

The Use of Retinoids for the Prevention and Treatment of Skin Cancers: An Updated Review
 , , ,
, , , 
Abstract
1. Introduction
2. The Biochemistry of Retinoids and Retinoid Receptors
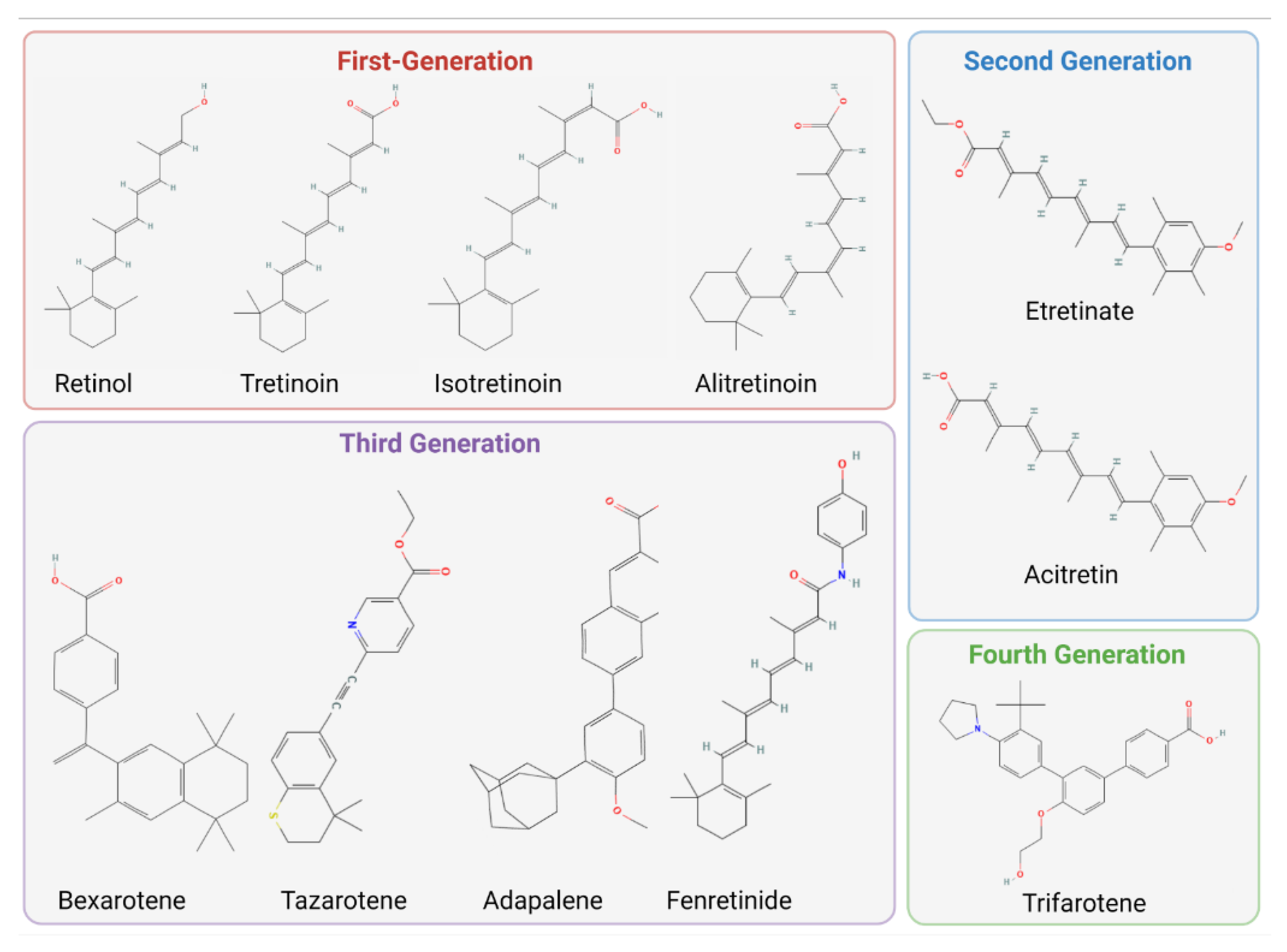
2.1. The Structures and Classifications of Retinoid Products
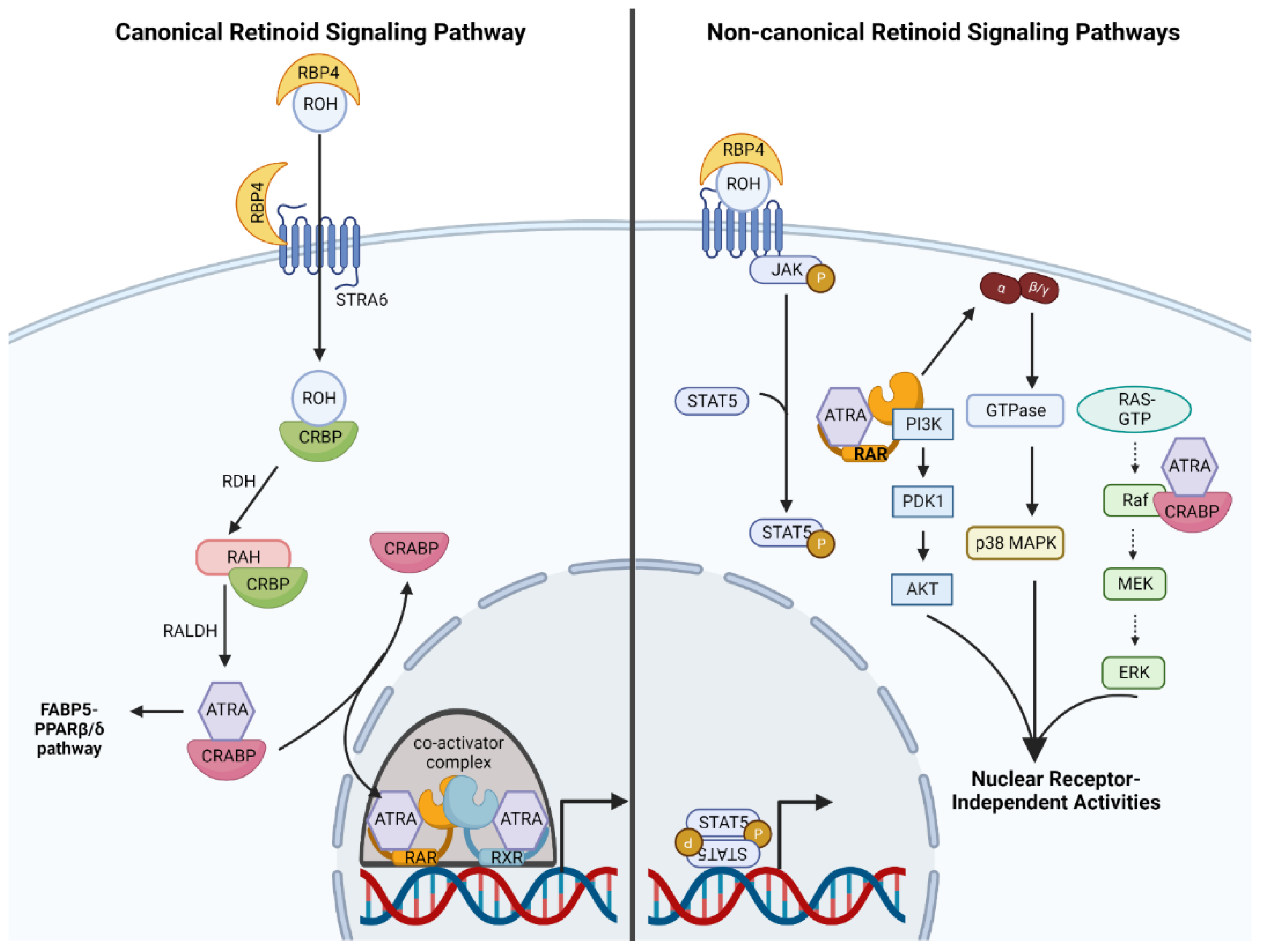
2.2. Molecular Mechanisms of Canonical Retinoid Signaling
2.3. Non-Canonical Retinoid Signaling: An Emerging Phenomenon
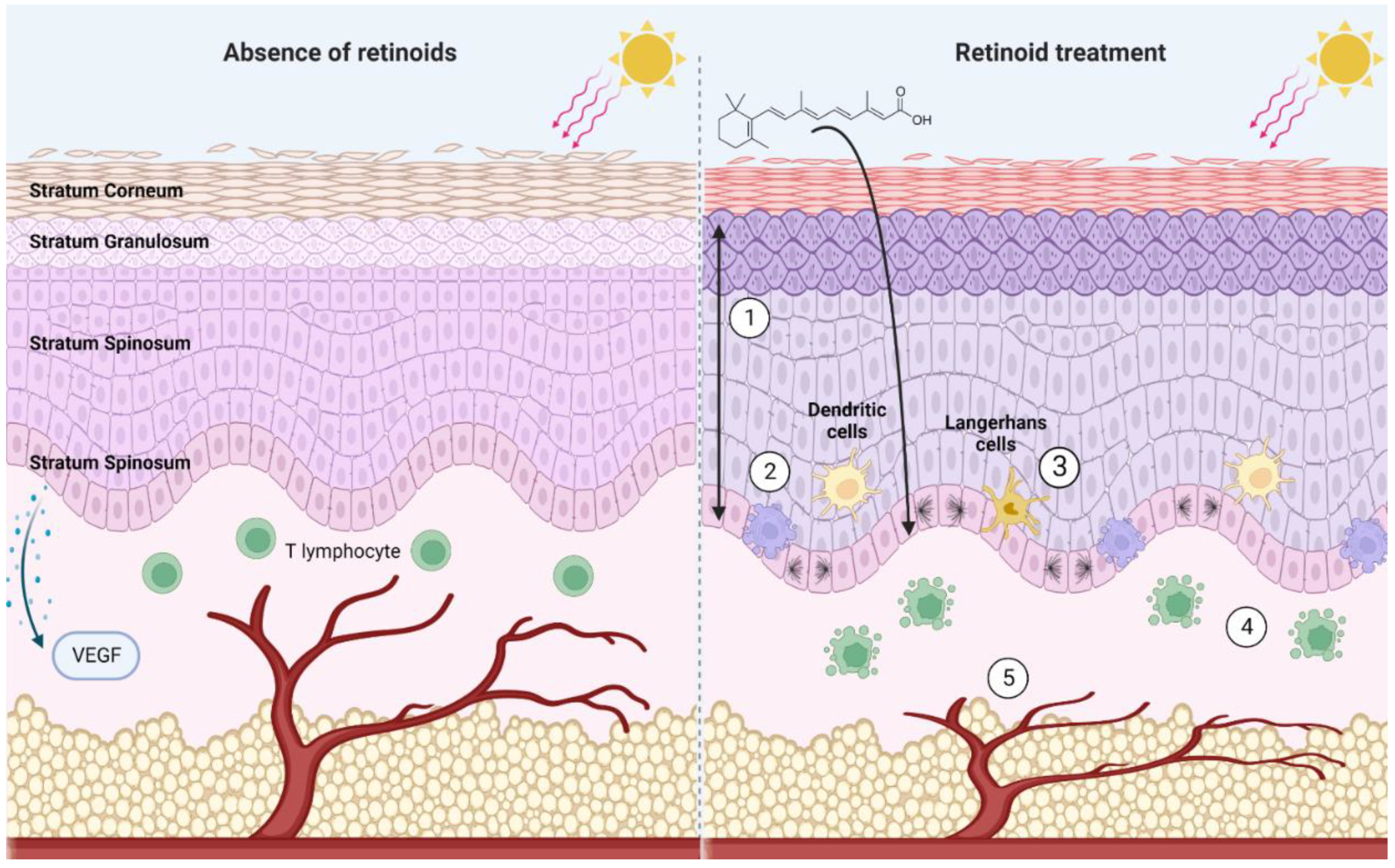
3. The Biological Effects of Retinoids in the Skin
3.1. Retinoids Control Epidermal Maturation and Turnover
3.2. Retinoids Influence the Immune Landscape of the Skin
3.3. Skin Structure and Vascularization Are Regulated by Retinoid Signaling
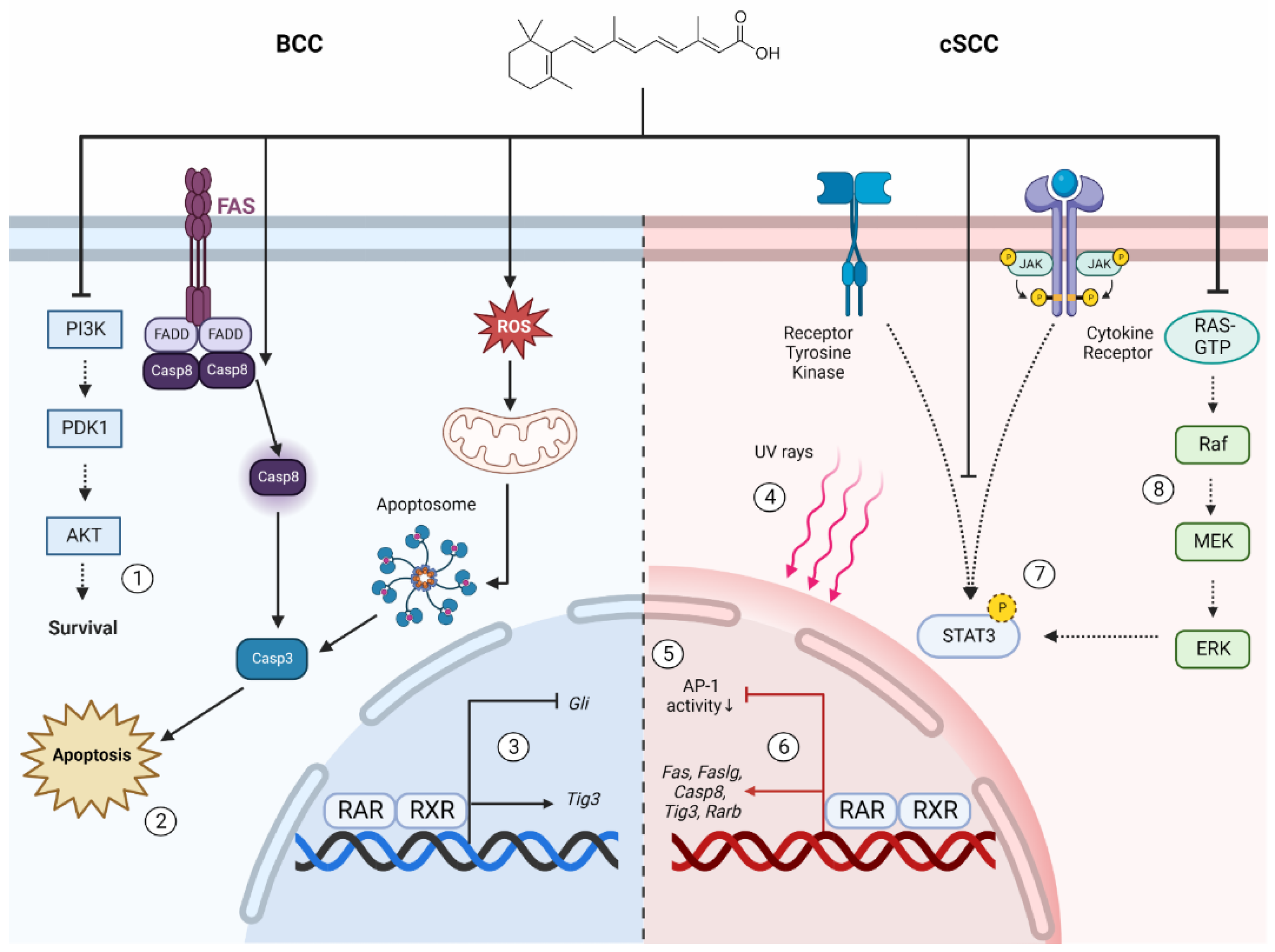
4. The Molecular Mechanisms of Retinoid Activity in Experimental Models of Keratinocyte Carcinomas (KCs)
4.1. Oncogenic Disruptions in Retinoid Signaling Contribute to Skin Carcinogenesis
4.2. The Prophylactic and Therapeutic Benefits of Retinoids in BCC Are Attributed to Apoptosis and Modulation of Oncogenic Signaling Pathways
4.3. Retinoids Suppress Several Hallmarks of Tumorigenesis in cSCC
4.4. Retinoids Exert Diverse Antineoplastic Effects in Models Representative of Other Skin Cancers
5. Clinical Applications of Retinoids for Keratinocyte Carcinoma Chemoprevention
6. The Use of Retinoids for Skin Cancer Treatment
6.1. The Prospect of Retinoids to Treat Keratinocyte Carcinomas and Premalignant Lesions
6.2. Retinoids Are Effective Treatments for CTCL and KS
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Wald, G.; Steven, D. An experiment in human vitamin A-deficiency. Proc. Natl. Acad. Sci. USA 1939, 25, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Tyson, M.D.; Smith, A.H. Tissue changes associated with vitamin A deficiency in the rat. Am. J. Pathol. 1929, 5, 57–70. [Google Scholar] [PubMed]
- Goodwin, G.P. A cutaneous manifestation of vitamin A deficiency. Br. Med. J. 1934, 2, 113–126. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hardy, M.K. Vitamin A deficiency producing follicular hyperkeratosis. Arch. Dermatol. Syphilol. 1946, 53, 392. [Google Scholar]
- Sabella, J.D.; Bern, H.A.; Kahn, R.H. Effects of locally applied vitamin A and estrogen on rat epidermis. Proc. Soc. Exp. Biol. Med. 1951, 76, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Dunlop, N.M.; Newton, D.L.; Henderson, W.R. Relationships between structure and activity of retinoids. Nature 1976, 263, 110–113. [Google Scholar] [CrossRef]
- Straumfjord, J.V. Vitamin A: Its effect on acne. Northwest Med. 1943, 42, 219–225. [Google Scholar]
- Stüttgen, G. Historical perspectives of tretinoin. J. Am. Acad. Dermatol. 1986, 15, 735–740. [Google Scholar] [CrossRef]
- Kligman, A.M.; Fulton, J.E., Jr.; Plewig, G. Topical vitamin A acid in acne vulgaris. Arch. Dermatol. 1969, 99, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Patton, T.; Ferris, L.K.; Wolverton, S.E. 22-Systemic retinoids. In Comprehensive Dermatologic Drug Therapy, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 245–262.e4. [Google Scholar] [CrossRef]
- Sami, N.; Feld, S.d.l.; Wolverton, S.E. 46-Topical retinoids. In Comprehensive Dermatologic Drug Therapy, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 528–540.e4. [Google Scholar] [CrossRef]
- Tang, X.H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Dobrotkova, V.; Chlapek, P.; Mazanek, P.; Sterba, J.; Veselska, R. Traffic lights for retinoids in oncology: Molecular markers of retinoid resistance and sensitivity and their use in the management of cancer differentiation therapy. BMC Cancer 2018, 18, 1059. [Google Scholar] [CrossRef] [PubMed]
- Peck, G.L. Topical tretinoin in actinic keratosis and basal cell carcinoma. J. Am. Acad. Dermatol. 1986, 15, 829–835. [Google Scholar] [CrossRef]
- Karimkhani, C.; Boyers, L.N.; Dellavalle, R.P.; Weinstock, M.A. It’s time for “keratinocyte carcinoma” to replace the term “nonmelanoma skin cancer”. J. Am. Acad. Dermatol. 2015, 72, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Cattelan, L.; Ghazawi, F.M.; Le, M.; Lagace, F.; Savin, E.; Zubarev, A.; Gantchev, J.; Tomaszewski, M.; Sasseville, D.; Waschke, K.; et al. Epidemiologic trends and geographic distribution of esophageal cancer in Canada: A national population-based study. Cancer Med. 2020, 9, 401–417. [Google Scholar] [CrossRef]
- Cattelan, L.; Ghazawi, F.M.; Le, M.; Savin, E.; Zubarev, A.; Lagace, F.; Sasseville, D.; Waschke, K.; Litvinov, I.V. Investigating epidemiologic trends and the geographic distribution of patients with anal squamous cell carcinoma throughout Canada. Curr. Oncol. 2020, 27, e294–e306. [Google Scholar] [CrossRef] [PubMed]
- Gantchev, J.; Martinez Villarreal, A.; Gunn, S.; Zetka, M.; Odum, N.; Litvinov, I.V. The ectopic expression of meiCT genes promotes meiomitosis and may facilitate carcinogenesis. Cell Cycle 2020, 19, 837–854. [Google Scholar] [CrossRef]
- Ghazawi, F.M.; Lu, J.; Savin, E.; Zubarev, A.; Chauvin, P.; Sasseville, D.; Zeitouni, A.; Litvinov, I.V. Epidemiology and patient distribution of oral cavity and oropharyngeal SCC in Canada. J. Cutan. Med. Surg. 2020, 24, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Lagace, F.; Ghazawi, F.M.; Le, M.; Savin, E.; Zubarev, A.; Powell, M.; Moreau, L.; Sasseville, D.; Popa, I.; Litvinov, I.V. Penile invasive squamous cell carcinoma: Analysis of incidence, mortality trends, and geographic distribution in Canada. J. Cutan. Med. Surg. 2020, 24, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, E.; Jfri, A.; Gefri, A.; O’Brien, E.; Litvinov, I.V.; Zubarev, A.; Savin, E.; Netchiporouk, E. Cutaneous squamous cell carcinoma in patients with hidradenitis suppurativa. Cancers 2021, 13, 1153. [Google Scholar] [CrossRef] [PubMed]
- Nehal, K.S.; Bichakjian, C.K. Update on keratinocyte carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Lefrancois, P.; Sasseville, D.; Parmentier, L.; Litvinov, I.V. Analysis of multiple basal cell carcinomas (BCCs) arising in one individual highlights genetic tumor heterogeneity and identifies novel driver mutations. J. Cell Commun. Signal. 2022. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Xie, P.; Gunn, S.; Sasseville, D.; Lefrancois, P. The transcriptional landscape analysis of basal cell carcinomas reveals novel signalling pathways and actionable targets. Life Sci. Alliance 2021, 4, e202000651. [Google Scholar] [CrossRef] [PubMed]
- Lefrancois, P.; Xie, P.; Gunn, S.; Gantchev, J.; Villarreal, A.M.; Sasseville, D.; Litvinov, I.V. In silico analyses of the tumor microenvironment highlight tumoral inflammation, a Th2 cytokine shift and a mesenchymal stem cell-like phenotype in advanced in basal cell carcinomas. J. Cell Commun. Signal. 2020, 14, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Youssef, K.K.; Van Keymeulen, A.; Lapouge, G.; Beck, B.; Michaux, C.; Achouri, Y.; Sotiropoulou, P.A.; Blanpain, C. Identification of the cell lineage at the origin of basal cell carcinoma. Nat. Cell Biol. 2010, 12, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Diz, V.; Solé-Sánchez, S.; Valdés-Gutiérrez, A.; Urpí, M.; Riba-Artés, D.; Penin, R.M.; Pascual, G.; González-Suárez, E.; Casanovas, O.; Viñals, F.; et al. Progeny of Lgr5-expressing hair follicle stem cell contributes to papillomavirus-induced tumor development in epidermis. Oncogene 2013, 32, 3732–3743. [Google Scholar] [CrossRef] [PubMed]
- van de Glind, G.C.; Out-Luiting, J.J.; Rebel, H.G.; Tensen, C.P.; de Gruijl, F.R. Lgr5+ stem cells and their progeny in mouse epidermis under regimens of exogenous skin carcinogenesis, and their absence in ensuing skin tumors. Oncotarget 2016, 7, 52085–52094. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Urban, K.; Mehrmal, S.; Uppal, P.; Giesey, R.L.; Delost, G.R. The global burden of skin cancer: A longitudinal analysis from the Global Burden of Disease Study, 1990–2017. JAAD Int. 2021, 2, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Muntyanu, A.; Ghazawi, F.M.; Nedjar, H.; Rahme, E.; Alakel, A.; Zubarev, A.; Netchiporouk, E.; Litvinov, I.V. Non-Melanoma skin cancer distribution in the Russian federation. Dermatology 2021, 237, 1007–1015. [Google Scholar] [CrossRef]
- Ramchatesingh, B.; Gantchev, J.; Villarreal, A.M.; Gill, R.P.K.; Lambert, M.; Sivachandran, S.; Lefrancois, P.; Litvinov, I.V. The contributions of cancer-testis and developmental genes to the pathogenesis of keratinocyte carcinomas. Cancers 2022, 14, 3630. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Lefracnois, P.; Litvinov, I.V. Cytotoxic T cells are replaced by novel clones after immune checkpoint blocker therapy. J. Cutan. Med. Surg. 2020, 24, 314–315. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Asgari, M.M.; Green, A.C.; Guhan, S.M.; Arron, S.T.; Proby, C.M.; Rollison, D.E.; Harwood, C.A.; Toland, A.E. Keratinocyte carcinomas: Current concepts and future research priorities. Clin. Cancer Res. 2019, 25, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Ahmady, S.; Jansen, M.H.E.; Nelemans, P.J.; Kessels, J.; Arits, A.; de Rooij, M.J.M.; Essers, B.A.B.; Quaedvlieg, P.J.F.; Kelleners-Smeets, N.W.J.; Mosterd, K. Risk of invasive cutaneous squamous cell carcinoma after different treatments for actinic keratosis: A secondary analysis of a randomized clinical trial. JAMA Dermatol. 2022, 158, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Otley, C.C. Skin cancer in organ transplant recipients: Epidemiology, pathogenesis, and management. J. Am. Acad. Dermatol. 2002, 47, 1–20; quiz 18–20. [Google Scholar] [CrossRef]
