
Thiosemicarbazones Can Act Synergistically with Anthracyclines to Downregulate CHEK1 Expression and Induce DNA Damage in Cell Lines Derived from Pediatric Solid Tumors

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. SH-SY5Y Cells: The Only Tested Cell Line with High Pgp Expression
2.2. Thiosemicarbazones Acted Mainly Synergistically with Anthracyclines
2.3. Thiosemicarbazones Downregulated CHEK1 Expression, Leading to Mitotic Catastrophe after These Drugs Were Combined with Anthracyclines
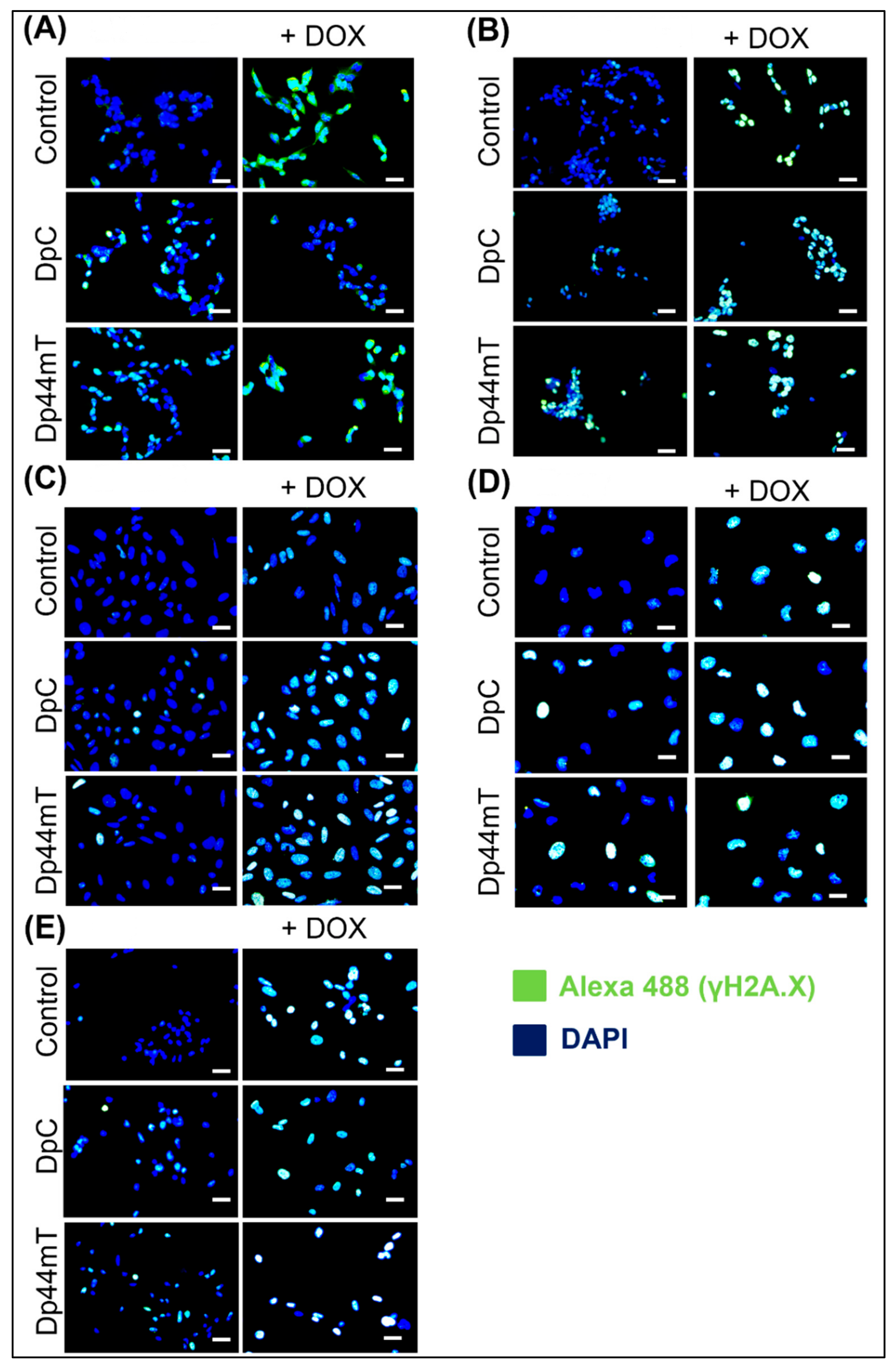
2.4. Thiosemicarbazones Can Induce DSBs in DNA
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Treatment Protocol
4.4. Cell Proliferation
4.5. Calculation of the CI
4.6. RT-qPCR
4.7. Immunoblotting
4.8. Indirect Immunofluorescence
4.9. Cell Cycle Analysis Using Propidium Iodide Staining
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martins-Teixeira, M.B.; Carvalho, I. Antitumour Anthracyclines: Progress and Perspectives. Chem. Med. Chem. 2020, 15, 933–948. [Google Scholar] [CrossRef] [PubMed]
- Nadas, J.; Sun, D. Anthracyclines as effective anticancer drugs. Expert Opin. Drug Discov. 2006, 1, 549–568. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Funes, H.; Coronado, C. Role of anthracyclines in the era of targeted therapy. Cardiovasc. Toxicol. 2007, 7, 56–60. [Google Scholar] [CrossRef]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.; Paz, O.G.; Chello, M.; Covino, E.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2012, 11 (Suppl. S1), S21–S36. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Capelôa, T.; Benyahia, Z.; Zampieri, L.X.; Blackman, M.C.N.M.; Sonveaux, P. Metabolic and non-metabolic pathways that control cancer resistance to anthracyclines. Semin. Cell Dev. Biol. 2020, 98, 181–191. [Google Scholar] [CrossRef]
- Raviv, Y.; Pollard, H.B.; Bruggemann, E.P.; Pastan, I.; Gottesman, M.M. Photosensitized labeling of a functional multidrug transporter in living drug–resistant tumor cells. J. Biol. Chem. 1990, 265, 3975–3980. [Google Scholar] [CrossRef]
- Nielsen, D.; Maare, C.; Poulsen, F.; Lauridsen, S.T.; Skovsgaard, T. Relationship between resistance, P-glycoprotein content and steady-state accumulation in five series of Ehrlich ascites tumour cell lines selected for resistance to daunorubicin. Cell Pharmacol. 1994, 1, 127–135. [Google Scholar]
- Nielsen, D.; Maare, C.; Skovsgaard, T. Kinetics of daunorubicin transport in Ehrlich ascites tumor cells with different expression of P-glycoprotein. Biochem. Pharmacol. 1994, 47, 2125–2135. [Google Scholar] [CrossRef]
- Shieh, M.J.; Hsu, C.Y.; Huang, L.Y.; Chen, H.Y.; Huang, F.H.; Lai, P.S. Reversal of doxorubicin-resistance by multifunctional nanoparticles in MCF-7/ADR cells. J. Control Release 2011, 152, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Cheah, H.Y.; Šarenac, O.; Arroyo, J.J.; Vasić, M.; Lozić, M.; Glumac, S.; Hoe, S.Z.; Hindmarch, C.C.; Murphy, D.; Kiew, L.V.; et al. Hemodynamic effects of HPMA copolymer based doxorubicin conjugate: A randomized controlled and comparative spectral study in conscious rats. Nanotoxicology 2017, 11, 210–222. [Google Scholar] [CrossRef]
