
A γ-Secretase Inhibitor Attenuates Cell Cycle Progression and Invasion in Human Oral Squamous Cell Carcinoma: An In Vitro Study

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
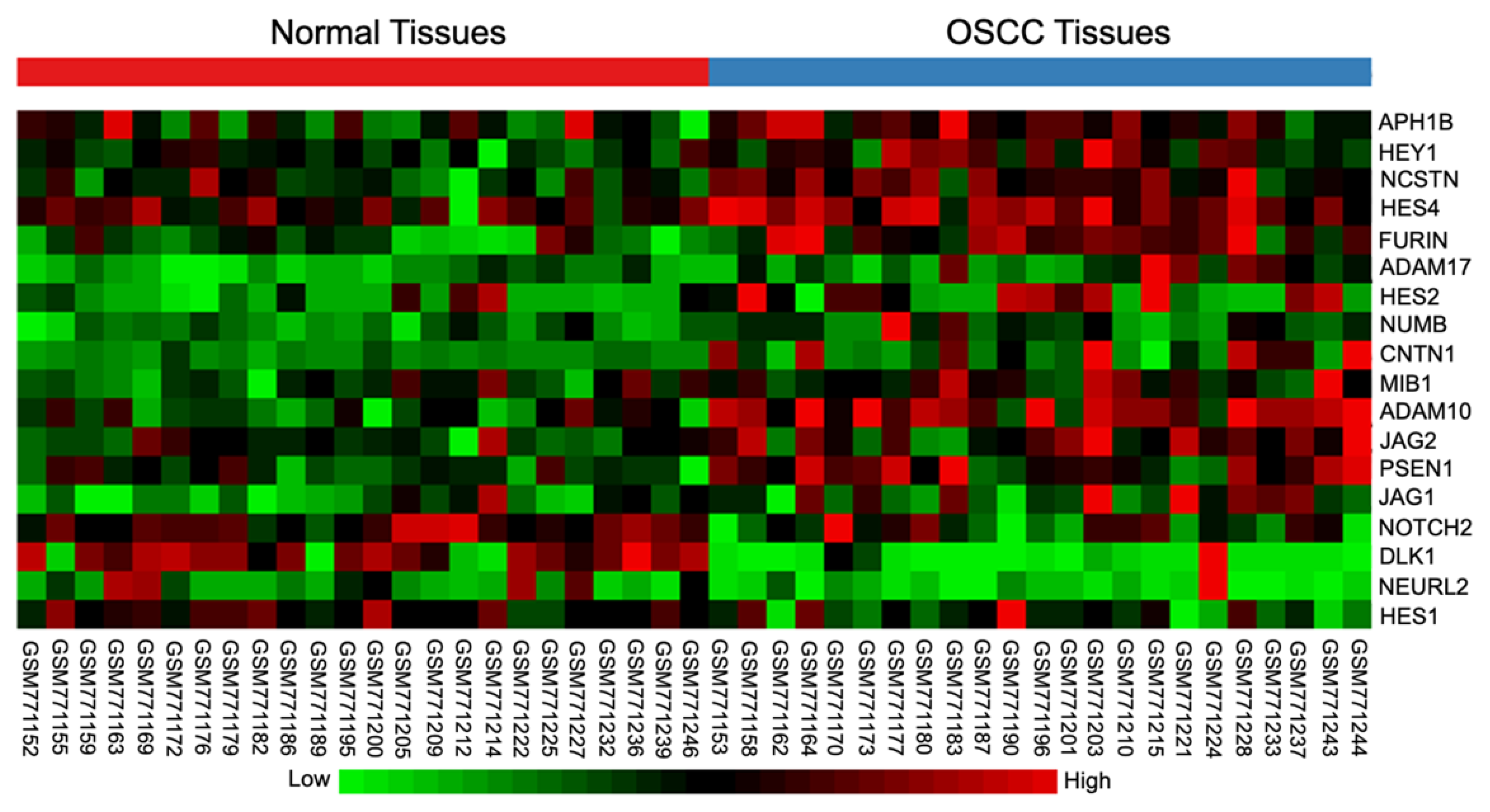
2.1. Differential Expression of Notch Signaling-Related Genes in OSCC Tissues
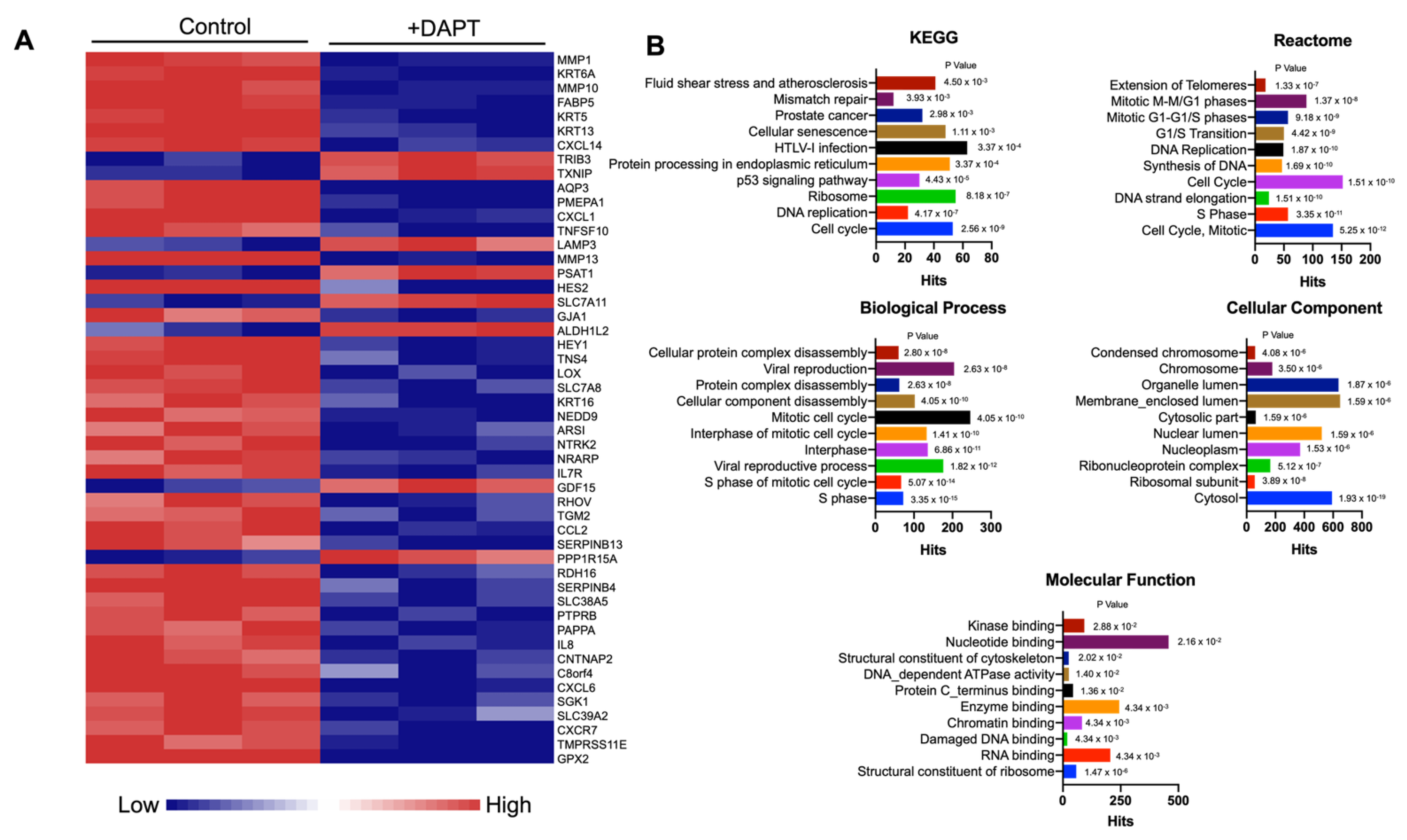
2.2. Differential Gene Expression of Notch Inhibitor-Treated HSC-4 Cells
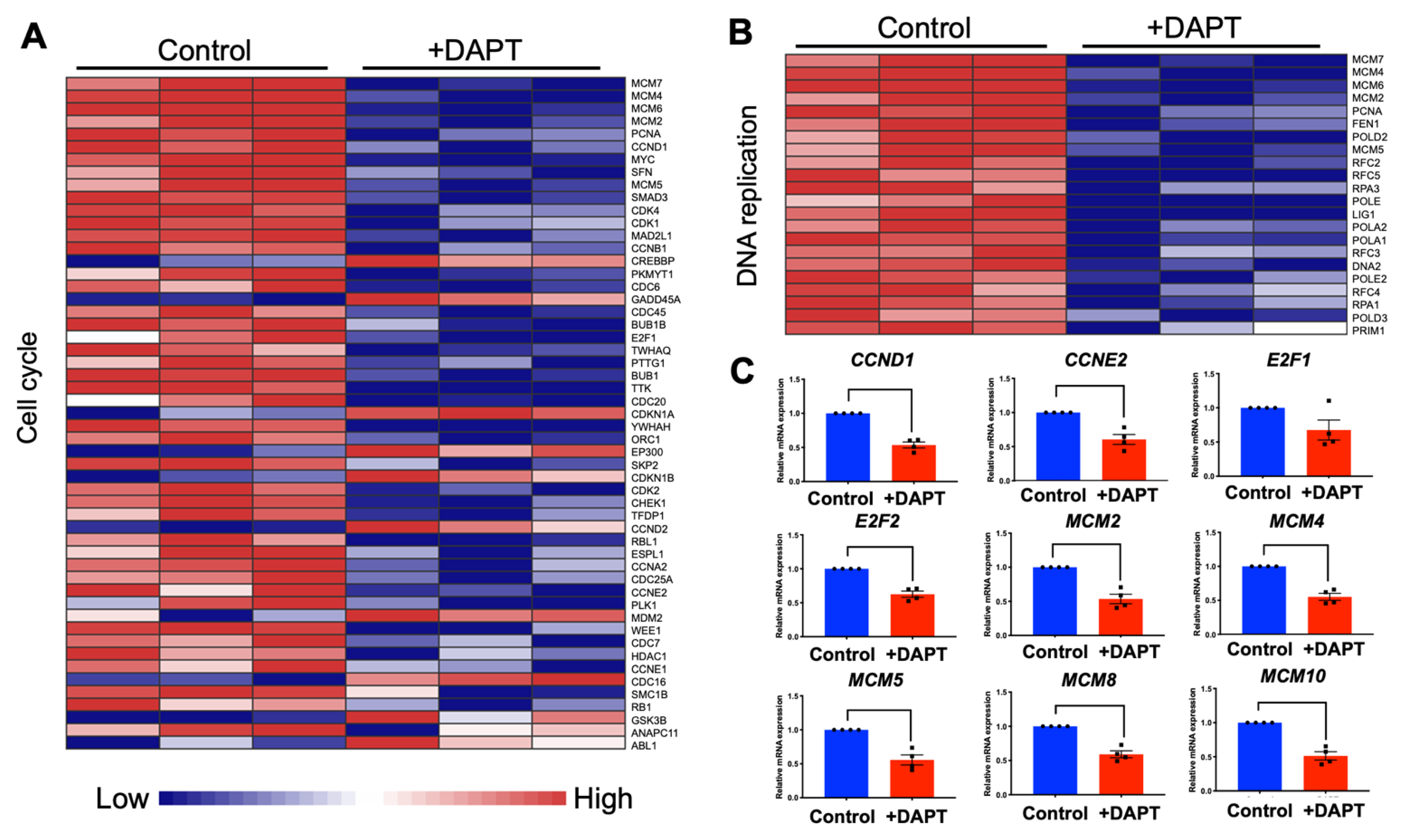
2.3. DAPT Downregulated Genes in the Cell Cycle and DNA Replication Pathways in HSC-4 Cells
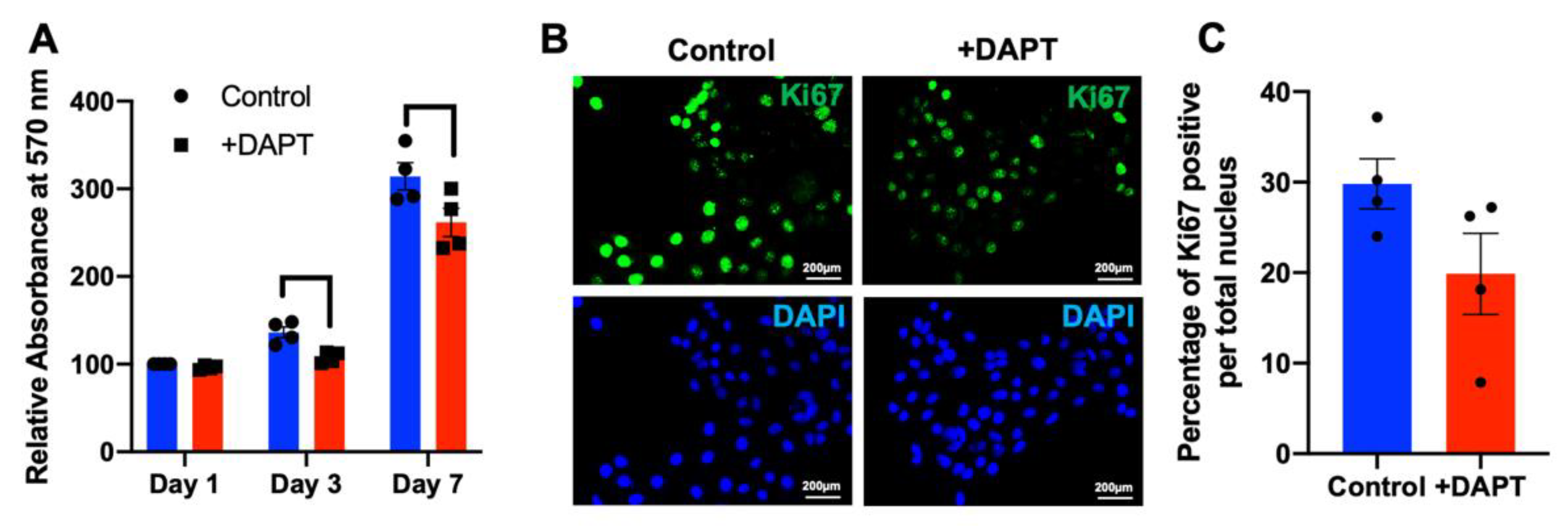
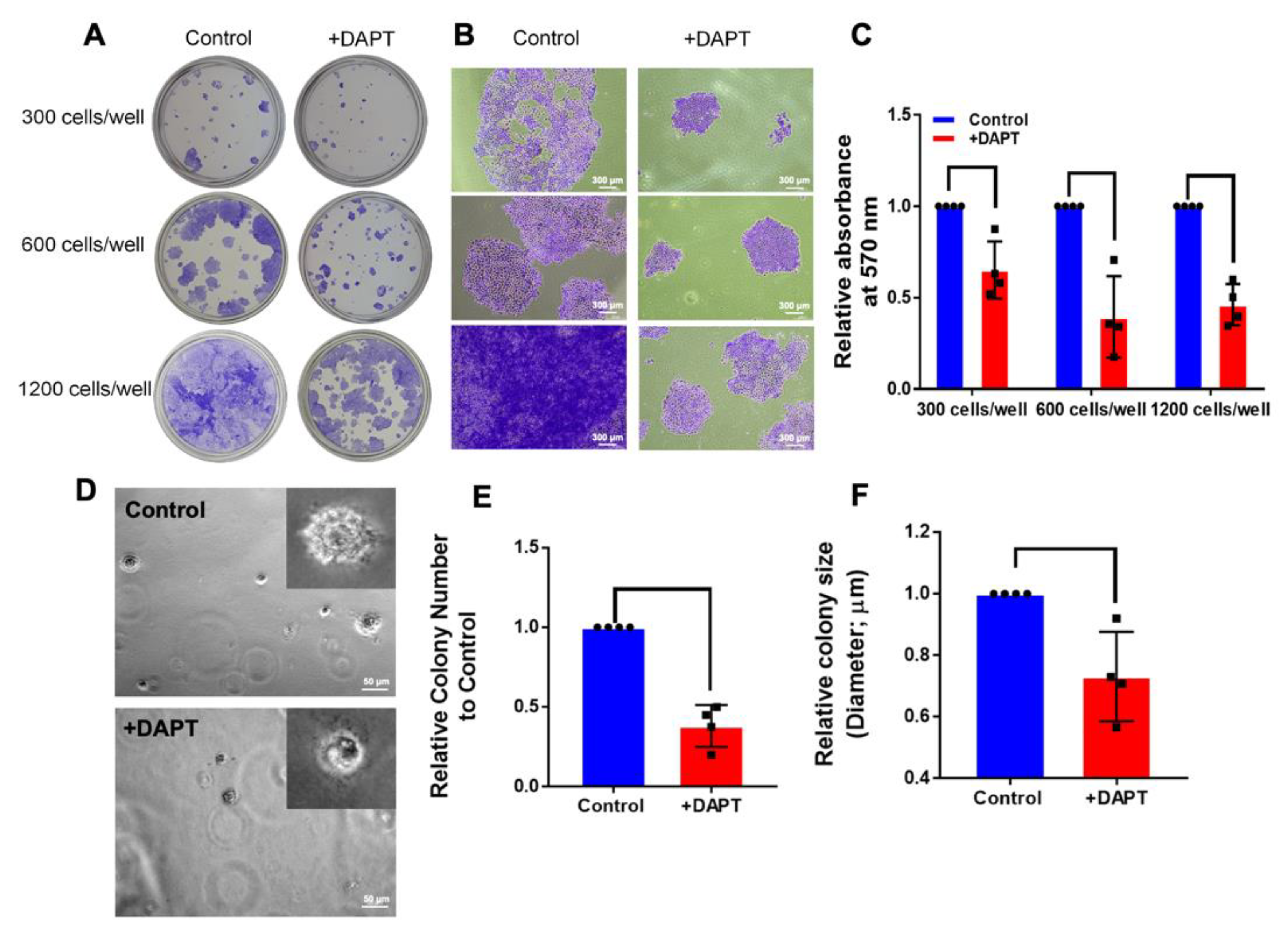
2.4. HSC-4 Proliferation Was Attenuated by DAPT Treatment
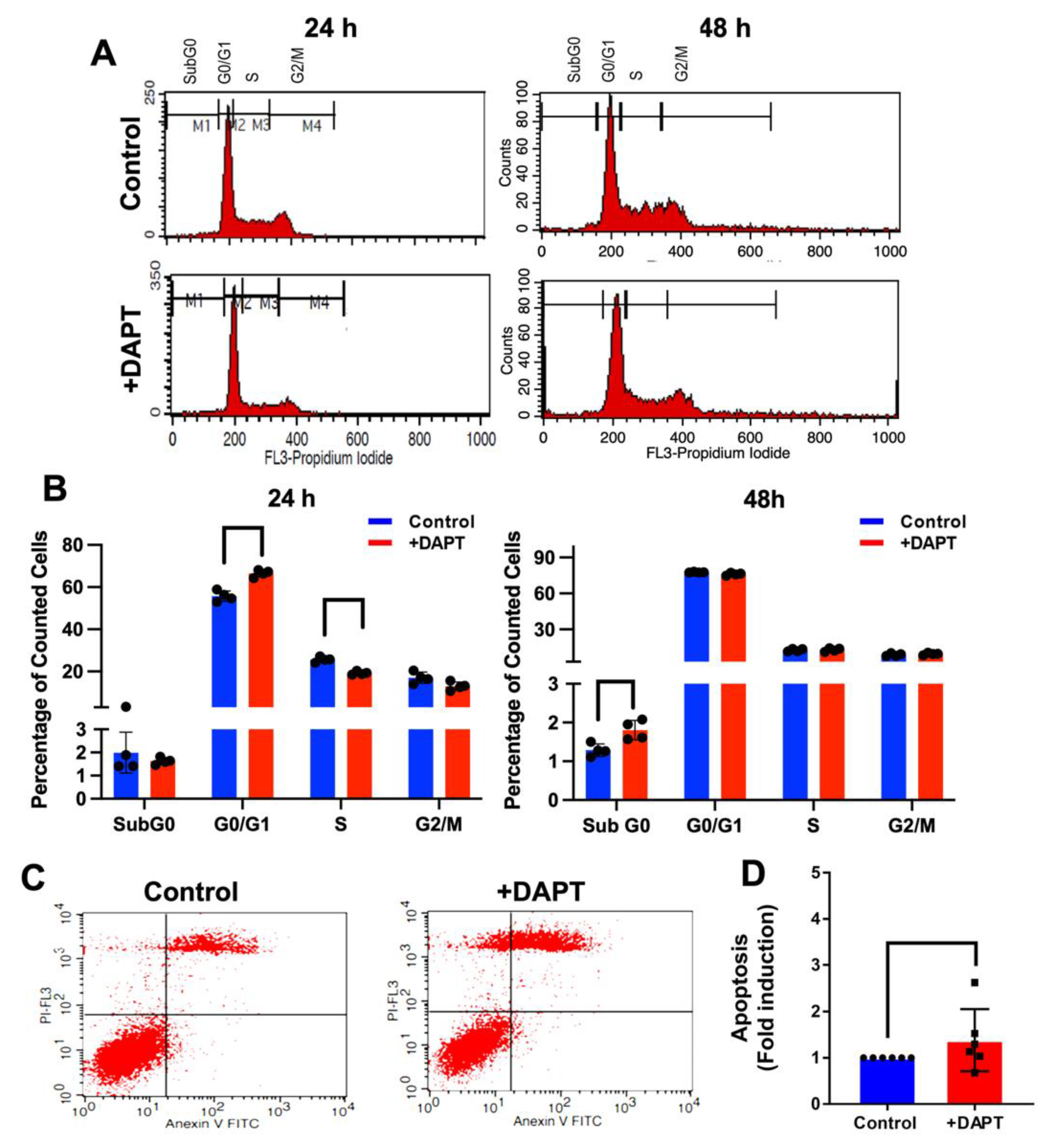
2.5. DAPT Induced Cell Cycle Arrest and Cell Apoptosis
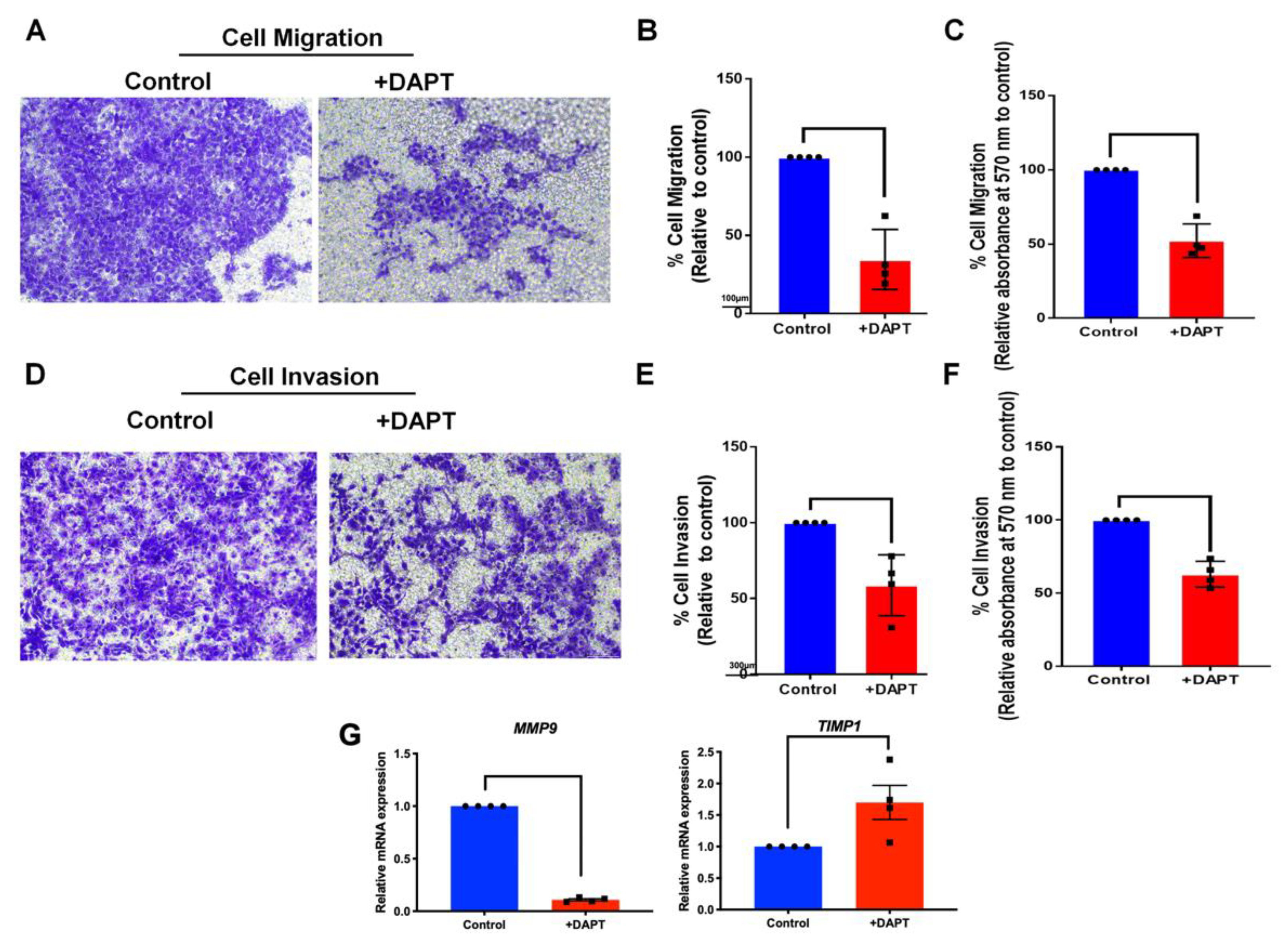
2.6. The Migration and Invasion Activities Were Abolished in DAPT-Treated HSC-4
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. RNA Sequencing
4.3. Bioinformatic Analysis
4.4. Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Cell Proliferation Assay
4.6. Immunofluorescence Staining
4.7. Colony Formation Assay
4.8. Cell Cycle Analysis
4.9. Annexin V FITC-PI Apoptosis Assay
4.10. Transwell Cell Migration
4.11. Transwell Invasion Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brouha, X.D.; Tromp, D.M.; Hordijk, G.J.; Winnubst, J.A.; de Leeuw, J.R. Oral and pharyngeal cancer: Analysis of patient delay at different tumor stages. Head Neck 2005, 27, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Maleki, D.; Ghojazadeh, M.; Mahmoudi, S.S.; Mahmoudi, S.M.; Pournaghi-Azar, F.; Torab, A.; Reza, P.; Saber, A.-A.; Naghavi-Behzad, M. Epidemiology of Oral Cancer in Iran: A Systematic Review. Asian Pac. J. Cancer Prev. 2015, 16, 5427–5432. [Google Scholar] [CrossRef] [PubMed]