- Jaju, P.D.; Ransohoff, K.J.; Tang, J.Y.; Sarin, K.Y. Familial skin cancer syndromes: Increased risk of nonmelanotic skin cancers and extracutaneous tumors. J. Am. Acad. Dermatol. 2016, 74, 437–451, quiz 452–434. [Google Scholar] [CrossRef]
- Bagherani, N.; Smoller, B.R. An overview of cutaneous T cell lymphomas. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Ghazawi, F.M.; Alghazawi, N.; Le, M.; Netchiporouk, E.; Glassman, S.J.; Sasseville, D.; Litvinov, I.V. Environmental and other extrinsic risk factors contributing to the pathogenesis of cutaneous T cell lymphoma (CTCL). Front. Oncol. 2019, 9, 300. [Google Scholar] [CrossRef]
- Ghazawi, F.M.; Litvinov, I.V. Distribution and clustering of cutaneous T-cell lymphoma (CTCL) cases in Canada: A response to a letter. J. Cutan. Med. Surg. 2018, 22, 657–658. [Google Scholar] [CrossRef]
- Ghazawi, F.M.; Netchiporouk, E.; Rahme, E.; Tsang, M.; Moreau, L.; Glassman, S.; Provost, N.; Gilbert, M.; Jean, S.E.; Pehr, K.; et al. Comprehensive analysis of cutaneous T-cell lymphoma (CTCL) incidence and mortality in Canada reveals changing trends and geographic clustering for this malignancy. Cancer 2017, 123, 3550–3567. [Google Scholar] [CrossRef]
- Ghazawi, F.M.; Netchiporouk, E.; Rahme, E.; Tsang, M.; Moreau, L.; Glassman, S.; Provost, N.; Gilbert, M.; Jean, S.E.; Roshdy, O.; et al. Distribution and clustering of cutaneous T-cell lymphoma (CTCL) cases in Canada during 1992 to 2010. J. Cutan. Med. Surg. 2018, 22, 154–165. [Google Scholar] [CrossRef]
- Olsen, E.; Vonderheid, E.; Pimpinelli, N.; Willemze, R.; Kim, Y.; Knobler, R.; Zackheim, H.; Duvic, M.; Estrach, T.; Lamberg, S.; et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007, 110, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Cordeiro, B.; Fredholm, S.; Odum, N.; Zargham, H.; Huang, Y.; Zhou, Y.; Pehr, K.; Kupper, T.S.; Woetmann, A.; et al. Analysis of STAT4 expression in cutaneous T-cell lymphoma (CTCL) patients and patient-derived cell lines. Cell Cycle 2014, 13, 2975–2982. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Cordeiro, B.; Huang, Y.; Zargham, H.; Pehr, K.; Dore, M.A.; Gilbert, M.; Zhou, Y.; Kupper, T.S.; Sasseville, D. Ectopic expression of cancer-testis antigens in cutaneous T-cell lymphoma patients. Clin. Cancer Res. 2014, 20, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Jones, D.A.; Sasseville, D.; Kupper, T.S. Transcriptional profiles predict disease outcome in patients with cutaneous T-cell lymphoma. Clin. Cancer Res. 2010, 16, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Kupper, T.S.; Sasseville, D. The role of AHI1 and CDKN1C in cutaneous T-cell lymphoma progression. Exp. Dermatol. 2012, 21, 964–966. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Dore, M.A.; Moreau, L.; Pehr, K.; Gilbert, M.; Zhou, Y.; Sasseville, D.; Kupper, T.S. The use of transcriptional profiling to improve personalized diagnosis and management of cutaneous T-cell lymphoma (CTCL). Clin. Cancer Res. 2015, 21, 2820–2829. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Zargham, H.; Pehr, K.; Gilbert, M.; Zhou, Y.; Moreau, L.; Woetmann, A.; Odum, N.; et al. Ectopic expression of embryonic stem cell and other developmental genes in cutaneous T-cell lymphoma. Oncoimmunology 2014, 3, e970025. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Pehr, K.; Sasseville, D. Connecting the dots in cutaneous T cell lymphoma (CTCL): STAT5 regulates malignant T cell proliferation via miR-155. Cell Cycle 2013, 12, 2172–2173. [Google Scholar] [CrossRef]
- Zhang, C.; Duvic, M. Treatment of cutaneous T-cell lymphoma with retinoids. Dermatol. Ther. 2006, 19, 264–271. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Prim. 2019, 5, 9. [Google Scholar] [CrossRef]
- Antman, K.; Chang, Y. Kaposi’s sarcoma. N. Engl. J. Med. 2000, 342, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Ruvolo, V.R.; Burns, W.H.; Sandford, G.; Wan, X.; Ciufo, D.; Hendrickson, S.B.; Guo, H.G.; Hayward, G.S.; Reitz, M.S. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 1997, 3, 287–292. [Google Scholar] [CrossRef] [PubMed]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature of retinoids: Recommendations 1981. Eur. J. Biochem. 1982, 129, 728–731. [Google Scholar]
- Rusu, A.; Tanase, C.; Pascu, G.A.; Todoran, N. Recent advances regarding the therapeutic potential of adapalene. Pharmaceuticals 2020, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Zasada, M.; Budzisz, E. Retinoids: Active molecules influencing skin structure formation in cosmetic and dermatological treatments. Postep. Dermatol. Alergol. 2019, 36, 392–397. [Google Scholar] [CrossRef]
- Aubert, J.; Piwnica, D.; Bertino, B.; Blanchet-Réthoré, S.; Carlavan, I.; Déret, S.; Dreno, B.; Gamboa, B.; Jomard, A.; Luzy, A.P.; et al. Nonclinical and human pharmacology of the potent and selective topical retinoic acid receptor-γ agonist trifarotene. Br. J. Dermatol. 2018, 179, 442–456. [Google Scholar] [CrossRef]
- Szymański, Ł.; Skopek, R.; Palusińska, M.; Schenk, T.; Stengel, S.; Lewicki, S.; Kraj, L.; Kamiński, P.; Zelent, A. Retinoic acid and its derivatives in skin. Cells 2020, 9, 2660. [Google Scholar] [CrossRef]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef]
- Kanai, M.; Raz, A.; Goodman, D.S. Retinol-binding protein: The transport protein for vitamin A in human plasma. J. Clin. Investig. 1968, 47, 2025–2044. [Google Scholar] [CrossRef]
- Schaefer, H. Penetration and percutaneous absorption of topical retinoids. A review. Ski. Pharmacol. Physiol. 1993, 6 (Suppl. 1), 17–23. [Google Scholar] [CrossRef]
- Chen, Y.; Clarke, O.B.; Kim, J.; Stowe, S.; Kim, Y.K.; Assur, Z.; Cavalier, M.; Godoy-Ruiz, R.; von Alpen, D.C.; Manzini, C.; et al. Structure of the STRA6 receptor for retinol uptake. Science 2016, 353, aad8266. [Google Scholar] [CrossRef] [PubMed]
- Bashor, M.M.; Chytil, F. Cellular retinol-binding protein. Biochim. Biophys. Acta 1975, 411, 87–96. [Google Scholar] [CrossRef]
- Levi, L.; Wang, Z.; Doud, M.K.; Hazen, S.L.; Noy, N. Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nat. Commun. 2015, 6, 8794. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors, RXR, and the big bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef]
- Huang, P.; Chandra, V.; Rastinejad, F. Retinoic acid actions through mammalian nuclear receptors. Chem. Rev. 2014, 114, 233–254. [Google Scholar] [CrossRef]
- Rieck, M.; Meissner, W.; Ries, S.; Müller-Brüsselbach, S.; Müller, R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) beta/delta: A comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol. Pharm. 2008, 74, 1269–1277. [Google Scholar] [CrossRef]
- Sanquer, S.; Gilchrest, B.A. Characterization of human cellular retinoic acid-binding proteins-I and -II: Ligand binding affinities and distribution in skin. Arch. Biochem. Biophys. 1994, 311, 86–94. [Google Scholar] [CrossRef]
- Xu, B.; Chen, L.; Zhan, Y.; Marquez, K.N.S.; Zhuo, L.; Qi, S.; Zhu, J.; He, Y.; Chen, X.; Zhang, H.; et al. The biological functions and regulatory mechanisms of fatty acid binding protein 5 in various diseases. Front. Cell Dev. Biol. 2022, 10, 857919. [Google Scholar] [CrossRef]
- Kozłowska, D.; Myśliwiec, H.; Harasim-Symbor, E.; Milewska, A.J.; Chabowski, A.; Flisiak, I. Serum fatty acid binding protein 5 (FABP5) as a potential biomarker of inflammation in psoriasis. Mol. Biol. Rep. 2021, 48, 4421–4429. [Google Scholar] [CrossRef]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Rosenauer, A.; Mann, K.; Chang, C.-P.B.; Rachez, C.; Freedman, L.P.; Miller, W.H. Ligand-inducible interaction of the DRIP/TRAP coactivator complex with retinoid receptors in retinoic acid–sensitive and –resistant acute promyelocytic leukemia cells. Blood 2000, 96, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.C.; Wyss, R.; Huselton, C.A.; Wiegand, U.W. A newly discovered xenobiotic metabolic pathway: Ethyl ester formation. Life Sci. 1991, 49, Pl169–Pl172. [Google Scholar] [CrossRef]
- Norris, A.W.; Cheng, L.; Giguère, V.; Rosenberger, M.; Li, E. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim. Biophys. Acta 1994, 1209, 10–18. [Google Scholar] [CrossRef]