- Ma, P.; Mumper, R.J. Anthracycline Nano-Delivery Systems to Overcome Multiple Drug Resistance: A Comprehensive Review. Nano Today 2013, 8, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Akasov, R.; Drozdova, M.; Zaytseva-Zotova, D.; Leko, M.; Chelushkin, P.; Marc, A.; Chevalot, I.; Burov, S.; Klyachko, N.; Vandamme, T.; et al. Novel Doxorubicin Derivatives: Synthesis and Cytotoxicity Study in 2D and 3D in Vitro Models. Adv. Pharm. Bull. 2017, 7, 593–601. [Google Scholar] [CrossRef]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Chhikara, B.S.; Mandal, D.; Parang, K. Synthesis, anticancer activities, and cellular uptake studies of lipophilic derivatives of doxorubicin succinate. J. Med. Chem. 2012, 55, 1500–1510. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, G.; Zhang, W.; Luo, H.; Qiu, L.; Liu, Q.; Sun, D.; Wang, P.G.; Wang, F. Synthesis and biological activities of a 3′-azido analogue of Doxorubicin against drug-resistant cancer cells. Int. J. Mol. Sci. 2012, 13, 3671–3684. [Google Scholar] [CrossRef]
- Piorecka, K.; Stanczyk, W.; Florczak, M. NMR analysis of antitumor drugs: Doxorubicin, daunorubicin and their functionalized derivatives. Tetrahedron Lett. 2017, 58, 152–155. [Google Scholar] [CrossRef]
- Kratz, F.; Beyer, U.; Roth, T.; Tarasova, N.; Collery, P.; Lechenault, F.; Cazabat, A.; Schumacher, P.; Unger, C.; Falken, U. Transferrin conjugates of doxorubicin: Synthesis, characterization, cellular uptake, and in vitro efficacy. J. Pharm. Sci. 1998, 87, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Cunnion, R.E.; Klecker, R.W., Jr.; Hamilton, T.C.; Ostchega, Y.; Parrillo, J.E.; Young, R.C. Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. J. Clin. Oncol. 1987, 5, 641–647. [Google Scholar] [CrossRef] [PubMed]
- List, A.F.; Kopecky, K.J.; Willman, C.L.; Head, D.R.; Persons, D.L.; Slovak, M.L.; Dorr, R.; Karanes, C.; Hynes, H.E.; Doroshow, J.H.; et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: A Southwest Oncology Group study. Blood 2011, 98, 3212–3220. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.S.; Grogan, T.M.; Meltzer, P.S.; Scheper, R.J.; Durie, B.G.; Taylor, C.W.; Miller, T.P.; Salmon, S.E. Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: Detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J. Clin. Oncol. 1989, 7, 415–424. [Google Scholar] [CrossRef]
- Kim, H.G.; Hien, T.T.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Kang, K.W.; Kwon, K.I.; Kim, B.H.; Kim, S.K.; Song, G.Y.; et al. Metformin inhibits P-glycoprotein expression via the NF-κB pathway and CRE transcriptional activity through AMPK activation. Br. J. Pharmacol. 2011, 162, 1096–1108. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Richardson, D.R.; Jansson, P.J. A mechanism for overcoming P-glycoprotein-mediated drug resistance: Novel combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. 2016, 7, e2510. [Google Scholar] [CrossRef]
