

SHP-1/STAT3-Signaling-Axis-Regulated Coupling between BECN1 and SLC7A11 Contributes to Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma

Abstract
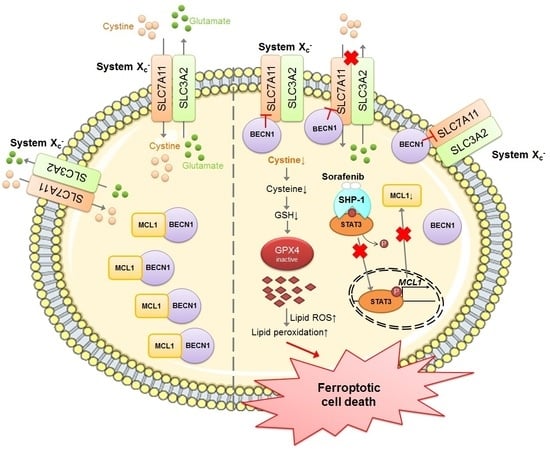
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
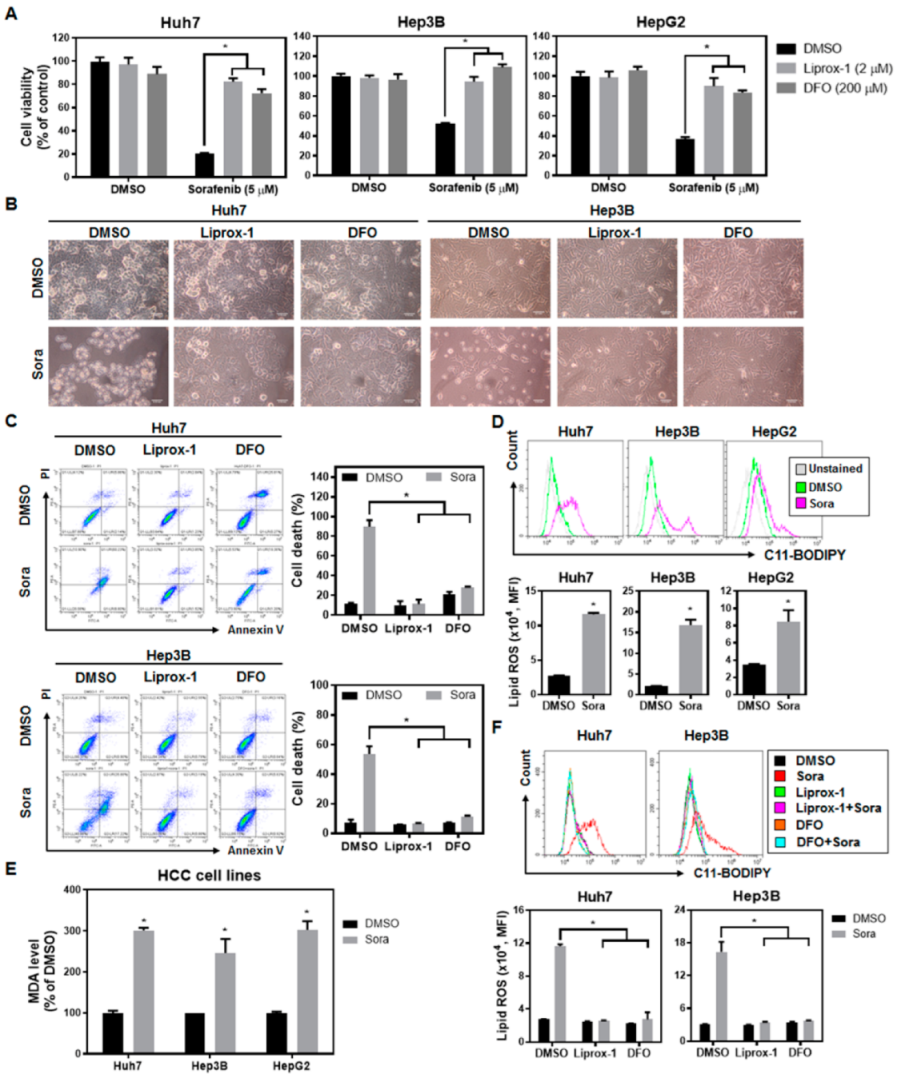
2.1. Sorafenib Treatment Enhances Lipid Reactive Oxygen Species (ROS) Levels and Induces Ferroptotic Cell Death in Hepatocellular Carcinoma (HCC) Cells
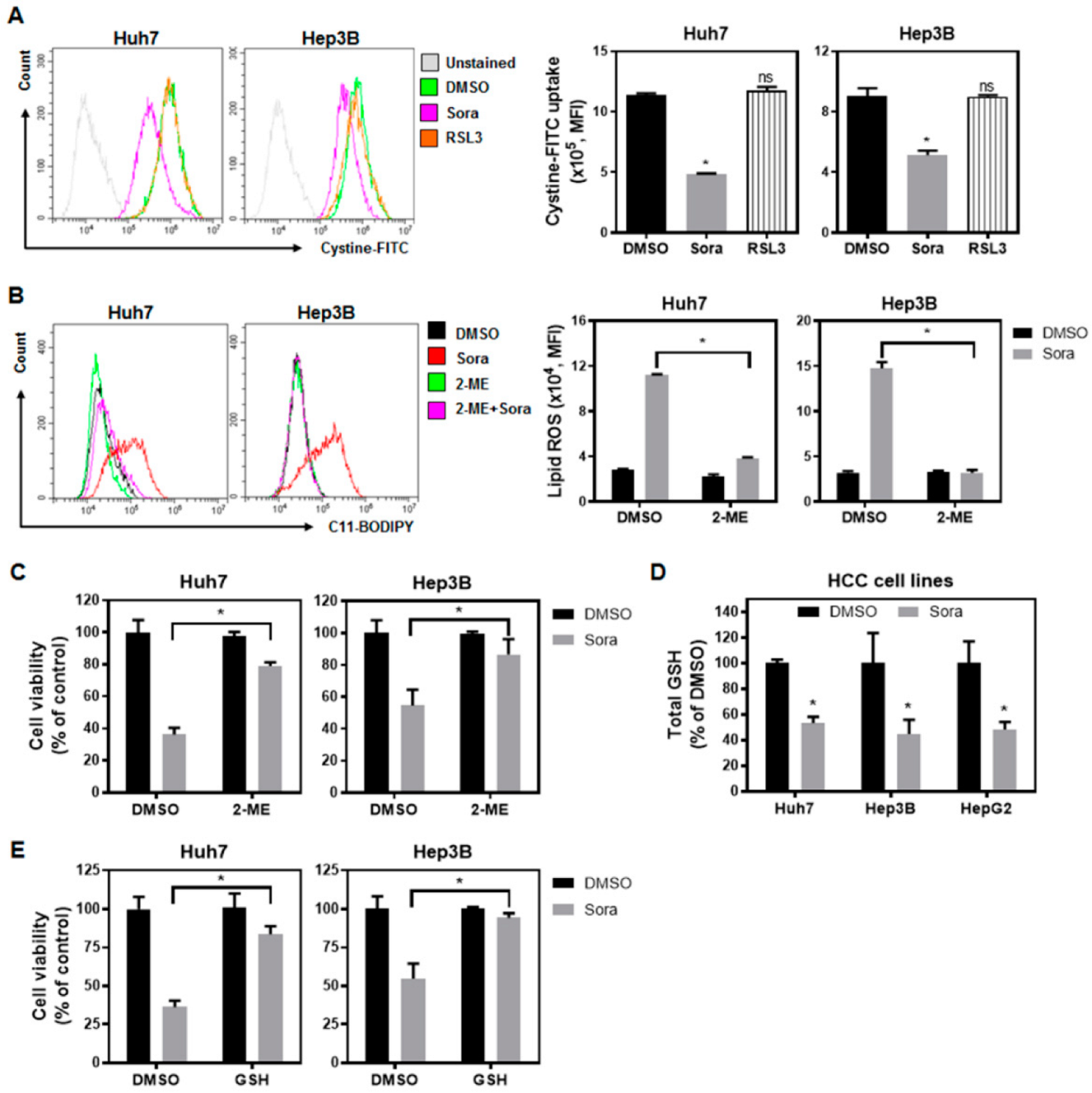
2.2. Sorafenib Inhibits System-Xc−-Mediated Cystine Intake and Enhances Lipid ROS Levels within HCC Cells, Contributing to Ferroptotic Cell Death
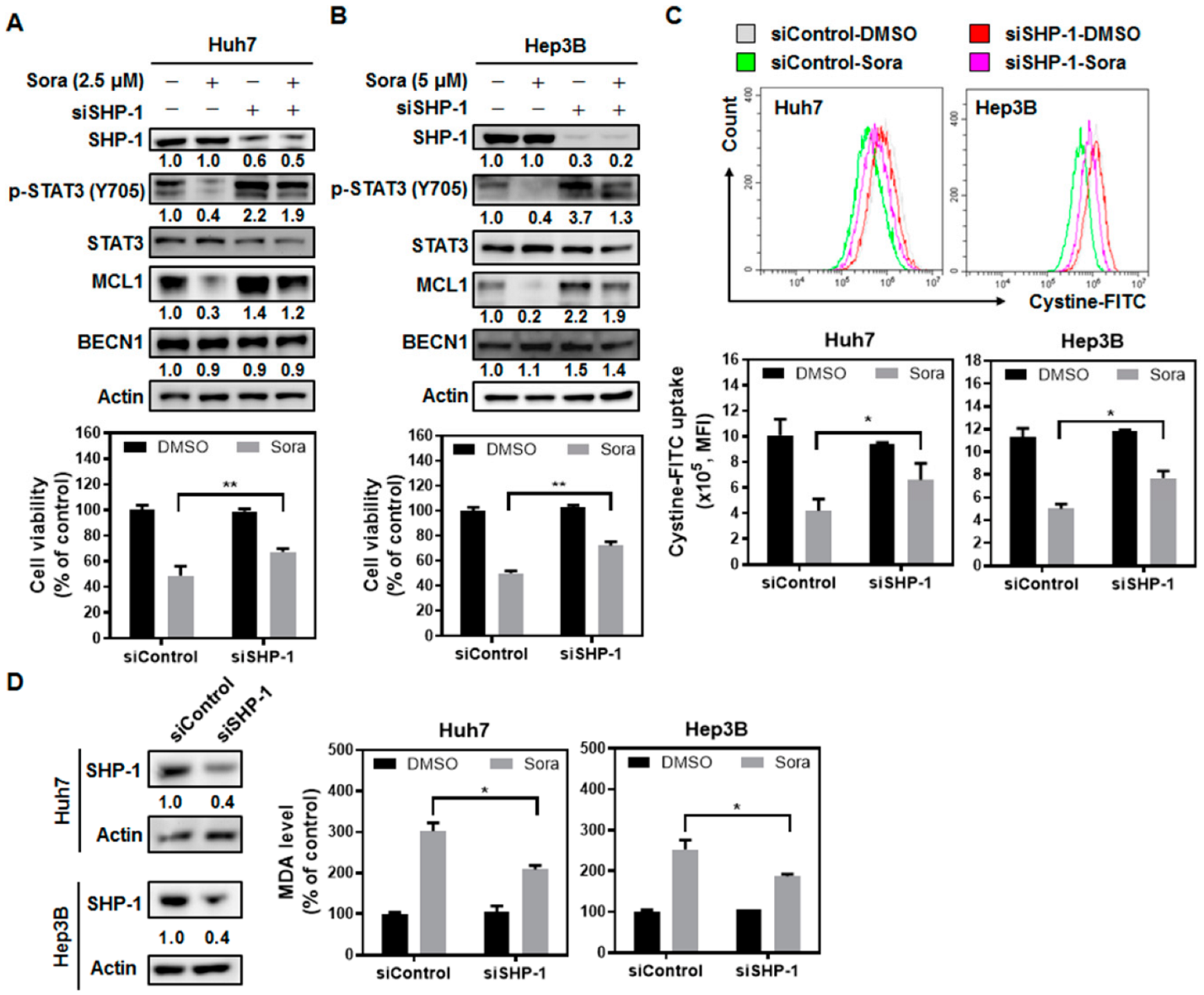
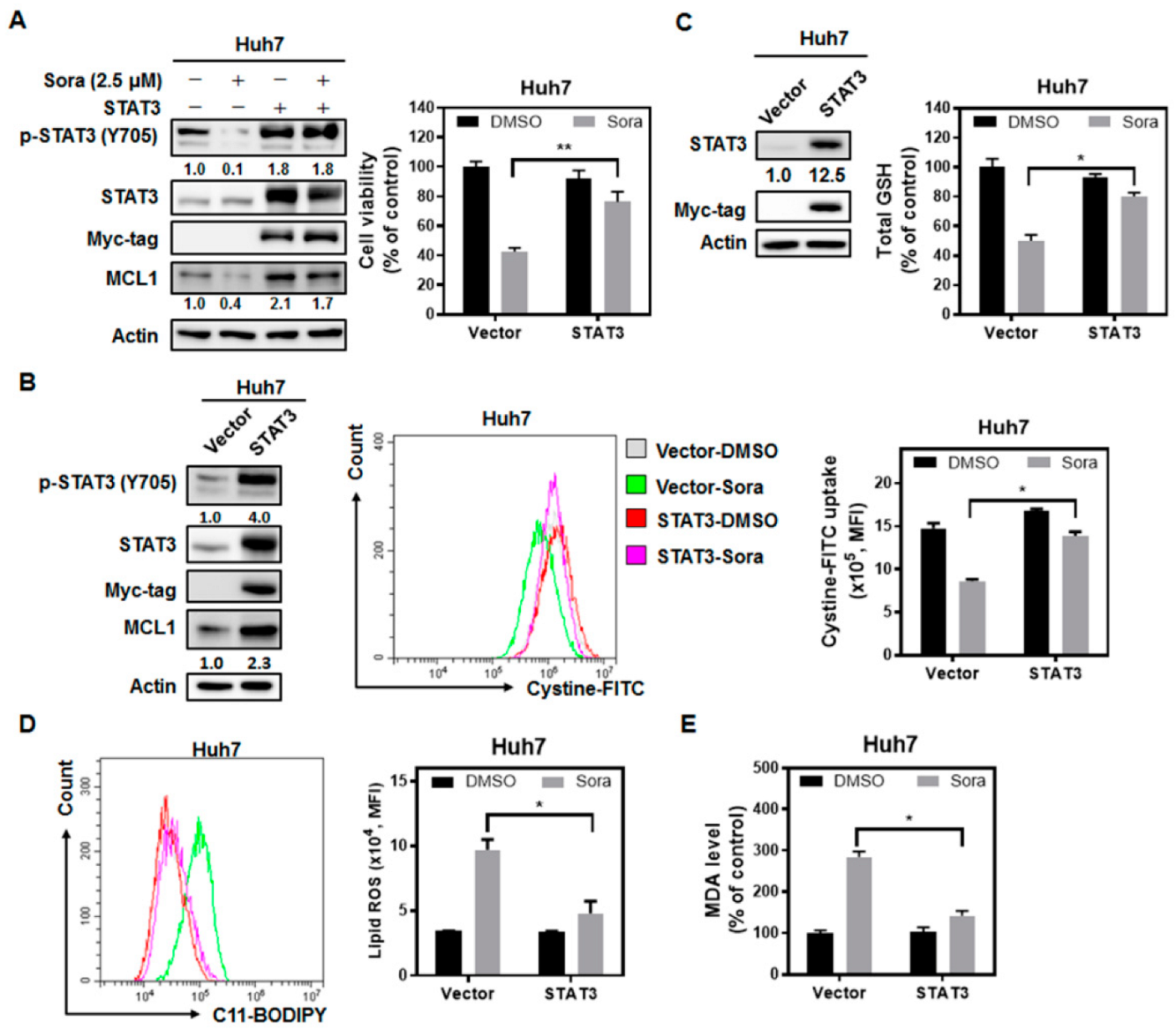
2.3. The Src Homology Region 2 Domain-Containing Phosphatase-1 (SHP-1)/STAT3 Signaling Pathway Plays a Pivotal Role in Sorafenib-Induced Ferroptosis in HCC
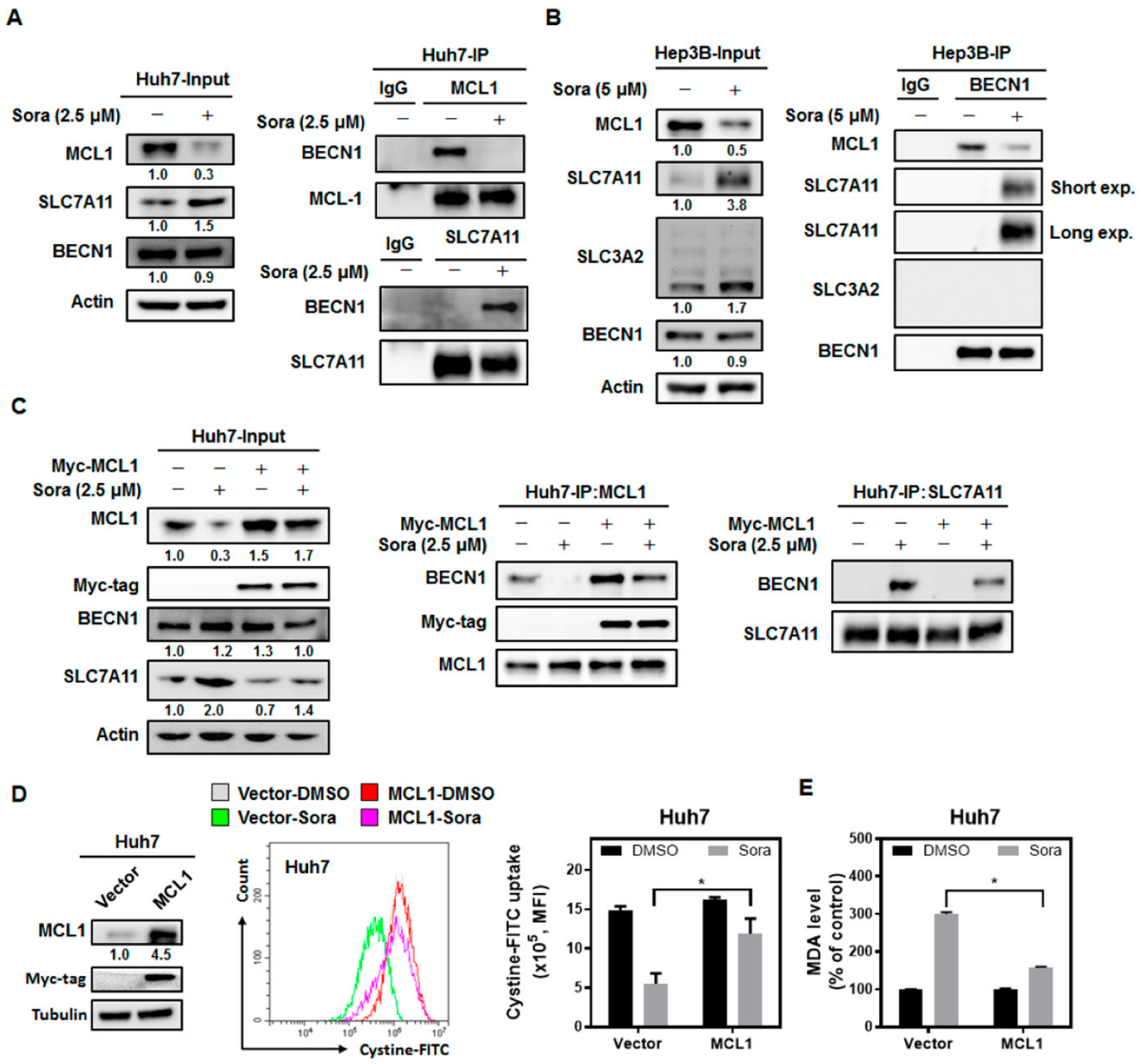
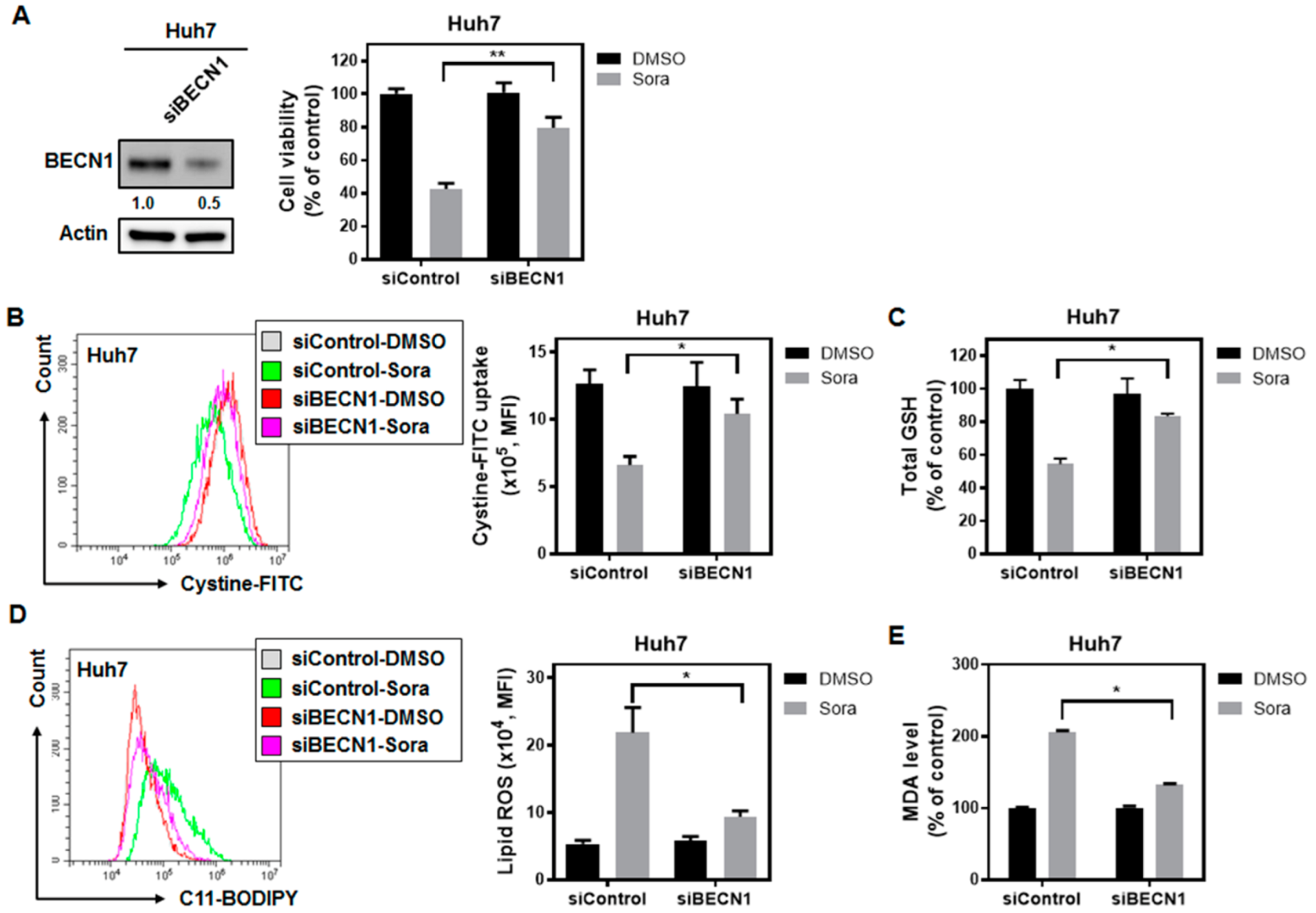
2.4. Sorafenib Treatment Dissociates Beclin 1(BECN1) from MCL1, and the Released BECN1 Inhibits Solute Carrier Family 7 Member 11 (SLC7A11), Culminating in Sorafenib-Triggered Ferroptosis in HCC Cells