- Tian, K.; Norris, A.W.; Lin, C.L.; Li, E. The isolation and characterization of purified heterocomplexes of recombinant retinoic acid receptor and retinoid X receptor ligand binding domains. Biochemistry 1997, 36, 5669–5676. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Ruiz, M.; Boddy, A.V.; Redfern, C.P.; Pearson, A.D.; Veal, G.J. Increasing the intracellular availability of all-trans retinoic acid in neuroblastoma cells. Br. J. Cancer 2005, 92, 696–704. [Google Scholar] [CrossRef]
- Nagpal, S.; Chandraratna, R.A. Recent developments in receptor-selective retinoids. Curr. Pharm. Des. 2000, 6, 919–931. [Google Scholar] [CrossRef]
- Chitranshi, N.; Dheer, Y.; Kumar, S.; Graham, S.L.; Gupta, V. Molecular docking, dynamics, and pharmacology studies on bexarotene as an agonist of ligand-activated transcription factors, retinoid X receptors. J. Cell Biochem. 2019, 120, 11745–11760. [Google Scholar] [CrossRef]
- Chandraratna, R.A. Tazarotene—First of a new generation of receptor-selective retinoids. Br. J. Dermatol. 1996, 135 (Suppl. 49), 18–25. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Shao, Z.M.; Li, X.S.; Ordonez, J.V.; Conley, B.A.; Wu, S.; Dawson, M.I.; Han, Q.X.; Chao, W.R.; Quick, T.; et al. N-(4-hydroxyphenyl)retinamide (4-HPR)-mediated biological actions involve retinoid receptor-independent pathways in human breast carcinoma. Carcinogenesis 1995, 16, 2477–2486. [Google Scholar] [CrossRef]
- Delia, D.; Aiello, A.; Lombardi, L.; Pelicci, P.G.; Grignani, F.; Grignani, F.; Formelli, F.; Menard, S.; Costa, A.; Veronesi, U.; et al. N-(4-hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res. 1993, 53, 6036–6041. [Google Scholar]
- Sabichi, A.L.; Xu, H.; Fischer, S.; Zou, C.; Yang, X.; Steele, V.E.; Kelloff, G.J.; Lotan, R.; Clifford, J.L. Retinoid receptor-dependent and independent biological activities of novel fenretinide analogues and metabolites. Clin. Cancer Res. 2003, 9, 4606–4613. [Google Scholar] [PubMed]
- Park, S.W.; Nhieu, J.; Persaud, S.D.; Miller, M.C.; Xia, Y.; Lin, Y.W.; Lin, Y.L.; Kagechika, H.; Mayo, K.H.; Wei, L.N. A new regulatory mechanism for Raf kinase activation, retinoic acid-bound Crabp1. Sci. Rep. 2019, 9, 10929. [Google Scholar] [CrossRef]
- Masiá, S.; Alvarez, S.; de Lera, A.R.; Barettino, D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol. Endocrinol. 2007, 21, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Aleshin, A.E.; Alitongbieke, G.; Zhou, Y.; Zhang, X.; Ye, X.; Hu, M.; Ren, G.; Chen, Z.; Ma, Y.; et al. Modulation of nongenomic activation of PI3K signalling by tetramerization of N-terminally-cleaved RXRα. Nat. Commun. 2017, 8, 16066. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Jin, H.; Majumdar, A.; Noy, N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 2011, 108, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Piskunov, A.; Rochette-Egly, C. A retinoic acid receptor RARα pool present in membrane lipid rafts forms complexes with G protein αQ to activate p38MAPK. Oncogene 2012, 31, 3333–3345. [Google Scholar] [CrossRef]
- Pawson, B.A. History of retinoids. J. Am. Acad. Dermatol. 1982, 6, 577–582. [Google Scholar] [CrossRef]
- Chapellier, B.; Mark, M.; Messaddeq, N.; Calléja, C.; Warot, X.; Brocard, J.; Gérard, C.; Li, M.; Metzger, D.; Ghyselinck, N.B.; et al. Physiological and retinoid-induced proliferations of epidermis basal keratinocytes are differently controlled. EMBO J. 2002, 21, 3402–3413. [Google Scholar] [CrossRef]
- Lee, D.D.; Stojadinovic, O.; Krzyzanowska, A.; Vouthounis, C.; Blumenberg, M.; Tomic-Canic, M. Retinoid-responsive transcriptional changes in epidermal keratinocytes. J. Cell. Physiol. 2009, 220, 427–439. [Google Scholar] [CrossRef]
- Abdelmalek, M.; Spencer, J. Retinoids and wound healing. Dermatol. Surg. 2006, 32, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Pino-Lagos, K.; Guo, Y.; Noelle, R.J. Retinoic acid: A key player in immunity. Biofactors 2010, 36, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. Biophys. Acta 2013, 1833, 3471–3480. [Google Scholar] [CrossRef]
- Chen, C.F.; Goyette, P.; Lohnes, D. RARγ acts as a tumor suppressor in mouse keratinocytes. Oncogene 2004, 23, 5350–5359. [Google Scholar] [CrossRef][Green Version]
- Saitou, M.; Sugai, S.; Tanaka, T.; Shimouchi, K.; Fuchs, E.; Narumiya, S.; Kakizuka, A. Inhibition of skin development by targeted expression of a dominant-negative retinoic acid receptor. Nature 1995, 374, 159–162. [Google Scholar] [CrossRef]
- Passeri, D.; Doldo, E.; Tarquini, C.; Costanza, G.; Mazzaglia, D.; Agostinelli, S.; Campione, E.; Di Stefani, A.; Giunta, A.; Bianchi, L.; et al. Loss of CRABP-II characterizes human skin poorly differentiated squamous cell carcinomas and favors DMBA/TPA-induced carcinogenesis. J. Investig. Dermatol. 2016, 136, 1255–1266. [Google Scholar] [CrossRef]
- Hatoum, A.; El-Sabban, M.E.; Khoury, J.; Yuspa, S.H.; Darwiche, N. Overexpression of retinoic acid receptors alpha and gamma into neoplastic epidermal cells causes retinoic acid-induced growth arrest and apoptosis. Carcinogenesis 2001, 22, 1955–1963. [Google Scholar] [CrossRef]
- Didierjean, L.; Carraux, P.; Grand, D.; Sass, J.O.; Nau, H.; Saurat, J.H. Topical retinaldehyde increases skin content of retinoic acid and exerts biologic activity in mouse skin. J. Investig. Dermatol. 1996, 107, 714–719. [Google Scholar] [CrossRef]
- Connor, M.J.; Ashton, R.E.; Lowe, N.J. A comparative study of the induction of epidermal hyperplasia by natural and synthetic retinoids. J. Pharm. Exp. 1986, 237, 31–35. [Google Scholar]
- Tsambaos, D.; Mahrle, G.; Orfanos, C.E. Epidermal changes induced by oral excess of aromatic retinoid in guinea pigs. Arch. Dermatol. Res. 1980, 267, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Maruyama, K.; Ono, I.; Nihei, Y.; Iwatsuki, K.; Kaneko, F. Effects of etretinate on keratinocyte proliferation and secretion of interleukin-1 α (IL-1 α) and IL-8. J. Dermatol. 1994, 21, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, S.; Backendorf, C.; Ponec, M. Regulation of keratinocyte proliferation and differentiation by all-trans-retinoic acid, 9-cis-retinoic acid and 1,25-dihydroxy vitamin D3. Arch. Dermatol. Res. 1996, 288, 729–738. [Google Scholar] [CrossRef]
- Schroeder, M.; Zouboulis, C.C. All-trans-retinoic acid and 13-cis-retinoic acid: Pharmacokinetics and biological activity in different cell culture models of human keratinocytes. Horm. Metab. Res. 2007, 39, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Duell, E.A.; Fisher, G.J.; Datta, S.C.; Wang, Z.Q.; Reddy, A.P.; Tavakkol, A.; Yi, J.Y.; Griffiths, C.E.; Elder, J.T.; et al. Application of retinol to human skin in vivo induces epidermal hyperplasia and cellular retinoid binding proteins characteristic of retinoic acid but without measurable retinoic acid levels or irritation. J. Investig. Dermatol. 1995, 105, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Pasonen-Seppänen, S.M.; Maytin, E.V.; Törrönen, K.J.; Hyttinen, J.M.; Hascall, V.C.; MacCallum, D.K.; Kultti, A.H.; Jokela, T.A.; Tammi, M.I.; Tammi, R.H. All-trans retinoic acid-induced hyaluronan production and hyperplasia are partly mediated by EGFR signaling in epidermal keratinocytes. J. Investig. Dermatol. 2008, 128, 797–807. [Google Scholar] [CrossRef]
- Rittié, L.; Varani, J.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Retinoid-induced epidermal hyperplasia is mediated by epidermal growth factor receptor activation via specific induction of its ligands heparin-binding EGF and amphiregulin in human skin in vivo. J. Investig. Dermatol. 2006, 126, 732–739. [Google Scholar] [CrossRef]
- Memezawa, A.; Takada, I.; Takeyama, K.; Igarashi, M.; Ito, S.; Aiba, S.; Kato, S.; Kouzmenko, A.P. Id2 gene-targeted crosstalk between Wnt and retinoid signaling regulates proliferation in human keratinocytes. Oncogene 2007, 26, 5038–5045. [Google Scholar] [CrossRef]
- Borland, M.G.; Foreman, J.E.; Girroir, E.E.; Zolfaghari, R.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Ross, A.C.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-beta/delta inhibits cell proliferation in human HaCaT keratinocytes. Mol. Pharm. 2008, 74, 1429–1442. [Google Scholar] [CrossRef]
- Popadic, S.; Ramic, Z.; Medenica, L.; Mostarica Stojkovic, M.; Trajković, V.; Popadic, D. Antiproliferative effect of vitamin A and D analogues on adult human keratinocytes in vitro. Ski. Pharmacol. Physiol. 2008, 21, 227–234. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Ma, P.; Sun, J.; Li, L.; Wei, J.; Tao, L.; Qian, K. The third-generation retinoid adapalene triggered DNA damage to induce S-phase arrest in HaCat cells. Fundam. Clin. Pharm. 2020, 34, 380–388. [Google Scholar] [CrossRef] [PubMed]