- Seebacher, N.; Lane, D.J.R.; Richardson, D.R.; Jansson, P.J. Turning the gun on cancer: Utilizing lysosomal P-glycoprotein as a new strategy to overcome multi-drug resistance. Free Radic. Biol. Med. 2016, 96, 432–445. [Google Scholar] [CrossRef]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef]
- Jansson, P.J.; Yamagishi, T.; Arvind, A.; Seebacher, N.; Gutierrez, E.; Stacy, A.; Maleki, S.; Sharp, D.; Sahni, S.; Richardson, D.R. Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes multidrug-resistance by a novel mechanism involving the hijacking of lysosomal P-glycoprotein (Pgp). J. Biol. Chem. 2015, 290, 9588–9603. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. Glucose modulation induces lysosome formation and increases lysosomotropic drug sequestration via the P-glycoprotein drug transporter. J. Biol. Chem. 2016, 291, 3796–3820. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, D.B.; Jansson, P.J.; Brunk, U.T.; Wong, J.; Ponka, P.; Richardson, D.R. Antitumor Activity of Metal-Chelating Compound Dp44mT Is Mediated by Formation of a Redox-Active Copper Complex That Accumulates in Lysosomes. Cancer Res. 2011, 71, 5871–5880. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.M.; Seebacher, N.A.; Arzuman, L.; Kovacevic, Z.; Lane, D.J.; Richardson, V.; Merlot, A.M.; Lok, H.; Kalinowski, D.S.; Sahni, S.; et al. Lysosomal membrane stability plays a major role in the cytotoxic activity of the anti-agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT). Biochim. Biophys. Acta 2016, 1863, 1665–1681. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.E.; Palanimuthu, D.; Bernhardt, P.V.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. Zinc(II)-thiosemicarbazone complexes are localized to the lysosomal compartment where they transmetallate with copper ions to induce cytotoxicity. J. Med. Chem. 2016, 59, 4965–4984. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Fruci, D.; Cho, W.C.S.; Nobili, V.; Locatelli, F.; Alisi, A. Drug Transporters and Multiple Drug Resistance in Pediatric Solid Tumors. Curr. Drug Metab. 2016, 17, 308–316. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, D.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The Iron Chelators Dp44mT and DFO Inhibit TGF-β-induced Epithelial-Mesenchymal Transition via Up-Regulation of N-Myc Downstream-regulated Gene 1 (NDRG1). J. Biol. Chem. 2012, 287, 17016–17028. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Stacy, A.E.; Palanimuthu, D.; Bernhardt, P.V.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. Structure-activity relationships of di-2-pyridylketone, 2-benzoylpyridine and 2-acetylpyridine thiosemicarbazones for overcoming Pgp-mediated drug resistance. J. Med. Chem. 2016, 59, 8601–8620. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Kurz, E.U.; Douglas, P.; Lees-Miller, S.P. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 2004, 279, 53272–53281. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef]
