.M.; Ha, K.S. Essential Role of Transglu-taminase 2 in Vascular Endothelial Growth Factor-Induced Vascular Leakage in the Retina of Diabetic Mice. Diabetes 2016, 65, 2414–2428. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.-Y.; Lee, Y.-J.; Kim, Y.-S.; Kim, S.-Y.; Han, E.-T.; Park, W.S.; Hong, S.-H.; Kim, Y.-M.; Ha, K.-S. Proinsulin C-peptide prevents hyperglycemia-induced vascular leakage and metastasis of melanoma cells in the lungs of diabetic mice. FASEB J. 2019, 33, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-Y.; Seo, J.-A.; Jung, S.-H.; Lee, Y.-J.; Han, E.-T.; Park, W.S.; Hong, S.-H.; Kim, Y.-M.; Ha, K.-S. Insulin prevents pulmonary vascular leakage by inhibiting transglutaminase 2 in diabetic mice. Life Sci. 2019, 233, 116711. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.-H.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olkkola, K.T.; Ahonen, J. Midazolam and Other Benzodiazepines. Neurotrophic Factors 2008, 182, 335–360. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Kim, M.; Lee, J.-Y.; Jung, S.-H.; Jeon, H.-Y.; Lee, S.A.; Kang, S.; Han, E.-T.; Park, W.S.; Hong, S.-H.; et al. The benzodiazepine anesthetic midazolam prevents hyperglycemia-induced microvascular leakage in the retinas of diabetic mice. FASEB J. 2018, 32, 6089–6099. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.; Liu, N.; Kuhn, L.A.; Garavito, R.M.; Ferguson-Miller, S. Translocator Protein 18 kDa (TSPO): An Old Protein with New Functions? Biochemistry 2016, 55, 2821–2831. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-Y.; Guo, F.; Wu, H.-L.; Wang, Y.; Liu, J.-S. Midazolam anesthesia protects neuronal cells from oxidative stress-induced death via activation of the JNK-ERK pathway. Mol. Med. Rep. 2016, 15, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; You, Q.; Tu, Y.; Li, Q.; Zheng, L.; Li, X.; Guonian, W.; Wang, G. Midazolam Inhibits the Apoptosis of Astrocytes Induced by Oxygen Glucose Deprivation via Targeting JAK2-STAT3 Signaling Pathway. Cell. Physiol. Biochem. 2015, 35, 126–136. [Google Scholar] [CrossRef]
- Seo, J.-A.; Jeon, H.-Y.; Kim, M.; Lee, Y.-J.; Han, E.-T.; Park, W.S.; Hong, S.-H.; Kim, Y.-M.; Ha, K.-S. Anti-metastatic effect of midazolam on melanoma B16F10 cells in the lungs of diabetic mice. Biochem. Pharmacol. 2020, 178, 114052. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Wang, Y.; Sun, X.; Jiang, X. Midazolam induces A549 cell apoptosis in vitro via the miR-520d-5p/STAT3 pathway. Int J Clin. Exp. Pathol. 2018, 11, 1365–1373. [Google Scholar] [PubMed]
- Joo, H.K.; Oh, S.C.; Cho, E.J.; Park, K.S.; Lee, J.Y.; Lee, E.J.; Lee, S.K.; Kim, H.S.; Park, J.B.; Jeon, B.H. Midazolam inhibits tumor necrosis factor-alpha-induced endothelial activation: Involvement of the peripheral benzodiazepine receptor. Anesthesiology 2009, 110, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Choi, S.S.; Ha, K.-S. Transglutaminase 2: A multi-functional protein in multiple subcellular compartments. Amino Acids 2010, 39, 619–631. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and Disease: Lessons From Genetically Engineered Mouse Models and Inherited Disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.-H.; Kwon, M.-H.; Han, E.-T.; Park, W.S.; Hong, S.-H.; Kim, Y.-M.; Ha, K.-S. Array-based Investigation of Amino Acids Responsible for Regulation of Transamidase and Kinase Activities of Transglutaminase 2. BioChip J. 2019, 13, 251–259. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.-C.; Hwang, J.; Na, S.; Kim, Y.-M.; Ha, K.