- Suwanwela, J.; Osathanon, T. Inflammation related genes are upregulated in surgical margins of advanced stage oral squamous cell carcinoma. J. Oral Biol. Craniofac. Res. 2017, 7, 193–197. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef]
- Kyzas, P.A.; Stefanou, D.; Batistatou, A.; Agnantis, N.J. Hypoxia-induced tumor angiogenic pathway in head and neck cancer: An in vivo study. Cancer Lett. 2005, 225, 297–304. [Google Scholar] [CrossRef]
- Li, T.; Xu, X.H.; Guo, X.; Yuan, T.; Tang, Z.H.; Jiang, X.M.; Xu, Y.L.; Zhang, L.L.; Chen, X.P.; Zhu, H.; et al. Activation of notch 3/c-MYC/CHOP axis regulates apoptosis and promotes sensitivity of lung cancer cells to mTOR inhibitor everolimus. Biochem. Pharmacol. 2020, 175, 113921. [Google Scholar] [CrossRef]
- Xie, J.; Lin, L.S.; Huang, X.Y.; Gan, R.H.; Ding, L.C.; Su, B.H.; Zhao, Y.; Lu, Y.G.; Zheng, D.L. The NOTCH1-HEY1 pathway regulates self-renewal and epithelial-mesenchymal transition of salivary adenoid cystic carcinoma cells. Int. J. Biol. Sci. 2020, 16, 598–610. [Google Scholar] [CrossRef]
- Wang, Z.; Da Silva, T.G.; Jin, K.; Han, X.; Ranganathan, P.; Zhu, X.; Sanchez-Mejias, A.; Bai, F.; Li, B.; Fei, D.L.; et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res. 2014, 74, 6364–6374. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Han, N.; Xu, H.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Notch signaling and EMT in non-small cell lung cancer: Biological significance and therapeutic application. J. Hematol. Oncol. 2014, 7, 87. [Google Scholar] [CrossRef]
- Wu, W.K.; Wang, X.J.; Cheng, A.S.; Luo, M.X.; Ng, S.S.; To, K.F.; Chan, F.K.L.; Cho, C.H.; Sung, J.J.Y.; Yu, J. Dysregulation and crosstalk of cellular signaling pathways in colon carcinogenesis. Crit. Rev. Oncol. Hematol. 2013, 86, 251–277. [Google Scholar] [CrossRef]