- DiSepio, D.; Ghosn, C.; Eckert, R.L.; Deucher, A.; Robinson, N.; Duvic, M.; Chandraratna, R.A.; Nagpal, S. Identification and characterization of a retinoid-induced class II tumor suppressor/growth regulatory gene. Proc. Natl. Acad. Sci. USA 1998, 95, 14811–14815. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Chen, C.; Zhang, Y.; Zhang, L.; Mei, Y.; Long, X.; Tan, R.; Liang, W.; Sun, L. Acitretin modulates HaCaT cells proliferation through STAT1- and STAT3-dependent signaling. Saudi Pharm. J. 2017, 25, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Tong, P.S.; Horowitz, N.N.; Wheeler, L.A. Trans retinoic acid enhances the growth response of epidermal keratinocytes to epidermal growth factor and transforming growth factor beta. J. Investig. Dermatol. 1990, 94, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, H.; Benischek, M. Vitamin A deficiency and metaplasia. J. Exp. Med. 1927, 46, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Indra, A.K.; Warot, X.; Brocard, J.; Messaddeq, N.; Kato, S.; Metzger, D.; Chambon, P. Skin abnormalities generated by temporally controlled RXRα mutations in mouse epidermis. Nature 2000, 407, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chiba, H.; Warot, X.; Messaddeq, N.; Gérard, C.; Chambon, P.; Metzger, D. RXR-alpha ablation in skin keratinocytes results in alopecia and epidermal alterations. Development 2001, 128, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Törmä, H. Regulation of keratin expression by retinoids. Dermato-Endocrinology 2011, 3, 136–140. [Google Scholar] [CrossRef]
- Brown, L.J.; Geesin, J.C.; Rothnagel, J.A.; Roop, D.R.; Gordon, J.S. Retinoic acid suppression of loricrin expression in reconstituted human skin cultured at the liquid-air interface. J. Investig. Dermatol. 1994, 102, 886–890. [Google Scholar] [CrossRef]
- Asselineau, D.; Dale, B.A.; Bernard, B.A. Filaggrin production by cultured human epidermal keratinocytes and its regulation by retinoic acid. Differentiation 1990, 45, 221–229. [Google Scholar] [CrossRef]
- Marvin, K.W.; George, M.D.; Fujimoto, W.; Saunders, N.A.; Bernacki, S.H.; Jetten, A.M. Cornifin, a cross-linked envelope precursor in keratinocytes that is down-regulated by retinoids. Proc. Natl. Acad. Sci. USA 1992, 89, 11026–11030. [Google Scholar] [CrossRef] [PubMed]
- Eichner, R.; Gendimenico, G.J.; Kahn, M.; Mallon, J.P.; Capetola, R.J.; Mezick, J.A. Effects of long-term retinoic acid treatment on epidermal differentiation in vivo: Specific modifications in the programme of terminal differentiation. Br. J. Dermatol. 1996, 135, 687–695. [Google Scholar] [PubMed]
- Moses, M.A.; George, A.L.; Sakakibara, N.; Mahmood, K.; Ponnamperuma, R.M.; King, K.E.; Weinberg, W.C. Molecular mechanisms of p63-mediated squamous cancer pathogenesis. Int. J. Mol. Sci 2019, 20, 3590. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Dellambra, E.; Golisano, O.; Martinelli, E.; Fantozzi, I.; Bondanza, S.; Ponzin, D.; McKeon, F.; De Luca, M. p63 identifies keratinocyte stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3156–3161. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.I.; Roop, D.R. The role of p63 in development and differentiation of the epidermis. J. Dermatol. Sci. 2004, 34, 3–9. [Google Scholar] [CrossRef]
- Bamberger, C.; Pollet, D.; Schmale, H. Retinoic acid inhibits downregulation of DeltaNp63alpha expression during terminal differentiation of human primary keratinocytes. J. Investig. Dermatol. 2002, 118, 133–138. [Google Scholar] [CrossRef]
- Törmä, H.; Bergström, A.; Ghiasifarahani, G.; Berne, B. The effect of two endogenous retinoids on the mRNA expression profile in human primary keratinocytes, focusing on genes causing autosomal recessive congenital ichthyosis. Arch. Dermatol. Res. 2014, 306, 739–747. [Google Scholar] [CrossRef]
- Mrass, P.; Rendl, M.; Mildner, M.; Gruber, F.; Lengauer, B.; Ballaun, C.; Eckhart, L.; Tschachler, E. Retinoic acid increases the expression of p53 and proapoptotic caspases and sensitizes keratinocytes to apoptosis: A possible explanation for tumor preventive action of retinoids. Cancer Res. 2004, 64, 6542–6548. [Google Scholar] [CrossRef]
- Islam, T.C.; Skarin, T.; Sumitran, S.; Toftgård, R. Retinoids induce apoptosis in cultured keratinocytes. Br. J. Dermatol. 2000, 143, 709–719. [Google Scholar] [CrossRef]
- Louafi, F.; Stewart, C.E.; Perks, C.M.; Thomas, M.G.; Holly, J.M. Role of the IGF-II receptor in mediating acute, non-genomic effects of retinoids and IGF-II on keratinocyte cell death. Exp. Dermatol. 2003, 12, 426–434. [Google Scholar] [CrossRef]
- Papoutsaki, M.; Lanza, M.; Marinari, B.; Nisticò, S.; Moretti, F.; Levrero, M.; Chimenti, S.; Costanzo, A. The p73 gene is an anti-tumoral target of the RARβ/γ-selective retinoid tazarotene. J. Investig. Dermatol. 2004, 123, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Gandarillas, A.; Goldsmith, L.A.; Gschmeissner, S.; Leigh, I.M.; Watt, F.M. Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Exp. Dermatol. 1999, 8, 71–79. [Google Scholar] [CrossRef]
- Denecker, G.; Ovaere, P.; Vandenabeele, P.; Declercq, W. Caspase-14 reveals its secrets. J. Cell Biol. 2008, 180, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Rendl, M.; Ban, J.; Mrass, P.; Mayer, C.; Lengauer, B.; Eckhart, L.; Declerq, W.; Tschachler, E. Caspase-14 expression by epidermal keratinocytes is regulated by retinoids in a differentiation-associated manner. J. Investig. Dermatol. 2002, 119, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; McKay, D.A. Topical retinoic acid inhibits changes in Langerhans cell density during carcinogenesis. In Vivo 1993, 7, 271–276. [Google Scholar]
- Ho, K.K.; Halliday, G.M.; Barnetson, R.S. Topical and oral retinoids protect Langerhans’ cells and epidermal Thy-1+ dendritic cells from being depleted by ultraviolet radiation. Immunology 1991, 74, 425–431. [Google Scholar] [PubMed]
- Szondy, Z.; Reichert, U.; Fésüs, L. Retinoic acids regulate apoptosis of T lymphocytes through an interplay between RAR and RXR receptors. Cell Death Differ. 1998, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef]
- Pirisi, L.; Batova, A.; Jenkins, G.R.; Hodam, J.R.; Creek, K.E. Increased sensitivity of human keratinocytes immortalized by human papillomavirus type 16 DNA to growth control by retinoids. Cancer Res. 1992, 52, 187–193. [Google Scholar]
- Khan, M.A.; Jenkins, G.R.; Tolleson, W.H.; Creek, K.E.; Pirisi, L. Retinoic acid inhibition of human papillomavirus type 16-mediated transformation of human keratinocytes. Cancer Res. 1993, 53, 905–909. [Google Scholar] [PubMed]
- Derstenfeld, A.; Cullingham, K.; Ran, Z.C.; Litvinov, I.V. Review of evidence and recommendation for human papillomavirus (HPV) vaccination of Canadian males over the age of 26 years. J. Cutan. Med. Surg. 2020, 24, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Muntyanu, A.; Nechaev, V.; Pastukhova, E.; Logan, J.; Rahme, E.; Netchiporouk, E.; Zubarev, A.; Litvinov, I.V. Risk factors and communities disproportionately affected by cervical cancer in the Russian Federation: A national population-based study. Lancet Reg. Health Eur. 2022, 20, 100454. [Google Scholar] [CrossRef]
- Caselli, E.; Galvan, M.; Santoni, F.; Alvarez, S.; de Lera, A.R.; Ivanova, D.; Gronemeyer, H.; Caruso, A.; Guidoboni, M.; Cassai, E.; et al. Retinoic acid analogues inhibit human herpesvirus 8 replication. Antivir Ther. 2008, 13, 199–209. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Geng, S. All-trans retinoic acid alters the expression of the tight junction proteins Claudin-1 and -4 and epidermal barrier function-associated genes in the epidermis. Int. J. Mol. Med. 2019, 43, 1789–1805. [Google Scholar] [CrossRef] [PubMed]
- Cosio, T.; Di Prete, M.; Gaziano, R.; Lanna, C.; Orlandi, A.; Di Francesco, P.; Bianchi, L.; Campione, E. Trifarotene: A current review and perspectives in dermatology. Biomedicines 2021, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Braungart, E.; Magdolen, V.; Degitz, K. Retinoic acid upregulates the plasminogen activator system in human epidermal keratinocytes. J. Investig. Dermatol. 2001, 116, 778–784. [Google Scholar] [CrossRef]
- Bailly, C.; Drèze, S.; Asselineau, D.; Nusgens, B.; Lapière, C.M.; Darmon, M. Retinoic acid inhibits the production of collagenase by human epidermal keratinocytes. J. Investig. Dermatol. 1990, 94, 47–51. [Google Scholar] [CrossRef]
- Kim, M.Y.; Lee, S.E.; Chang, J.Y.; Kim, S.C. Retinoid induces the degradation of corneodesmosomes and downregulation of corneodesmosomal cadherins: Implications on the mechanism of retinoid-induced desquamation. Ann. Dermatol. 2011, 23, 439–447. [Google Scholar] [CrossRef]