-S. C-Peptide Prevents Hyperglycemia-Induced Endothelial Apoptosis Through Inhibition of Reactive Oxygen Species–Mediated Transglutaminase 2 Activation. Diabetes 2012, 62, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurminskaya, M.V.; Belkin, A.M. Cellular Functions of Tissue Transglutaminase. Cell Death Regul. Health Dis. Part C 2012, 294, 1–97. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Feldman, E.; Pennathur, S.; Kretzler, M.; Brosius, F.C. 3rd, From fibrosis to sclerosis: Mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 2008, 57, 1439–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.-K.; Kim, S.-I.; Oh, S.Y.; Na, W.; Kang, Y.-H. Tangeretin Ameliorates Glucose-Induced Podocyte Injury through Blocking Epithelial to Mesenchymal Transition Caused by Oxidative Stress and Hypoxia. Int. J. Mol. Sci. 2020, 21, 8577. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Y.; Li, C.; Wang, X.; Zhu, R.; Liu, C.; Liu, H.; Wang, L.; Ma, R.; Fu, M.; et al. Recent Advances of Curcumin in the Prevention and Treatment of Renal Fibrosis. BioMed Res. Int. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Goldschmeding, R.; Floege, J.; Boor, P. Treatment of Renal Fibrosis—Turning Challenges into Opportunities. Adv. Chronic Kidney Dis. 2017, 24, 117–129. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Chertow, G.M.; Pergola, P.E.; Chen, F.; Kirby, B.J.; Sundy, J.S.; Patel, U.D.; GS-US-223-1015 Investigators. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Zaporowska-Stachowiak, I.; Szymanski, K.; Oduah, M.T.; Stachowiak-Szymczak, K.; Luczak, J.; Sopata, M. Midazolam: Safety of use in palliative care A systematic critical review. Biomed. Pharmacother. 2019, 114, 108838. [Google Scholar] [CrossRef]
- Patel, I.; Soni, P.; Fukuda, E.; Smith, D.; Leier, C.; Boudoulas, H. The pharmacokinetics of midazolam in patients with congestive heart failure. Br. J. Clin. Pharmacol. 1990, 29, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Lee, Y.; Jeon, H.; Han, E.; Park, W.S.; Hong, S.; Kim, Y.; Ha, K. The vicious cycle between transglutaminase 2 and reactive oxygen species in hyperglycemic memory–induced endothelial dysfunction. FASEB J. 2019, 33, 12655–12667. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.-Y.; Jung, S.-H.; Jung, Y.M.; Kim, Y.-M.; Ghandehari, H.; Ha, K.-S. Array-Based High-Throughput Analysis of Silk-Elastinlike Protein Polymer Degradation and C-Peptide Release by Proteases. Anal. Chem. 2016, 88, 5398–5405. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, J.-A.; Sayyed, N.D.; Lee, Y.-J.; Jeon, H.-Y.; Kim, E.-B.; Hong, S.-H.; Cho, S.; Kim, M.; Ha, K.-S. Midazolam Ameliorates Hyperglycemia-Induced Glomerular Endothelial Dysfunction by Inhibiting Transglutaminase 2 in Diabetes. Int. J. Mol. Sci. 2022, 23, 753. https://doi.org/10.3390/ijms23020753
Seo J-A, Sayyed ND, Lee Y-J, Jeon H-Y, Kim E-B, Hong S-H, Cho S, Kim M, Ha K-S. Midazolam Ameliorates Hyperglycemia-Induced Glomerular Endothelial Dysfunction by Inhibiting Transglutaminase 2 in Diabetes. International Journal of Molecular Sciences. 2022; 23(2):753. https://doi.org/10.3390/ijms23020753
Chicago/Turabian StyleSeo, Jae-Ah, Nilofar Danishmalik Sayyed, Yeon-Ju Lee, Hye-Yoon Jeon, Eun-Bin Kim, Seok-Ho Hong, Soyeon Cho, Minsoo Kim, and Kwon-Soo Ha. 2022. "Midazolam Ameliorates Hyperglycemia-Induced Glomerular Endothelial Dysfunction by Inhibiting Transglutaminase 2 in Diabetes" International Journal of Molecular Sciences 23, no. 2: 753. https://doi.org/10.3390/ijms23020753
APA StyleSeo, J.-A., Sayyed, N. D., Lee, Y.-J., Jeon, H.-Y., Kim, E.-B., Hong, S.-H., Cho, S., Kim, M., & Ha, K.-S. (2022). Midazolam Ameliorates Hyperglycemia-Induced Glomerular Endothelial Dysfunction by Inhibiting Transglutaminase 2 in Diabetes. International Journal of Molecular Sciences, 23(2), 753. https://doi.org/10.3390/ijms23020753