- Mustofa, M.K.; Tanoue, Y.; Tateishi, C.; Vaziri, C.; Tateishi, S. Roles of CHEK2/CHEK2 in guarding against environmentally induced DNA damage and replication-stress. Environ. Mol. Mutagenes. 2020, 61, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.A.; Klein, S.R.; Agama, K.K.; Toyoda, E.; Adachi, N.; Pommier, Y.; Shacter, E.B. The Iron Chelator Dp44mT Causes DNA Damage and Selective Inhibition of Topoisomerase IIα in Breast Cells. Cancer Res. 2009, 69, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Banáth, J.P.; Olive, P.L. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003, 63, 4347–4350. [Google Scholar]
- Olive, P.L. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004, 75, 355–373. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohnishi, T. Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett. 2005, 229, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Xue, J.; Sowin, T.J.; Rosenberg, S.H.; Zhang, H. A novel mechanism of checkpoint abrogation conferred by CHEK1 downregulation. Oncogene 2005, 24, 1403–1411. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Richardson, D.R.; Tran, E.H.; Ponka, P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood 1995, 86, 4295–4306. [Google Scholar] [CrossRef]
- Krzyzankova, M.; Chovanova, S.; Chlapek, P.; Radsetoulal, M.; Neradil, J.; Zitterbart, K.; Sterba, J.; Veselska, R. LOX/COX inhibitors enhance the antineoplastic effects of all-trans retinoic acid in osteosarcoma cell lines. Tumor Biol. 2014, 35, 7617–7627. [Google Scholar] [CrossRef]
- Lovejoy, D.B.; Sharp, D.M.; Seebacher, N.; Obeidy, P.; Prichard, T.; Stefani, C.; Basha, M.T.; Sharpe, P.C.; Jansson, P.J.; Kalinowski, D.S.; et al. Novel Second-Generation Di-2-Pyridylketone Thiosemicarbazones Show Synergism with Standard Chemotherapeutics and Demonstrate Potent Activity against Lung Cancer Xenografts after Oral and Intravenous Administration in Vivo. J. Med. Chem. 2012, 55, 7230–7244. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Mikulenkova, E.; Neradil, J.; Zitterbart, K.; Sterba, J.; Veselska, R. Overexpression of the ∆Np73 isoform is associated with centrosome amplification in brain tumor cell lines. Tumor. Biol. 2015, 36, 7483–7491. [Google Scholar] [CrossRef]
- Vindeløv, L.L.; Christensen, I.J.; Nissen, N.I. A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry 1983, 3, 323–327. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | DOX [nM] 3D | DAU [nM] 3D | MIT [nM] 3D | DpC [nM] 3D | Dp44m [nM] 3D | DpC [µM] 1D | Dp44mT [µM] 1D |
|---|---|---|---|---|---|---|---|
| SH-SY5Y | 29.7 ± 4.9 | 40.2 ± 13.9 | 20.1 ± 3.6 | 3.8 ± 0.3 | 1.9 ± 0.8 | 11.1 ± 2.2 | 30.1 ± 1.5 |
| SK-N-BE(2) | 40.8 ± 8.1 | 31.9 ± 8.7 | 27.0 ± 1.7 | 7.2 ± 1.2 | 4.2 ± 1.1 | 14.8 ± 2.3 | 31.1 ± 5.2 |