- Hijioka, H.; Setoguchi, T.; Miyawaki, A.; Gao, H.; Ishida, T.; Komiya, S.; Nakamura, N. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int. J. Oncol. 2010, 36, 817–822. [Google Scholar]
- Joo, Y.H.; Jung, C.K.; Kim, M.S.; Sun, D.I. Relationship between vascular endothelial growth factor and Notch1 expression and lymphatic metastasis in tongue cancer. Otolaryngol. Head Neck Surg. 2009, 140, 512–518. [Google Scholar] [CrossRef]
- Izumchenko, E.; Sun, K.; Jones, S.; Brait, M.; Agrawal, N.; Koch, W.; McCord, C.L.; Riley, D.R.; Angiuoli, S.V.; Velculescu, V.E.; et al. Notch1 mutations are drivers of oral tumorigenesis. Cancer Prev. Res. (Phila) 2015, 8, 277–286. [Google Scholar] [CrossRef]
- Song, X.; Xia, R.; Li, J.; Long, Z.; Ren, H.; Chen, W.; Mao, L. Common and complex Notch1 mutations in Chinese oral squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 701–710. [Google Scholar] [CrossRef]
- Lee, S.H.; Hong, H.S.; Liu, Z.X.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. TNFalpha enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 424, 58–64. [Google Scholar] [CrossRef]
- Osathanon, T.; Nowwarote, N.; Pavasant, P. Expression and influence of Notch signaling in oral squamous cell carcinoma. J. Oral. Sci. 2016, 58, 283–294. [Google Scholar] [CrossRef]
- Hatano, K.; Saigo, C.; Kito, Y.; Shibata, T.; Takeuchi, T. Overexpression of JAG2 is related to poor outcomes in oral squamous cell carcinoma. Clin. Exp. Dent. Res. 2020, 6, 174–180. [Google Scholar] [CrossRef]
- Sardesai, S.; Badawi, M.; Mrozek, E.; Morgan, E.; Phelps, M.; Stephens, J.; Wei, L.; Kassem, M.; Ling, Y.; Lustberg, M.; et al. A phase I study of an oral selective gamma secretase (GS) inhibitor RO4929097 in combination with neoadjuvant paclitaxel and carboplatin in triple negative breast cancer. Investig. New Drugs. 2020, 38, 1400–1410. [Google Scholar] [CrossRef]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef]