- Weninger, W.; Rendl, M.; Mildner, M.; Tschachler, E. Retinoids downregulate vascular endothelial growth factor/vascular permeability factor production by normal human keratinocytes. J. Investig. Dermatol. 1998, 111, 907–911. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, Y.K.; Eun, H.C.; Cho, K.H.; Chung, J.H. All-trans retinoic acid antagonizes UV-induced VEGF production and angiogenesis via the inhibition of ERK activation in human skin keratinocytes. J. Investig. Dermatol. 2006, 126, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Skazik, C.; Amann, P.M.; Heise, R.; Marquardt, Y.; Czaja, K.; Kim, A.; Rühl, R.; Kurschat, P.; Merk, H.F.; Bickers, D.R.; et al. Downregulation of STRA6 expression in epidermal keratinocytes leads to hyperproliferation-associated differentiation in both in vitro and in vivo skin models. J. Investig. Dermatol. 2014, 134, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.K.; Castaneda, E.; Antal, M.C.; Jiang, M.; Messaddeq, N.; Meng, X.; Loehr, C.V.; Gariglio, P.; Kato, S.; Wahli, W.; et al. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor α in epidermal keratinocytes. J. Investig. Dermatol. 2007, 127, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- So, P.L.; Lee, K.; Hebert, J.; Walker, P.; Lu, Y.; Hwang, J.; Kopelovich, L.; Athar, M.; Bickers, D.; Aszterbaum, M.; et al. Topical tazarotene chemoprevention reduces Basal cell carcinoma number and size in Ptch1+/− mice exposed to ultraviolet or ionizing radiation. Cancer Res. 2004, 64, 4385–4389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- So, P.L.; Fujimoto, M.A.; Epstein, E.H., Jr. Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis. Mol. Cancer 2008, 7, 1275–1284. [Google Scholar] [CrossRef]
- Wu, C.S.; Chen, G.S.; Lin, P.Y.; Pan, I.H.; Wang, S.T.; Lin, S.H.; Yu, H.S.; Lin, C.C. Tazarotene induces apoptosis in human basal cell carcinoma via activation of caspase-8/t-Bid and the reactive oxygen species-dependent mitochondrial pathway. DNA Cell Biol. 2014, 33, 652–666. [Google Scholar] [CrossRef]
- Scharadin, T.M.; Jiang, H.; Jans, R.; Rorke, E.A.; Eckert, R.L. TIG3 tumor suppressor-dependent organelle redistribution and apoptosis in skin cancer cells. PLoS ONE 2011, 6, e23230. [Google Scholar] [CrossRef]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the molecular genetics of basal cell carcinoma. Int. J. Mol. Sci 2017, 18, 2485. [Google Scholar] [CrossRef]
- Goyette, P.; Allan, D.; Peschard, P.; Chen, C.F.; Wang, W.; Lohnes, D. Regulation of gli activity by all-trans retinoic acid in mouse keratinocytes. Cancer Res. 2000, 60, 5386–5389. [Google Scholar]
- Chow, R.Y.; Jeon, U.S.; Levee, T.M.; Kaur, G.; Cedeno, D.P.; Doan, L.T.; Atwood, S.X. PI3K promotes basal cell carcinoma growth through kinase-induced p21 degradation. Front. Oncol. 2021, 11, 668247. [Google Scholar] [CrossRef]
- So, P.L.; Wang, G.Y.; Wang, K.; Chuang, M.; Chiueh, V.C.; Kenny, P.A.; Epstein, E.H., Jr. PI3K-AKT signaling is a downstream effector of retinoid prevention of murine basal cell carcinogenesis. Cancer Prev. Res. 2014, 7, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Sly, L.; De Luca, L.M. High dietary retinoic acid prevents malignant conversion of skin papillomas induced by a two-stage carcinogenesis protocol in female SENCAR mice. Carcinogenesis 1994, 15, 2383–2386. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Tarone, R.; Huynh, M.; De Luca, L.M. High dietary retinoic acid inhibits tumor promotion and malignant conversion in a two-stage skin carcinogenesis protocol using 7,12-dimethylbenz[a]anthracene as the initiator and mezerein as the tumor promoter in female SENCAR mice. Cancer Lett. 1995, 95, 113–118. [Google Scholar] [CrossRef]
- Huang, P.Y.; Balmain, A. Modeling cutaneous squamous carcinoma development in the mouse. Cold Spring Harb. Perspect. Med. 2014, 4, a013623. [Google Scholar] [CrossRef]
- De Luca, L.M.; Tarone, R.; Huynh, M.; Jones, C.S.; Chen, L.C. Dietary retinoic acid inhibits mouse skin carcinogenesis irrespective of age at initiation. Nutr. Cancer 1996, 25, 249–257. [Google Scholar] [CrossRef]
- Ponnamperuma, R.M.; Shimizu, Y.; Kirchhof, S.M.; De Luca, L.M. beta-Carotene fails to act as a tumor promoter, induces RAR expression, and prevents carcinoma formation in a two-stage model of skin carcinogenesis in male Sencar mice. Nutr. Cancer 2000, 37, 82–88. [Google Scholar] [CrossRef]
- Kabbout, M.; Hatoum, A.; Abou-Lteif, G.; Chakroun, I.; Homaidan, F.R.; Darwiche, N. Stage-specific effect of N-(4-hydroxyphenyl)retinamide on cell growth in squamous cell carcinogenesis. Mol. Carcinog. 2004, 40, 12–23. [Google Scholar] [CrossRef]
- Tennenbaum, T.; Lowry, D.; Darwiche, N.; Morgan, D.L.; Gartsbein, M.; Hansen, L.; De Luca, L.M.; Hennings, H.; Yuspa, S.H. Topical retinoic acid reduces skin papilloma formation but resistant papillomas are at high risk for malignant conversion. Cancer Res. 1998, 58, 1435–1443. [Google Scholar]
- Verma, A.K.; Duvick, L.; Ali, M. Modulation of mouse skin tumor promotion by dietary 13-cis-retinoic acid and α-difluoromethylornithine. Carcinogenesis 1986, 7, 1019–1023. [Google Scholar] [CrossRef]
- McCormick, D.L.; Moon, R.C. Antipromotional activity of dietary N-(4-hydroxyphenyl)retinamide in two-stage skin tumorigenesis in CD-1 and SENCAR mice. Cancer Lett. 1986, 31, 133–138. [Google Scholar] [CrossRef]
- Sultana, S.; Alam, A.; Sharma, S.; Khan, N. 13-cis Retinoic acid ameliorates benzoyl peroxide-induced oxidative stress and hyperproliferative response in murine skin: A chemopreventive study. Cancer Detect. Prev. 2004, 28, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ma, W.Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. USA 1997, 94, 5826–5830. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Young, M.R.; Li, J.J.; Rincón, M.; Flavell, R.A.; Sathyanarayana, B.K.; Hunziker, R.; Colburn, N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. USA 1999, 96, 9827–9832. [Google Scholar] [CrossRef] [PubMed]
- Syed, Z.; Cheepala, S.B.; Gill, J.N.; Stein, J.; Nathan, C.; Digiovanni, J.; Batra, V.; Adegboyega, P.; Kleiner, H.E.; Clifford, J.L. All-trans retinoic acid suppresses Stat3 signaling during skin carcinogenesis. Cancer Prev. Res. 2009, 2, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Sano, S.; Kataoka, K.; Abel, E.; Carbajal, S.; Beltran, L.; Clifford, J.; Peavey, M.; Shen, J.; Digiovanni, J. Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene 2008, 27, 1087–1094. [Google Scholar] [CrossRef]
- Sano, S.; Chan, K.S.; Kira, M.; Kataoka, K.; Takagi, S.; Tarutani, M.; Itami, S.; Kiguchi, K.; Yokoi, M.; Sugasawa, K.; et al. Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 2005, 65, 5720–5729. [Google Scholar] [CrossRef]
- Sorg, O.; Tran, C.; Carraux, P.; Grand, D.; Hügin, A.; Didierjean, L.; Saurat, J.H. Spectral properties of topical retinoids prevent DNA damage and apoptosis after acute UV-B exposure in hairless mice. Photochem. Photobiol. 2005, 81, 830–836. [Google Scholar] [CrossRef]
- Wang, Z.; Boudjelal, M.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation of human skin causes functional vitamin A deficiency, preventable by all-trans retinoic acid pre-treatment. Nat. Med. 1999, 5, 418–422. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Rudnicka, L.; Marczak, M.; Szmurło, A.; Makieła, B.; Skiendzielewska, A.; Skopinska, M.; Majewski, S.; Jabłonska, S. Acitretin decreases tumor cell-induced angiogenesis. Ski. Pharmacol. Physiol. 1991, 4, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Frey, J.R.; Peck, R.; Bollag, W. Antiproliferative activity of retinoids, interferon alpha and their combination in five human transformed cell lines. Cancer Lett. 1991, 57, 223–227. [Google Scholar] [CrossRef]
- Zhang, M.L.; Tao, Y.; Zhou, W.Q.; Ma, P.C.; Cao, Y.P.; He, C.D.; Wei, J.; Li, L.J. All-trans retinoic acid induces cell-cycle arrest in human cutaneous squamous carcinoma cells by inhibiting the mitogen-activated protein kinase-activated protein 1 pathway. Clin. Exp. Dermatol. 2014, 39, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Cheepala, S.B.; Yin, W.; Syed, Z.; Gill, J.N.; McMillian, A.; Kleiner, H.E.; Lynch, M.; Loganantharaj, R.; Trutschl, M.; Cvek, U.; et al. Identification of the B-Raf/Mek/Erk MAP kinase pathway as a target for all-trans retinoic acid during skin cancer promotion. Mol. Cancer 2009, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.Y.; He, C.D.; Xiao, T.; Jin, X.; Chen, J.; Wang, Y.K.; Liu, M.; Wang, K.B.; Jiang, Y.; Wei, H.C.; et al. Acitretin induces apoptosis through CD95 signalling pathway in human cutaneous squamous cell carcinoma cell line SCL-1. J. Cell. Mol. Med. 2009, 13, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Ulukaya, E.; Kurt, A.; Wood, E.J. 4-(N-hydroxyphenyl)retinamide can selectively induce apoptosis in human epidermoid carcinoma cells but not in normal dermal fibroblasts. Cancer Investig. 2001, 19, 145–154. [Google Scholar] [CrossRef]
- Ulukaya, E.; Pirianov, G.; Kurt, M.A.; Wood, E.J.; Mehmet, H. Fenretinide induces cytochrome c release, caspase 9 activation and apoptosis in the absence of mitochondrial membrane depolarisation. Cell Death Differ. 2003, 10, 856–859. [Google Scholar] [CrossRef]
- Hail, N., Jr.; Kim, H.J.; Lotan, R. Mechanisms of fenretinide-induced apoptosis. Apoptosis 2006, 11, 1677–1694. [Google Scholar] [CrossRef]
- Sokolowska-Wojdylo, M.; Lugowska-Umer, H.; Maciejewska-Radomska, A. Oral retinoids and rexinoids in cutaneous T-cell lymphomas. Postep. Dermatol. Alergol. 2013, 30, 19–29. [Google Scholar] [CrossRef]