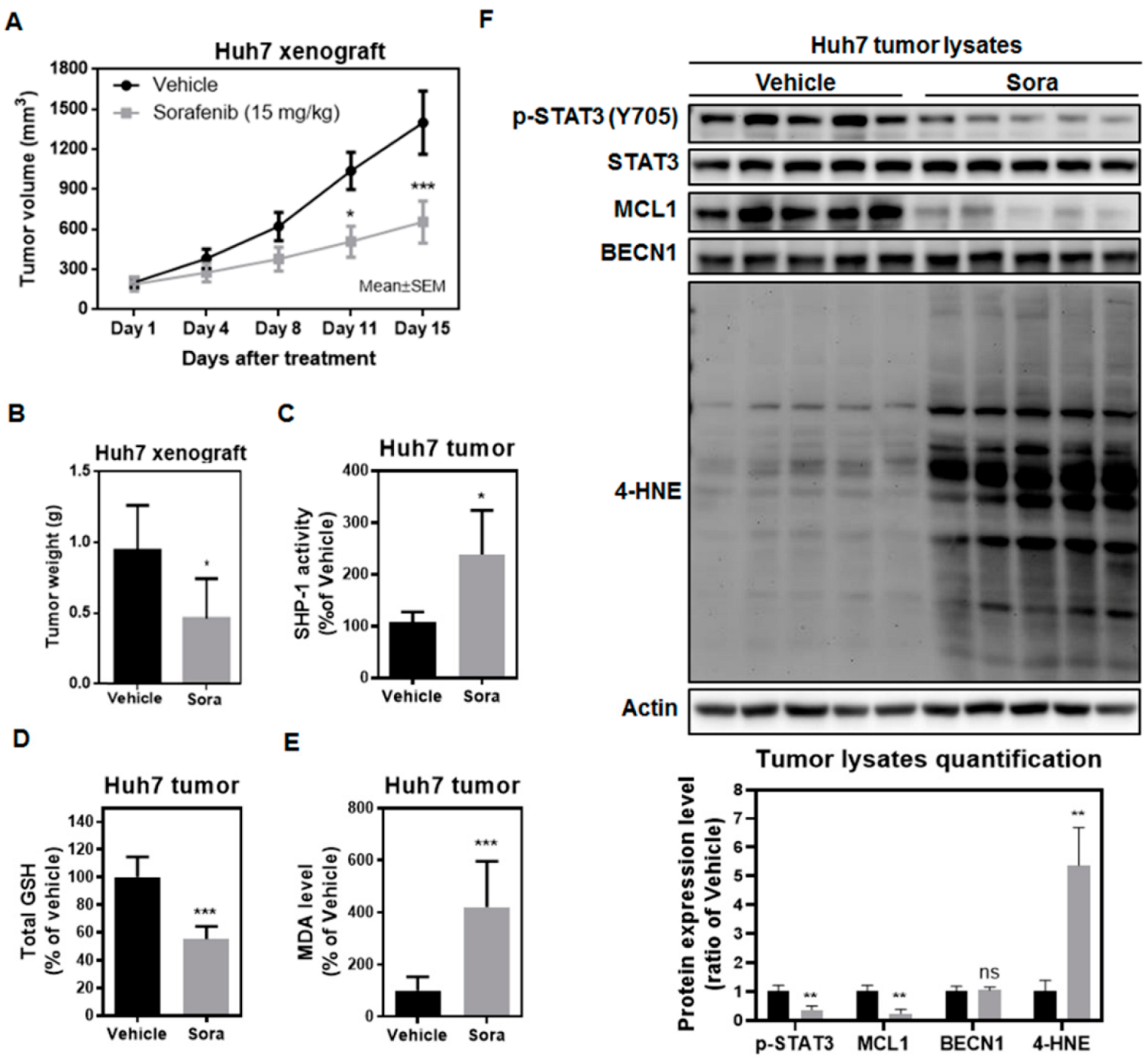
2.5. Sorafenib Induces Ferroptosis in the Huh7 Murine Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Reagents, Chemicals, and Antibodies
4.2. Cell Culture
4.3. Flow Cytometry Analysis
4.4. GSH Assay
4.5. Lipid Peroxidation Assay
4.6. Western Blot Analyses
4.7. Immunoprecipitation Analysis
4.8. Reverse-Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
4.9. SHP-1 Activity Measurement
4.10. Xenograft Tumor Model
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Steegmann-Olmedillas, J.L. The role of iron in tumour cell proliferation. Clin. Transl. Oncol. 2011, 13, 71–76. [Google Scholar] [CrossRef]
- Jung, M.; Mertens, C.; Tomat, E.; Brune, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Puisieux, M.F.; Pellat, A.; Assaf, A.; Ginestet, C.; Brezault, C.; Dhooge, M.; Soyer, P.; Coriat, R. Therapeutic Management of Advanced Hepatocellular Carcinoma: An Updated Review. Cancers 2022, 14, 2357. [Google Scholar] [CrossRef]