- Cook, N.; Basu, B.; Smith, D.M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 793–801. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids. Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Rathinam, M.K.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef]
- Kushwah, R.; Guezguez, B.; Lee, J.B.; Hopkins, C.I.; Bhatia, M. Pleiotropic roles of Notch signaling in normal, malignant, and developmental hematopoiesis in the human. EMBO Rep. 2014, 15, 1128–1138. [Google Scholar] [CrossRef]
- Miele, L.; Osborne, B. Arbiter of differentiation and death: Notch signaling meets apoptosis. J. Cell. Physiol. 1999, 181, 393–409. [Google Scholar] [CrossRef]
- Wang, S.; Fan, H.; Xu, J.; Zhao, E. Prognostic implication of NOTCH1 in early stage oral squamous cell cancer with occult metastases. Clin. Oral. Investig. 2018, 22, 1131–1138. [Google Scholar] [CrossRef]
- Rasul, S.; Balasubramanian, R.; Filipović, A.; Slade, M.; Yagüe, E.; Coombes, R. Inhibition of γ-secretase induces G2/M arrest and triggers apoptosis in breast cancer cells. Br. J. Cancer 2009, 100, 1879–1888. [Google Scholar] [CrossRef]
- Patrad, E.; Niapour, A.; Farassati, F.; Amani, M. Combination treatment of all-trans retinoic acid (ATRA) and γ-secretase inhibitor (DAPT) cause growth inhibition and apoptosis induction in the human gastric cancer cell line. Cytotechnology 2018, 70, 865–877. [Google Scholar] [CrossRef]
- Grottkau, B.E.; Chen, X.R.; Friedrich, C.C.; Yang, X.M.; Jing, W.; Wu, Y.; Cai, X.X.; Liu, Y.; Huang, Y.; Lin, Y. DAPT enhances the apoptosis of human tongue carcinoma cells. Int. J. Oral Sci. 2009, 1, 81–89. [Google Scholar] [CrossRef]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Lanner, F.; Main, H.; Andersson, E.R.; Bergmann, O.; Sahlgren, C.; Heldring, N.; Hermanson, O.; Hansson, E.M.; Lendahl, U. Notch induces cyclin-D1-dependent proliferation during a specific temporal window of neural differentiation in ES cells. Dev. Biol. 2010, 348, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Zindy, F.; Sherr, C.J. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997, 11, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.W.; Liu, C.H.; Lu, K.; Huang, Y.Q.; Wang, Y.D.; Xing, L.; Zhang, L.J.; Liu, N.; Jiang, H.; et al. miRNA-30a functions as a tumor suppressor by downregulating cyclin E2 expression in castration-resistant prostate cancer. Mol. Med. Rep. 2016, 14, 2077–2084. [Google Scholar] [CrossRef]
- Barbieri, F.; Lorenzi, P.; Ragni, N.; Schettini, G.; Bruzzo, C.; Pedullà, F.; Alama, A. Overexpression of cyclin D1 is associated with poor survival in epithelial ovarian cancer. Oncology 2004, 66, 310–315. [Google Scholar] [CrossRef]
- Cohen, B.; Shimizu, M.; Izrailit, J.; Ng, N.F.; Buchman, Y.; Pan, J.G.; Dering, J.; Reedijk, M. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res. Treat. 2010, 123, 113–124. [Google Scholar] [CrossRef]
- Tanaka, M.; Setoguchi, T.; Hirotsu, M.; Gao, H.; Sasaki, H.; Matsunoshita, Y.; Komiya, S. Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br. J. Cancer 2009, 100, 1957–1965. [Google Scholar] [CrossRef]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef]
- Noseda, M.; Niessen, K.; McLean, G.; Chang, L.; Karsan, A. Notch-dependent cell cycle arrest is associated with downregulation of minichromosome maintenance proteins. Circ. Res. 2005, 97, 102–104. [Google Scholar] [CrossRef]
- Yu, S.; Wang, G.; Shi, Y.; Xu, H.; Zheng, Y.; Chen, Y. MCMs in cancer: Prognostic potential and mechanisms. Anal. Cell. Pathol. 2020, 2020, 3750294. [Google Scholar] [CrossRef]
- Lei, M. The MCM complex: Its role in DNA replication and implications for cancer therapy. Curr. Cancer Drug Targets 2005, 5, 365–380. [Google Scholar] [CrossRef]