- Zhang, C.; Hazarika, P.; Ni, X.; Weidner, D.A.; Duvic, M. Induction of apoptosis by bexarotene in cutaneous T-cell lymphoma cells: Relevance to mechanism of therapeutic action. Clin. Cancer Res. 2002, 8, 1234–1240. [Google Scholar]
- Nieto-Rementería, N.; Pérez-Yarza, G.; Boyano, M.D.; Apraiz, A.; Izu, R.; Díaz-Pérez, J.L.; Asumendi, A. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br. J. Dermatol. 2009, 160, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tao, Y.; Zhang, M.; Ma, P.; Li, L.; Diao, Q. Effects of 9-cis-retinoic acid on the proliferation and apoptosis of cutaneous T-cell lymphoma cells. Anti-Cancer Drugs 2019, 30, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Netchiporouk, E.; Gantchev, J.; Tsang, M.; Thibault, P.; Watters, A.K.; Hughes, J.M.; Ghazawi, F.M.; Woetmann, A.; Odum, N.; Sasseville, D.; et al. Analysis of CTCL cell lines reveals important differences between mycosis fungoides/Sezary syndrome vs. HTLV-1+ leukemic cell lines. Oncotarget 2017, 8, 95981–95998. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Egusa, C.; Maeda, T.; Tsuboi, R. Combination of retinoid and histone deacetylase inhibitor produced an anti-tumor effect in cutaneous T-cell lymphoma by restoring tumor suppressor gene, retinoic acid receptorβ2, via histone acetylation. J. Dermatol. Sci. 2016, 81, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.K.; Newton, S.B.; Bach, T.L.; Budgin, J.B.; Benoit, B.M.; Lin, J.H.; Yoon, J.S.; Wysocka, M.; Abrams, C.S.; Rook, A.H. Bexarotene blunts malignant T-cell chemotaxis in Sezary syndrome: Reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am. J. Hematol. 2007, 82, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Tanita, K.; Fujimura, T.; Sato, Y.; Lyu, C.; Kambayashi, Y.; Ogata, D.; Fukushima, S.; Miyashita, A.; Nakajima, H.; Nakamura, M.; et al. Bexarotene reduces production of CCL22 from tumor-associated macrophages in cutaneous T-cell lymphoma. Front. Oncol. 2019, 9, 907. [Google Scholar] [CrossRef]
- Ferenczi, K.; Fuhlbrigge, R.C.; Pinkus, J.; Pinkus, G.S.; Kupper, T.S. Increased CCR4 expression in cutaneous T cell lymphoma. J. Investig. Dermatol. 2002, 119, 1405–1410. [Google Scholar] [CrossRef]
- Liu, X.; Jin, S.; Hu, S.; Li, R.; Pan, H.; Liu, Y.; Lai, P.; Xu, D.; Sun, J.; Liu, Z.; et al. Single-cell transcriptomics links malignant T cells to the tumor immune landscape in cutaneous T cell lymphoma. Nat. Commun. 2022, 13, 1158. [Google Scholar] [CrossRef]
- Gorgun, G.; Foss, F. Immunomodulatory effects of RXR rexinoids: Modulation of high-affinity IL-2R expression enhances susceptibility to denileukin diftitox. Blood 2002, 100, 1399–1403. [Google Scholar] [CrossRef][Green Version]
- Corbeil, J.; Rapaport, E.; Richman, D.D.; Looney, D.J. Antiproliferative effect of retinoid compounds on Kaposi’s sarcoma cells. J. Clin. Investig. 1994, 93, 1981–1986. [Google Scholar] [CrossRef]
- Sakakibara, S.; Tosato, G. Viral interleukin-6: Role in Kaposi’s sarcoma-associated herpesvirus: Associated malignancies. J. Interferon Cytokine Res. 2011, 31, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, S.; Cai, J.; Zheng, T.; Patel, S.; Masood, R.; Lin, G.Y.; Friant, S.; Johnson, A.; Smith, D.L.; Chandraratna, R.A.; et al. Retinoid antagonism of NF-IL6: Insight into the mechanism of antiproliferative effects of retinoids in Kaposi’s sarcoma. Mol. Cell. Biol. 1997, 17, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; Morini, M.; Pfeffer, U.; Minghelli, S.; Noonan, D.M.; Albini, A. Inhibition of Kaposi’s sarcoma in vivo by fenretinide. Clin. Cancer Res. 2003, 9, 6020–6029. [Google Scholar] [PubMed]
- Kim, J.; Park, M.K.; Li, W.Q.; Qureshi, A.A.; Cho, E. Association of vitamin A intake with cutaneous squamous cell carcinoma risk in the United States. JAMA Dermatol. 2019, 155, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Lenouvel, C.M.; Litvinov, I.V.; Netchiporouk, E. Dietary vitamin A intake is shown to decrease the risk of cutaneous squamous cell carcinomas. J. Cutan. Med. Surg. 2020, 24, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Schmults, C.D.; Blitzblau, R.; Aasi, S.Z.; Alam, M.; Andersen, J.S.; Baumann, B.C.; Bordeaux, J.; Chen, P.L.; Chin, R.; Contreras, C.M.; et al. NCCN Guidelines® insights: Squamous cell skin cancer, version 1.2022. J. Natl. Compr. Cancer Netw. 2021, 19, 1382–1394. [Google Scholar] [CrossRef]
- Bavinck, J.N.; Tieben, L.M.; Van der Woude, F.J.; Tegzess, A.M.; Hermans, J.; ter Schegget, J.; Vermeer, B.J. Prevention of skin cancer and reduction of keratotic skin lesions during acitretin therapy in renal transplant recipients: A double-blind, placebo-controlled study. J. Clin. Oncol. 1995, 13, 1933–1938. [Google Scholar] [CrossRef]
- Harwood, C.A.; Leedham-Green, M.; Leigh, I.M.; Proby, C.M. Low-dose retinoids in the prevention of cutaneous squamous cell carcinomas in organ transplant recipients: A 16-year retrospective study. Arch. Dermatol. 2005, 141, 456–464. [Google Scholar] [CrossRef]
- de Sévaux, R.G.; Smit, J.V.; de Jong, E.M.; van de Kerkhof, P.C.; Hoitsma, A.J. Acitretin treatment of premalignant and malignant skin disorders in renal transplant recipients: Clinical effects of a randomized trial comparing two doses of acitretin. J. Am. Acad. Dermatol. 2003, 49, 407–412. [Google Scholar] [CrossRef]
- Solomon-Cohen, E.; Reiss-Huss, S.; Hodak, E.; Davidovici, B. Low-dose acitretin for secondary prevention of keratinocyte carcinomas in solid-organ transplant recipients. Dermatology 2022, 238, 161–166. [Google Scholar] [CrossRef]
- Nijsten, T.E.; Stern, R.S. Oral retinoid use reduces cutaneous squamous cell carcinoma risk in patients with psoriasis treated with psoralen-UVA: A nested cohort study. J. Am. Acad. Dermatol. 2003, 49, 644–650. [Google Scholar] [CrossRef]
- Stern, R.S.; Lunder, E.J. Risk of squamous cell carcinoma and methoxsalen (psoralen) and UV-A radiation (PUVA). A meta-analysis. Arch. Dermatol. 1998, 134, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- Anforth, R.; Blumetti, T.C.; Clements, A.; Kefford, R.; Long, G.V.; Fernandez-Peñas, P. Systemic retinoids for the chemoprevention of cutaneous squamous cell carcinoma and verrucal keratosis in a cohort of patients on BRAF inhibitors. Br. J. Dermatol. 2013, 169, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sikorski, D.; Xu, H.; Zubarev, A.; Chergui, M.; Lagace, F.; Miller, W.H., Jr.; Redpath, M.; Ghazal, S.; Butler, M.O.; et al. Defining the criteria for reflex testing for BRAF mutations in cutaneous melanoma patients. Cancers 2021, 13, 2282. [Google Scholar] [CrossRef]
- Zhou, S.; Sivachandran, S.; Sikorski, D.; Xu, H.H.; Lagace, F.; Claveau, J.; Salopek, T.G.; Gniadecki, R.; Litvinov, I.V. Reflex molecular testing in melanoma diagnosis: When should BRAF mutation testing be ordered and who should order it? J. Cutan. Med. Surg. 2022, 26, 201–202. [Google Scholar] [CrossRef]
- Brewster, A.M.; Lee, J.J.; Clayman, G.L.; Clifford, J.L.; Reyes, M.J.; Zhou, X.; Sabichi, A.L.; Strom, S.S.; Collins, R.; Meyers, C.A.; et al. Randomized trial of adjuvant 13-cis-retinoic acid and interferon alfa for patients with aggressive skin squamous cell carcinoma. J. Clin. Oncol. 2007, 25, 1974–1978. [Google Scholar] [CrossRef]
- Kadakia, K.C.; Barton, D.L.; Loprinzi, C.L.; Sloan, J.A.; Otley, C.C.; Diekmann, B.B.; Novotny, P.J.; Alberts, S.R.; Limburg, P.J.; Pittelkow, M.R. Randomized controlled trial of acitretin versus placebo in patients at high-risk for basal cell or squamous cell carcinoma of the skin (North Central Cancer Treatment Group Study 969251). Cancer 2012, 118, 2128–2137. [Google Scholar] [CrossRef]
- Weinstock, M.A.; Bingham, S.F.; Digiovanna, J.J.; Rizzo, A.E.; Marcolivio, K.; Hall, R.; Eilers, D.; Naylor, M.; Kirsner, R.; Kalivas, J.; et al. Tretinoin and the prevention of keratinocyte carcinoma (Basal and squamous cell carcinoma of the skin): A veterans affairs randomized chemoprevention trial. J. Investig. Dermatol. 2012, 132, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, K.; Liu, J.; Powers, J.G. Skin cancer in the military: A systematic review of melanoma and nonmelanoma skin cancer incidence, prevention, and screening among active duty and veteran personnel. J. Am. Acad. Dermatol. 2018, 78, 1185–1192. [Google Scholar] [CrossRef]
- Kraemer, K.H.; DiGiovanna, J.J.; Moshell, A.N.; Tarone, R.E.; Peck, G.L. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N. Engl. J. Med. 1988, 318, 1633–1637. [Google Scholar] [CrossRef]
- Berth-Jones, J.; Graham-Brown, R.A. Xeroderma pigmentosum variant: Response to etretinate. Br. J. Dermatol. 1990, 122, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.; Lazarov, A.; Halevy, S. Treatment of xeroderma pigmentosum variant with low-dose etretinate. Br. J. Dermatol. 1996, 134, 815–816. [Google Scholar] [CrossRef] [PubMed]
- Peck, G.L.; DiGiovanna, J.J.; Sarnoff, D.S.; Gross, E.G.; Butkus, D.; Olsen, T.G.; Yoder, F.W. Treatment and prevention of basal cell carcinoma with oral isotretinoin. J. Am. Acad. Dermatol. 1988, 19, 176–185. [Google Scholar] [CrossRef]