- Hage, C.; Hoves, S.; Strauss, L.; Bissinger, S.; Prinz, Y.; Poschinger, T.; Kiessling, F.; Ries, C.H. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell-Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology 2019, 70, 1280–1297. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Maziere, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, F. Propofol induces the ferroptosis of colorectal cancer cells by downregulating STAT3 expression. Oncol. Lett. 2021, 22, 767. [Google Scholar] [CrossRef]
- Chen, K.F.; Tai, W.T.; Liu, T.H.; Huang, H.P.; Lin, Y.C.; Shiau, C.W.; Li, P.K.; Chen, P.J.; Cheng, A.L. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin. Cancer Res. 2010, 16, 5189–5199. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Li, H.; Lou, L.; Huang, Q.; Zhang, Z.; Mo, J.; Li, M.; Lu, J.; Zhu, K.; Chu, Y.; et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. 2022, 52, 102317. [Google Scholar] [CrossRef]
- Yang, L.X.; Wu, Y.N.; Wang, P.W.; Su, W.C.; Shieh, D.B. Iron Release Profile of Silica-Modified Zero-Valent Iron NPs and Their Implication in Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4336. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Investig. 2021, 131, e143691. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc− Activity. Curr. Biol. 2018, 28, 2388–2399.e2385. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Nguyen, A.P.; le Grand, J.N.; Arbour, N.; Vanderluit, J.L.; Park, D.S.; Opferman, J.T.; Slack, R.S. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011, 30, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Mazumder, S.; Hill, B.T.; Kalaycio, M.; Almasan, A. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013, 4, e628. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Bekric, D.; Ocker, M.; Mayr, C.; Stintzing, S.; Ritter, M.; Kiesslich, T.; Neureiter, D. Ferroptosis in Hepatocellular Carcinoma: Mechanisms, Drug Targets and Approaches to Clinical Translation. Cancers 2022, 14, 1826. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Chen, P.J.; Chu, P.Y.; Huang, H.P.; Liu, C.Y.; Huang, J.W.; Chen, K.F. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology 2014, 59, 190–201. [Google Scholar] [CrossRef]