- Peng, Y.-P.; Zhu, Y.; Yin, L.-D.; Zhang, J.-J.; Guo, S.; Fu, Y.; Miao, Y.; Wei, J.-S. The expression and prognostic roles of MCMs in pancreatic cancer. PLoS ONE 2016, 11, e0164150. [Google Scholar] [CrossRef]
- Li, X.; He, X.; Tian, W.; Wang, J. Short hairpin RNA targeting Notch2 inhibits U87 human glioma cell proliferation by inducing cell cycle arrest and apoptosis in vitro and in vivo. Mol. Med. Rep. 2014, 10, 2843–2850. [Google Scholar] [CrossRef][Green Version]
- Dimova, D.K.; Dyson, N.J. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 2005, 24, 2810–2826. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef]
- Tarangelo, A.; Lo, N.; Teng, R.; Kim, E.; Le, L.; Watson, D.; Furth, E.E.; Raman, P.; Ehmer, U.; Viatour, P. Recruitment of Pontin/Reptin by E2f1 amplifies E2f transcriptional response during cancer progression. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Noda, N.; Honma, S.; Ohmiya, Y. Hes1 is required for contact inhibition of cell proliferation in 3T3-L1 preadipocytes. Genes Cells 2011, 16, 704–713. [Google Scholar] [CrossRef]
- Hartman, J.; Müller, P.; Foster, J.S.; Wimalasena, J.; Gustafsson, J.-A. HES-1 inhibits 17β-estradiol and heregulin-β1-mediated upregulation of E2F–1. Oncogene 2004, 23, 8826–8833. [Google Scholar] [CrossRef]
- Chen, L.; Yu, J.H.; Lu, Z.H.; Zhang, W. E2F2 induction in related to cell proliferation and poor prognosis in non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 10545. [Google Scholar]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Böttinger, E.P. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Hijioka, H.; Kume, K.; Miyawaki, A.; Nakamura, N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol. Lett. 2013, 6, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Roche, J. The epithelial-to-mesenchymal transition in cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef]