- Peck, G.L.; Gross, E.G.; Butkus, D.; DiGiovanna, J.J. Chemoprevention of basal cell carcinoma with isotretinoin. J. Am. Acad Dermatol. 1982, 6, 815–823. [Google Scholar] [CrossRef]
- Tang, J.Y.; Chiou, A.S.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Chanana, A.M.; Lee, W.; Lindgren, J.A.; Raphael, M.A.; Thompson, B.J.; Bickers, D.R.; et al. Tazarotene: Randomized, double-blind, vehicle-controlled, and open-label concurrent trials for basal cell carcinoma prevention and therapy in patients with basal cell nevus syndrome. Cancer Prev. Res. 2014, 7, 292–299. [Google Scholar] [CrossRef]
- Joshi, T.P.; Farr, M.A.; Lewis, D.J. Isotretinoin as chemoprophylaxis for cutaneous malignancies in Muir-Torre syndrome: A novel concept. Dermatol. Ther. 2022, 35, e15540. [Google Scholar] [CrossRef]
- Bollag, W.; Ott, F. Retinoic acid: Topical treatment of senile or actinic keratoses and basal cell carcinomas. Agents Actions 1970, 1, 172–175. [Google Scholar] [CrossRef]
- Zhang, X.B.; Zhang, S.Q.; Li, C.X.; Huang, Z.M.; Luo, Y.W. Acitretin systemic and retinoic acid 0.1% cream supression of basal cell carcinoma. Derm. Rep. 2010, 2, e4. [Google Scholar] [CrossRef]
- Giannotti, B.; Vanzi, L.; Difonzo, E.M.; Pimpinelli, N. The treatment of basal cell carcinomas in a patient with xeroderma pigmentosum with a combination of imiquimod 5% cream and oral acitretin. Clin. Exp. Dermatol. 2003, 28 (Suppl. 1), 33–35. [Google Scholar] [CrossRef]
- Hughes, B.R.; Marks, R.; Pearse, A.D.; Gaskell, S.A. Clinical response and tissue effects of etretinate treatment of patients with solar keratoses and basal cell carcinoma. J. Am. Acad. Dermatol. 1988, 18, 522–529. [Google Scholar] [CrossRef]
- Brenner, S.; Wolf, R.; Dascalu, D.I. Topical tretinoin treatment in basal cell carcinoma. J. Dermatol. Surg. Oncol. 1993, 19, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Peris, K.; Fargnoli, M.C.; Chimenti, S. Preliminary observations on the use of topical tazarotene to treat basal-cell carcinoma. N. Engl. J. Med. 1999, 341, 1767–1768. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Ni, X.; Talpur, R.; Herne, K.; Schulz, C.; Sui, D.; Ward, S.; Joseph, A.; Hazarika, P. Tazarotene-induced gene 3 is suppressed in basal cell carcinomas and reversed in vivo by tazarotene application. J. Investig. Dermatol. 2003, 121, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Ianhez, M.; Fleury, L.F., Jr.; Miot, H.A.; Bagatin, E. Retinoids for prevention and treatment of actinic keratosis. Bras. Dermatol. 2013, 88, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Goldfarb, M.T.; Weiss, J.S.; Metz, R.D.; Hamilton, T.A.; Voorhees, J.J.; Griffiths, C.E. Assessment of adapalene gel for the treatment of actinic keratoses and lentigines: A randomized trial. J. Am. Acad. Dermatol. 2003, 49, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, S.; Sharma, V.K. Successful treatment of multiple premalignant and malignant lesions in arsenical keratosis with a combination of acitretin and intralesional 5-fluorouracil. J. Dermatol. 2003, 30, 730–734. [Google Scholar] [CrossRef]
- Galitzer, B.I. Photodynamic therapy for actinic keratoses of the upper extremities using 10% aminolevulinic acid gel, red light, and adapalene pretreatment. J. Clin. Aesthet. Dermatol. 2021, 14, 19–24. [Google Scholar]
- Bardazzi, F.; Bianchi, F.; Parente, G.; Guareschi, E.; Landi, C. A pilot study on the use of topical tazarotene to treat squamous cell carcinoma in situ. J. Am. Acad. Dermatol. 2005, 52, 1102–1104. [Google Scholar] [CrossRef]
- Lippman, S.M.; Meyskens, F.L., Jr. Treatment of advanced squamous cell carcinoma of the skin with isotretinoin. Ann. Intern. Med. 1987, 107, 499–502. [Google Scholar] [CrossRef]
- Levine, N.; Miller, R.C.; Meyskens, F.L., Jr. Oral isotretinoin therapy. Use in a patient with multiple cutaneous squamous cell carcinomas and keratoacanthomas. Arch. Dermatol. 1984, 120, 1215–1217. [Google Scholar] [CrossRef]
- Lippman, S.M.; Parkinson, D.R.; Itri, L.M.; Weber, R.S.; Schantz, S.P.; Ota, D.M.; Schusterman, M.A.; Krakoff, I.H.; Gutterman, J.U.; Hong, W.K. 13-cis-retinoic acid and interferon α-2a: Effective combination therapy for advanced squamous cell carcinoma of the skin. J. Natl. Cancer Inst. 1992, 84, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, Y.; Wang, H.; Ji, C. Case report: Successful treatment of cutaneous squamous cell carcinoma in three patients with a combination of acitretin and clarithromycin. Front. Oncol. 2021, 11, 650974. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.F.; Jones, S.E.; Levine, N.; Lynch, P.J.; Booth, A.R.; Meyskens, F.L., Jr. Isotretinoin and cutaneous helper T-cell lymphoma (mycosis fungoides). Arch. Dermatol. 1987, 123, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Neely, S.M.; Mehlmauer, M.; Feinstein, D.I. The effect of isotretinoin in six patients with cutaneous T-cell lymphoma. Arch. Intern. Med. 1987, 147, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.P.K.; Gantchev, J.; Martinez Villarreal, A.; Ramchatesingh, B.; Netchiporouk, E.; Akilov, O.E.; Odum, N.; Gniadecki, R.; Koralov, S.B.; Litvinov, I.V. Understanding cell lines, patient-derived xenograft and genetically engineered mouse models used to study cutaneous T-cell lymphoma. Cells 2022, 11, 593. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous T-cell lymphoma-a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef]
- Tsang, M.; Gantchev, J.; Netchiporouk, E.; Moreau, L.; Ghazawi, F.M.; Glassman, S.; Sasseville, D.; Litvinov, I.V. A study of meiomitosis and novel pathways of genomic instability in cutaneous T-cell lymphomas (CTCL). Oncotarget 2018, 9, 37647–37661. [Google Scholar] [CrossRef]
- Molin, L.; Thomsen, K.; Volden, G.; Aronsson, A.; Hammar, H.; Hellbe, L.; Wantzin, G.L.; Roupe, G. Oral retinoids in mycosis fungoides and Sézary syndrome: A comparison of isotretinoin and etretinate. A study from the Scandinavian Mycosis Fungoides Group. Acta Derm. Venereol. 1987, 67, 232–236. [Google Scholar]
- Jones, G.; McLean, J.; Rosenthal, D.; Roberts, J.; Sauder, D.N. Combined treatment with oral etretinate and electron beam therapy in patients with cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome). J. Am. Acad. Dermatol. 1992, 26, 960–967. [Google Scholar] [CrossRef]
- Cheeley, J.; Sahn, R.E.; DeLong, L.K.; Parker, S.R. Acitretin for the treatment of cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 2013, 68, 247–254. [Google Scholar] [CrossRef]
- Duvic, M.; Hymes, K.; Heald, P.; Breneman, D.; Martin, A.G.; Myskowski, P.; Crowley, C.; Yocum, R.C. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: Multinational phase II-III trial results. J. Clin. Oncol. 2001, 19, 2456–2471. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Martin, A.G.; Kim, Y.; Olsen, E.; Wood, G.S.; Crowley, C.A.; Yocum, R.C. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch. Dermatol. 2001, 137, 581–593. [Google Scholar] [PubMed]
- Prince, H.M.; McCormack, C.; Ryan, G.; Baker, C.; Rotstein, H.; Davison, J.; Yocum, R. Bexarotene capsules and gel for previously treated patients with cutaneous T-cell lymphoma: Results of the Australian patients treated on phase II trials. Australas. J. Dermatol. 2001, 42, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Sugaya, M.; Tokura, Y.; Ohtsuka, M.; Tsuboi, R.; Nagatani, T.; Tani, M.; Setoyama, M.; Matsushita, S.; Kawai, K.; et al. Phase I/II study of the oral retinoid X receptor agonist bexarotene in Japanese patients with cutaneous T-cell lymphomas. J. Dermatol. 2017, 44, 135–142. [Google Scholar] [CrossRef]
- Breneman, D.; Duvic, M.; Kuzel, T.; Yocum, R.; Truglia, J.; Stevens, V.J. Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphoma. Arch. Dermatol. 2002, 138, 325–332. [Google Scholar] [CrossRef]
- Heald, P.; Mehlmauer, M.; Martin, A.G.; Crowley, C.A.; Yocum, R.C.; Reich, S.D. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: Results of the phase III clinical trial. J. Am. Acad. Dermatol. 2003, 49, 801–815. [Google Scholar] [CrossRef]
- Geskin, L.; Malone, D.C. An exploratory cost-effectiveness analysis of systemic treatments for cutaneous T-cell lymphoma. J. Dermatol. Treat. 2018, 29, 522–530. [Google Scholar] [CrossRef]
- Dummer, R.; Beyer, M.; Hymes, K.; Epping, M.T.; Bernards, R.; Steinhoff, M.; Sterry, W.; Kerl, H.; Heath, K.; Ahern, J.D.; et al. Vorinostat combined with bexarotene for treatment of cutaneous T-cell lymphoma: In Vitro and phase I clinical evidence supporting augmentation of retinoic acid receptor/retinoid X receptor activation by histone deacetylase inhibition. Leuk. Lymphoma 2012, 53, 1501–1508. [Google Scholar] [CrossRef]
- Lokitz, M.L.; Wong, H.K. Bexarotene and narrowband ultraviolet B phototherapy combination treatment for mycosis fungoides. Photodermatol. Photoimmunol. Photomed. 2007, 23, 255–257. [Google Scholar] [CrossRef]