- Maiuri, M.C.; le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers 2019, 11, 1646. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Aishima, S.; Fujita, N.; Tanaka, Y.; Kubo, Y.; Motomura, T.; Taketomi, A.; Shirabe, K.; Maehara, Y.; Oda, Y. Tumor-associated macrophage promotes tumor progression via STAT3 signaling in hepatocellular carcinoma. Pathobiology 2013, 80, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, J.; Liu, W.N.; Fong, S.Y.; Shuen, T.W.H.; Liu, M.; Harden, S.; Tan, S.Y.; Cheng, J.Y.; Tan, W.W.S.; et al. Analysis and Validation of Human Targets and Treatments Using a Hepatocellular Carcinoma-Immune Humanized Mouse Model. Hepatology 2021, 74, 1395–1410. [Google Scholar] [CrossRef]
- Li, Y.; Song, Z.; Han, Q.; Zhao, H.; Pan, Z.; Lei, Z.; Zhang, J. Targeted inhibition of STAT3 induces immunogenic cell death of hepatocellular carcinoma cells via glycolysis. Mol. Oncol. 2022, 16, 2861–2880. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Lin, Y.; Lu, X.; Zhang, C.; Chen, S.; Jian, Z. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell Biochem. 2018, 119, 9419–9432. [Google Scholar] [CrossRef]
- Su, T.H.; Shiau, C.W.; Jao, P.; Liu, C.H.; Liu, C.J.; Tai, W.T.; Jeng, Y.M.; Yang, H.C.; Tseng, T.C.; Huang, H.P.; et al. Sorafenib and its derivative SC-1 exhibit antifibrotic effects through signal transducer and activator of transcription 3 inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 7243–7248. [Google Scholar] [CrossRef]
- Chen, C.H.; Chen, M.C.; Wang, J.C.; Tsai, A.C.; Chen, C.S.; Liou, J.P.; Pan, S.L.; Teng, C.M. Synergistic interaction between the HDAC inhibitor, MPT0E028, and sorafenib in liver cancer cells in vitro and in vivo. Clin. Cancer Res. 2014, 20, 1274–1287. [Google Scholar] [CrossRef]
- Yao, X.; Zhao, C.R.; Yin, H.; Wang, K.; Gao, J.J. Synergistic antitumor activity of sorafenib and artesunate in hepatocellular carcinoma cells. Acta Pharmacol. Sin. 2002, 41, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Guo, W.; Zhao, Y.; Zhang, Z.; Tan, N.; Zhao, F.; Ge, C.; Liang, L.; Jia, D.; Chen, T.; Yao, M.; et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011, 312, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Iseda, N.; Itoh, S.; Toshida, K.; Tomiyama, T.; Morinaga, A.; Shimokawa, M.; Shimagaki, T.; Wang, H.; Kurihara, T.; Toshima, T.; et al. Ferroptosis is induced by lenvatinib through fibroblast growth factor receptor-4 inhibition in hepatocellular carcinoma. Cancer Sci. 2022, 113, 2272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wei, C.; Liu, G.; Zhang, L.; Li, J.; Li, L.; Cai, S.; Fang, L. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell Prolif. 2022, 55, e13158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- Yao, F.; Deng, Y.; Zhao, Y.; Mei, Y.; Zhang, Y.; Liu, X.; Martinez, C.; Su, X.; Rosato, R.R.; Teng, H.; et al. A targetable LIFR–NF-κB–LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat. Commun. 2021, 12, 7333. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-Y.; Chen, L.-J.; Chen, G.; Chao, T.-I.; Wang, C.-Y. SHP-1/STAT3-Signaling-Axis-Regulated Coupling between BECN1 and SLC7A11 Contributes to Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 11092. https://doi.org/10.3390/ijms231911092
Huang C-Y, Chen L-J, Chen G, Chao T-I, Wang C-Y. SHP-1/STAT3-Signaling-Axis-Regulated Coupling between BECN1 and SLC7A11 Contributes to Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(19):11092. https://doi.org/10.3390/ijms231911092
Chicago/Turabian StyleHuang, Chao-Yuan, Li-Ju Chen, Grace Chen, Tzu-I Chao, and Cheng-Yi Wang. 2022. "SHP-1/STAT3-Signaling-Axis-Regulated Coupling between BECN1 and SLC7A11 Contributes to Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 19: 11092. https://doi.org/10.3390/ijms231911092
APA StyleHuang, C.-Y., Chen, L.-J., Chen, G., Chao, T.-I., & Wang, C.-Y. (2022). SHP-1/STAT3-Signaling-Axis-Regulated Coupling between BECN1 and SLC7A11 Contributes to Sorafenib-Induced Ferroptosis in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 23(19), 11092. https://doi.org/10.3390/ijms231911092