- Zhang, L.; Sha, J.; Yang, G.; Huang, X.; Bo, J.; Huang, Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer cells. Cell Cycle 2017, 16, 999–1007. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, G.; Zhou, L.; Li, P.; Yun, M.; Shi, Q.; Wang, T.L.; Wu, X.T. Notch signalling induces epithelial-mesenchymal transition to promote metastasis in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 42, 2276–2284. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2018. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 March 2018).
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Marin-Garcia, P.; Ping, P.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome diagram viewer: Data structures and strategies to boost performance. Bioinformatics 2018, 34, 1208–1214. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Fabregat, A.; Korninger, F.; Viteri, G.; Sidiropoulos, K.; Marin-Garcia, P.; Ping, P.; Wu, G.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome graph database: Efficient access to complex pathway data. PLoS Comput. Biol. 2018, 14, e1005968. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Maraver, A.; Fernandez-Marcos, P.J.; Herranz, D.; Munoz-Martin, M.; Gomez-Lopez, G.; Canamero, M.; Mulero, F.; Megías, D.; Sanchez-Carbayo, M.; Shen, J.; et al. Therapeutic effect of gamma-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell 2012, 22, 222–234. [Google Scholar] [CrossRef]
- Reis, P.P.; Waldron, L.; Perez-Ordonez, B.; Pintilie, M.; Galloni, N.N.; Xuan, Y.; Cervigne, N.K.; Warner, G.C.; Makitie, A.A.; Simpson, C.; et al. A gene signature in histologically normal surgical margins is predictive of oral carcinoma recurrence. BMC Cancer 2011, 11, 437. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Li, P.; Xiao, L.Y.; Tan, H. Muc-1 promotes migration and invasion of oral squamous cell carcinoma cells via PI3K-Akt signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 10365–10374. [Google Scholar]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pongjantarasatian, S.; Nowwarote, N.; Rotchanakitamnuai, V.; Srirodjanakul, W.; Saehun, R.; Janebodin, K.; Manokawinchoke, J.; Fournier, B.P.J.; Osathanon, T. A γ-Secretase Inhibitor Attenuates Cell Cycle Progression and Invasion in Human Oral Squamous Cell Carcinoma: An In Vitro Study. Int. J. Mol. Sci. 2022, 23, 8869. https://doi.org/10.3390/ijms23168869
Pongjantarasatian S, Nowwarote N, Rotchanakitamnuai V, Srirodjanakul W, Saehun R, Janebodin K, Manokawinchoke J, Fournier BPJ, Osathanon T. A γ-Secretase Inhibitor Attenuates Cell Cycle Progression and Invasion in Human Oral Squamous Cell Carcinoma: An In Vitro Study. International Journal of Molecular Sciences. 2022; 23(16):8869. https://doi.org/10.3390/ijms23168869
Chicago/Turabian StylePongjantarasatian, Sarai, Nunthawan Nowwarote, Varumporn Rotchanakitamnuai, Watcharee Srirodjanakul, Ritmongkol Saehun, Kajohnkiart Janebodin, Jeeranan Manokawinchoke, Benjamin P. J. Fournier, and Thanaphum Osathanon. 2022. "A γ-Secretase Inhibitor Attenuates Cell Cycle Progression and Invasion in Human Oral Squamous Cell Carcinoma: An In Vitro Study" International Journal of Molecular Sciences 23, no. 16: 8869. https://doi.org/10.3390/ijms23168869
APA StylePongjantarasatian, S., Nowwarote, N., Rotchanakitamnuai, V., Srirodjanakul, W., Saehun, R., Janebodin, K., Manokawinchoke, J., Fournier, B. P. J., & Osathanon, T. (2022). A γ-Secretase Inhibitor Attenuates Cell Cycle Progression and Invasion in Human Oral Squamous Cell Carcinoma: An In Vitro Study. International Journal of Molecular Sciences, 23(16), 8869. https://doi.org/10.3390/ijms23168869