- D’Acunto, C.; Gurioli, C.; Neri, I. Plaque stage mycosis fungoides treated with bexarotene at low dosage and UVB-NB. J. Dermatol. Treat. 2010, 21, 45–48. [Google Scholar] [CrossRef]
- Foss, F.; Demierre, M.F.; DiVenuti, G. A phase-1 trial of bexarotene and denileukin diftitox in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood 2005, 106, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Kannangara, A.P.; Levitan, D.; Fleischer, A.B., Jr. Evaluation of the efficacy of the combination of oral bexarotene and methotrexate for the treatment of early stage treatment-refractory cutaneous T-cell lymphoma. J. Dermatol. Treat. 2009, 20, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Ortiz, P.; Dummer, R.; Ranki, A.; Hasan, B.; Meulemans, B.; Gellrich, S.; Knobler, R.; Stadler, R.; Karrasch, M. Efficacy and safety of bexarotene combined with psoralen-ultraviolet A (PUVA) compared with PUVA treatment alone in stage IB-IIA mycosis fungoides: Final results from the EORTC Cutaneous Lymphoma Task Force phase III randomized clinical trial (NCT00056056). Br. J. Dermatol. 2012, 167, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Alhusayen, R.; Vu, T.T.; Almuhanna, N.; Wohlmuth-Wieser, I.; Hardin, J.; Hughes, J.M.; Chia, J.; Street, L.; Shear, N.H.; Walsh, S.R.; et al. Evaluation of alitretinoin for the treatment of mycosis fungoides and sezary syndrome. Dermatology 2021, 237, 479–485. [Google Scholar] [CrossRef]
- Kaemmerer, T.; Stadler, P.C.; Helene Frommherz, L.; Guertler, A.; Einar French, L.; Reinholz, M. Alitretinoin in the treatment of cutaneous T-cell lymphoma. Cancer Med. 2021, 10, 7071–7078. [Google Scholar] [CrossRef]
- Besner Morin, C.; Roberge, D.; Turchin, I.; Petrogiannis-Haliotis, T.; Popradi, G.; Pehr, K. Tazarotene 0.1% Cream as monotherapy for early-stage cutaneous T-cell lymphoma. J. Cutan. Med. Surg. 2016, 20, 244–248. [Google Scholar] [CrossRef]
- Apisarnthanarax, N.; Talpur, R.; Ward, S.; Ni, X.; Kim, H.W.; Duvic, M. Tazarotene 0.1% gel for refractory mycosis fungoides lesions: An open-label pilot study. J. Am. Acad. Dermatol. 2004, 50, 600–607. [Google Scholar] [CrossRef]
- Bailey, J.; Pluda, J.M.; Foli, A.; Saville, M.W.; Bauza, S.; Adamson, P.C.; Murphy, R.F.; Cohen, R.B.; Broder, S.; Yarchoan, R. Phase I/II study of intermittent all-trans-retinoic acid, alone and in combination with interferon alfa-2a, in patients with epidemic Kaposi’s sarcoma. J. Clin. Oncol. 1995, 13, 1966–1974. [Google Scholar] [CrossRef]
- Duvic, M.; Friedman-Kien, A.E.; Looney, D.J.; Miles, S.A.; Myskowski, P.L.; Scadden, D.T.; Von Roenn, J.; Galpin, J.E.; Groopman, J.; Loewen, G.; et al. Topical treatment of cutaneous lesions of acquired immunodeficiency syndrome-related Kaposi sarcoma using alitretinoin gel: Results of phase 1 and 2 trials. Arch. Dermatol. 2000, 136, 1461–1469. [Google Scholar] [CrossRef]
- Walmsley, S.; Northfelt, D.W.; Melosky, B.; Conant, M.; Friedman-Kien, A.E.; Wagner, B. Treatment of AIDS-related cutaneous Kaposi’s sarcoma with topical alitretinoin (9-cis-retinoic acid) gel. Panretin Gel North American Study Group. J. Acquir. Immune Defic. Syndr. 1999, 22, 235–246. [Google Scholar] [CrossRef]
- Bodsworth, N.J.; Bloch, M.; Bower, M.; Donnell, D.; Yocum, R. Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi’s sarcoma. Am. J. Clin. Dermatol. 2001, 2, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Aboulafia, D.M.; Norris, D.; Henry, D.; Grossman, R.J.; Thommes, J.; Bundow, D.; Yocum, R.C.; Stevens, V. 9-cis-retinoic acid capsules in the treatment of AIDS-related Kaposi sarcoma: Results of a phase 2 multicenter clinical trial. Arch. Dermatol. 2003, 139, 178–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morganroth, G.S. Topical 0.1% alitretinoin gel for classic Kaposi sarcoma. Arch. Dermatol. 2002, 138, 542–543. [Google Scholar] [CrossRef] [PubMed]
- Daadaa, N.; Souissi, A.; Chaabani, M.; Chelly, I.; Ben Salem, M.; Mokni, M. Involution of classic Kaposi sarcoma lesions under acitretin treatment Kaposi sarcoma treated with acitretin. Clin. Case Rep. 2020, 8, 3340–3343. [Google Scholar] [CrossRef]
- Collins, M.D.; Mao, G.E. Teratology of retinoids. Annu. Rev. Pharm. Toxicol. 1999, 39, 399–430. [Google Scholar] [CrossRef]
- Kraemer, K.H.; DiGiovanna, J.J.; Peck, G.L. Chemoprevention of skin cancer in xeroderma pigmentosum. J. Dermatol. 1992, 19, 715–718. [Google Scholar] [CrossRef]
- De Graaf, Y.G.; Euvrard, S.; Bouwes Bavinck, J.N. Systemic and topical retinoids in the management of skin cancer in organ transplant recipients. Dermatol. Surg. 2004, 30, 656–661. [Google Scholar] [CrossRef]
- Ferreira, R.; Napoli, J.; Enver, T.; Bernardino, L.; Ferreira, L. Advances and challenges in retinoid delivery systems in regenerative and therapeutic medicine. Nat. Commun. 2020, 11, 4265. [Google Scholar] [CrossRef]
- Epping, M.T.; Wang, L.; Edel, M.J.; Carlée, L.; Hernandez, M.; Bernards, R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 2005, 122, 835–847. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (s) | Trade Names (Manufacturer) | Generation | FDA-Approved Uses for Skin Cancer Management | Off-Label Uses for Skin Cancer Management | Proposed Mechanisms of Action | Adverse Events/Warnings | References |
|---|---|---|---|---|---|---|---|
| Acitretin | Soriatane (Roche) | Second | None | Oral dosing for KC and AK chemoprevention in SOTR and other at-risk individuals. | High affinity for CRABPII displaces RA and potentiates endogenous RA signaling. | Teratogenicity skeletal abnormalities Hypertriglyceridemia Hyperlipidemia Hepatotoxicity Xerosis | [10,76,209,211] |
| Adapalene | Differin (Galderma) | Third | None | 0.1–0.3% topical formulation for AK treatment | Selective RARγ agonist DNA damage and S-phase arrest in immortalized keratinocytes | Irritation, erythema, burning | [11,113,237] |
| Alitretinoin 9-cis retinoic acid | Toctino (Basilea) | First | 0.1% topical formulation for AIDS-related KS | 0.1% topical formulation for non-AIDS related KS. 0.1% topical formulation for CTCL second-line therapy. Oral dosing of 10–30 mg daily used off label for the treatment of CTCL. | RAR-RXR activation Induction of apoptosis in KS cells. Angiogenesis inhibition Inhibition of KSHV replication. G0/G1 cell cycle arrest, apoptosis and modulation of the JAK/STAT pathway. | Teratogenicity Irritation Dermatitis Scaling | [11,202,204,273,275] |
| Bexarotene Targretin® | Targretin (Ligand) | First | Oral dosing and topical 0.1–1.0% for advance-stage or treatment refractory MF resistant to at least one therapy. | None | Pan-RXR agonist Induction of apoptosis via p53/p73 activation Possible control over skin infiltration colonization by T cells | Teratogenicity irritation, erythema, burning dermatitis | [10,11,197,198,253] |
| Isotretinoin 13-cis retinoic acid Accutane® | Accutane (Roche), Claravis (Barr), Sotret (Ranbaxy), Amnesteem (Bertek), Absorica (Ranbaxy), Myorisan (VersaPharm) | First | None | Oral dosing for BCC/cSCC chemoprevention in NBCCS and XP. | RAR-RXR activation | Teratogenicity Skeletal abnormalities Hepatotoxicity | [10,11,225,226,278] |
| Tazarotene Tazorac® | Tazorac (Almirall) | Third | None | 0.1% topical formulation for MF treatment as second line therapy | Selective RARγ agonist | Teratogenicity Irritation, erythema, burning | [11,268,269] |
| Tretinoin, All-trans retinoic acid (ATRA) | Vesanoid (Roche) | First | None | 0.1% topical tretinoin to treat AKs. | RAR-RXR activation Induction of keratinocyte apoptosis. Control of hyperproliferative STAT signaling UV protection | Teratogenicity Irritation, erythema | [11,130,131,177,181,279] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramchatesingh, B.; Martínez Villarreal, A.; Arcuri, D.; Lagacé, F.; Setah, S.A.; Touma, F.; Al-Badarin, F.; Litvinov, I.V. The Use of Retinoids for the Prevention and Treatment of Skin Cancers: An Updated Review. Int. J. Mol. Sci. 2022, 23, 12622. https://doi.org/10.3390/ijms232012622
Ramchatesingh B, Martínez Villarreal A, Arcuri D, Lagacé F, Setah SA, Touma F, Al-Badarin F, Litvinov IV. The Use of Retinoids for the Prevention and Treatment of Skin Cancers: An Updated Review. International Journal of Molecular Sciences. 2022; 23(20):12622. https://doi.org/10.3390/ijms232012622
Chicago/Turabian StyleRamchatesingh, Brandon, Amelia Martínez Villarreal, Domenico Arcuri, François Lagacé, Samy Abu Setah, Fadi Touma, Faris Al-Badarin, and Ivan V. Litvinov. 2022. "The Use of Retinoids for the Prevention and Treatment of Skin Cancers: An Updated Review" International Journal of Molecular Sciences 23, no. 20: 12622. https://doi.org/10.3390/ijms232012622
APA StyleRamchatesingh, B., Martínez Villarreal, A., Arcuri, D., Lagacé, F., Setah, S. A., Touma, F., Al-Badarin, F., & Litvinov, I. V. (2022). The Use of Retinoids for the Prevention and Treatment of Skin Cancers: An Updated Review. International Journal of Molecular Sciences, 23(20), 12622. https://doi.org/10.3390/ijms232012622