| Saos-2 | 23.9 ± 2.8 | 20.8 ± 0.9 | 6.1 ± 0.6 | 4.4 ± 0.9 | 6.6 ± 0.9 | 20.0 ± 4.2 | 61.6 ± 14.9 |
| Daoy | 9.8 ± 2.0 | 17.6 ± 3.3 | 7.5 ± 0.2 | 17.0 ± 3.4 | 7.9 ± 1.9 | 3.9 ± 0.6 | 0.9 ± 0.1 |
| RD | 18.2 ± 3.8 | 15.7 ± 1.0 | 26.3 ± 4.4 | 13.8 ± 2.4 | 9.4 ± 0.8 | 11.5 ± 1.5 | 55.8 ± 7.7 |
| Cell Line | Drug Combination | Treatment Design | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Consecutive | Consecutive + Valspodar | Concomitant | ||||||||
| CI | SD | CI | SD | CI | SD | |||||
| SH-SY5Y | DOX | Dp44mT | 0.739 | 0.064 | 0.795 | 0.062 | 0.779 | 0.079 | ||
| DpC | 0.793 | 0.127 | 0.492 | 0.068 | 0.684 | 0.062 | ||||
| DAU | Dp44mT | 0.655 | 0.174 | 0.828 | 0.066 | 0.842 | 0.124 | |||
| DpC | 0.599 | 0.077 | 0.588 | 0.051 | 0.647 | 0.161 | ||||
| MIT | Dp44mT | 0.507 | 0.099 | 0.758 | 0.121 | 0.921 | 0.091 | |||
| DpC | 0.527 | 0.081 | 0.898 | 0.054 | 0.620 | 0.222 | ||||
| SK-N-BE(2) | DOX | Dp44mT | 0.722 | 0.053 | 0.567 | 0.117 | 0.712 | 0175 | ||
| DpC | 0.974 | 0.050 | 0.635 | 0.062 | 0.795 | 0,161 | ||||
| DAU | Dp44mT | 0.562 | 0.118 | 0.534 | 0.117 | 0.732 | 0.202 | |||
| DpC | 0.640 | 0.140 | 0.409 | 0.074 | 0.638 | 0.154 | ||||
| MIT | Dp44mT | 0.304 | 0.107 | 0.524 | 0.049 | 0.458 | 0.157 | |||
| DpC | 0.590 | 0.137 | 0.531 | 0.133 | 0.569 | 0.177 | ||||
| Saos-2 | DOX | Dp44mT | 1.105 | 0.102 | 0.710 | 0.125 | 0.657 | 0.051 | ||
| DpC | 1.205 | 0.066 | 0.596 | 0.057 | 0.831 | 0.101 | ||||
| DAU | Dp44mT | 1.244 | 0.289 | 0.712 | 0.149 | 0.813 | 0.135 | |||
| DpC | 1.422 | 0.085 | 0.611 | 0.172 | 0.672 | 0.177 | ||||
| MIT | Dp44mT | 1.108 | 0.182 | 0.610 | 0.056 | 0.828 | 0.112 | |||
| DpC | 1.439 | 0.255 | 0.723 | 0.160 | 0.527 | 0.108 | ||||
| Daoy | DOX | Dp44mT | 0.720 | 0.210 | 0.591 | 0.078 | 1.055 | 0.081 | ||
| DpC | 0.578 | 0.138 | 0.611 | 0.015 | 0.637 | 0.113 | ||||
| DAU | Dp44mT | 0.474 | 0.033 | 0.602 | 0.118 | 1.275 | 0.280 | |||
| DpC | 0.789 | 0.167 | 0.788 | 0.157 | 0.854 | 0.103 | ||||
| MIT | Dp44mT | 0.663 | 0.277 | 0.564 | 0.137 | 0.501 | 0.238 | |||
| DpC | 0.439 | 0.029 | 0.746 | 0.035 | 0.700 | 0.175 | ||||
| RD | DOX | Dp44mT | 0.807 | 0.126 | 0.472 | 0.108 | 0.648 | 0.043 | ||
| DpC | 0.770 | 0.008 | 0.830 | 0.057 | 0.712 | 0.312 | ||||
| DAU | Dp44mT | 0.458 | 0.143 | 0.383 | 0.059 | 0.773 | 0.211 | |||
| DpC | 0.609 | 0.097 | 0.671 | 0.173 | 0.764 | 0.118 | ||||
| MIT | Dp44mT | 0.501 | 0.099 | 0.366 | 0.031 | 0.493 | 0.108 | |||
| DpC | 0.641 | 0.081 | 0.541 | 0.134 | 0.638 | 0.127 | ||||
| Categories of Interactions | ||||||||||
| 0.31–0.70 | synergism | 0.91–1.10 | nearly additive | |||||||
| 0.71–0.85 | moderate synergism | 1.11–1.20 | slight antagonism | |||||||
| 0.86–0.90 | slight synergism | 1.21–1.45 | moderate antagonism | |||||||
| Cell Line | Type of Medium | Glucose | FCS | Atb | Glu | NEAA |
|---|---|---|---|---|---|---|
| Daoy | DMEM | Low | 10% | + | + | + |
| KB-3-1 | DMEM | High | 10% | + | + | - |
| KB-V1 | DMEM + VBL | High | 10% | + | + | + |
| RD | DMEM | High | 10% | + | + | + |
| Saos-2 | DMEM | Low | 10% | + | + | - |
| SH-SY5Y | DMEM:F12 (1:1) | High | 20% | + | + | + |
| SK-N-BE(2) | DMEM:F12 (1:1) | High | 20% | + | + | + |
| Gene | Primer Sequence | Product Length (bp) |
|---|---|---|
| ABCB1 | F: 5′-CTTTAGTGGAAAGACCACAGATGA-3′ R: 5′-CTTTAGTGGAAAGACCACAGATGA-3′ | 228 |
| GAPDH | F: 5′-AGC CAC ATC GCT CAG ACA CC-3′ R: 5′-GTA CTC AGC GCC AGC ATC G-3′ | 302 |
| Primary antibodies | |||||
|---|---|---|---|---|---|
| Antigen | Type/Host | Clone | Catalog No. | Manufacturer | Dilution |
| GAPDH | Mono/Rb | 14C10 | 2118S | CST | 1:10,000 |
| CHEK1 | Mono/Mo | 2G1D5 | 2360S | CST | 1:1000 |
| CHEK2 | Mono/Rb | D9C6 | 6334S | CST | 1:1000 |
| Pgp | Mono/Mo | F4 | P7965 | Sigma-Aldrich | 1:5000 |
| Secondary antibodies | |||||
| Host | Specificity | Conjugate | Catalog No. | Manufacturer | Dilution |
| Goat | Anti-Rb IgG | HRP | 7074 | CST | 1:5000 |
| Horse | Anti-Mo IgG | HRP | 7076 | CST | 1:5000 |
| Primary antibodies | |||||
|---|---|---|---|---|---|
| Antigen | Type/Host | Clone | Catalog No. | Manufacturer | Dilution |
| Pgp | Mono/Mo | F4 | P7965 | Sigma-Aldrich | 1:100 |
| LAMP-2 | Mono/Mo | H4B4 | ab25631 | Abcam | 1:100 |
| Phospho-γH2A.X | Mono/Mo | 3F2 | MA1-2022 | Invitrogen | 1:100 |
| Secondary antibodies | |||||
| Host | Specificity | Conjugate | Catalog No. | Manufacturer | Dilution |
| Donkey | Anti-Mo IgG | AF-488 | A21202 | Invitrogen | 1:200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paukovcekova, S.; Krchniakova, M.; Chlapek, P.; Neradil, J.; Skoda, J.; Veselska, R. Thiosemicarbazones Can Act Synergistically with Anthracyclines to Downregulate CHEK1 Expression and Induce DNA Damage in Cell Lines Derived from Pediatric Solid Tumors. Int. J. Mol. Sci. 2022, 23, 8549. https://doi.org/10.3390/ijms23158549
Paukovcekova S, Krchniakova M, Chlapek P, Neradil J, Skoda J, Veselska R. Thiosemicarbazones Can Act Synergistically with Anthracyclines to Downregulate CHEK1 Expression and Induce DNA Damage in Cell Lines Derived from Pediatric Solid Tumors. International Journal of Molecular Sciences. 2022; 23(15):8549. https://doi.org/10.3390/ijms23158549
Chicago/Turabian StylePaukovcekova, Silvia, Maria Krchniakova, Petr Chlapek, Jakub Neradil, Jan Skoda, and Renata Veselska. 2022. "Thiosemicarbazones Can Act Synergistically with Anthracyclines to Downregulate CHEK1 Expression and Induce DNA Damage in Cell Lines Derived from Pediatric Solid Tumors" International Journal of Molecular Sciences 23, no. 15: 8549. https://doi.org/10.3390/ijms23158549
APA StylePaukovcekova, S., Krchniakova, M., Chlapek, P., Neradil, J., Skoda, J., & Veselska, R. (2022). Thiosemicarbazones Can Act Synergistically with Anthracyclines to Downregulate CHEK1 Expression and Induce DNA Damage in Cell Lines Derived from Pediatric Solid Tumors. International Journal of Molecular Sciences, 23(15), 8549. https://doi.org/10.3390/ijms23158549