
The Effect of Conjugation of Ciprofloxacin and Moxifloxacin with Fatty Acids on Their Antibacterial and Anticancer Activity

 , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
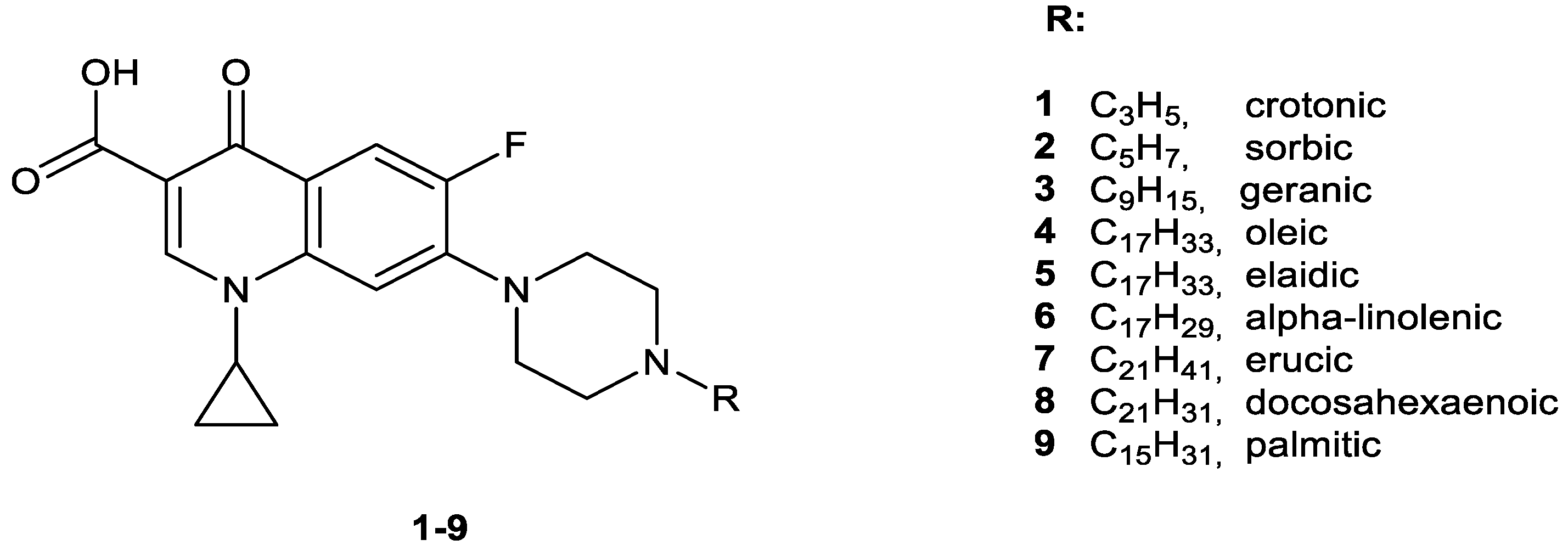
2.1. Chemistry
2.2. Cytotoxic Activity
2.3. In Vitro Antibacterial Studies
2.4. In Vitro Antimycobacterial Activity
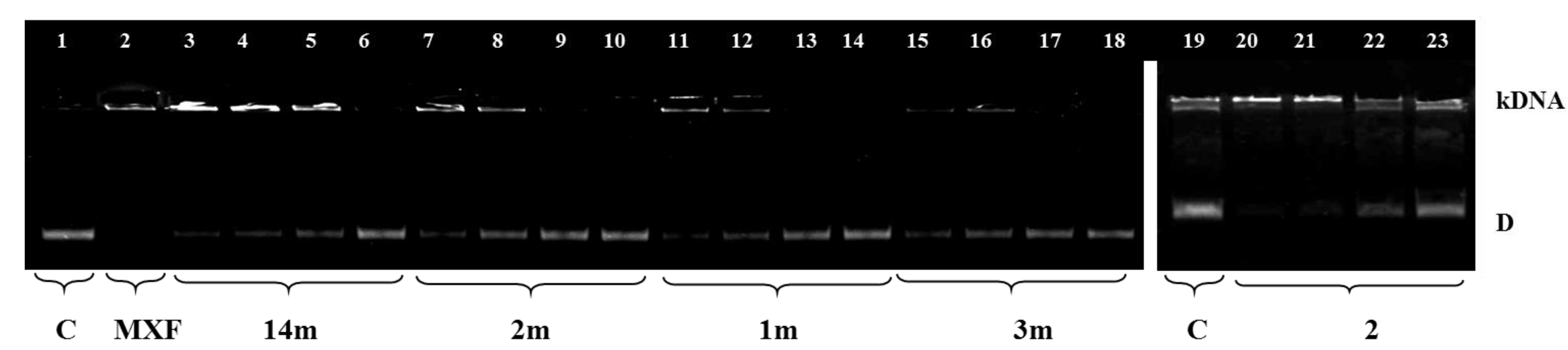
2.5. Inhibition of Bacterial DNA Topoisomerases
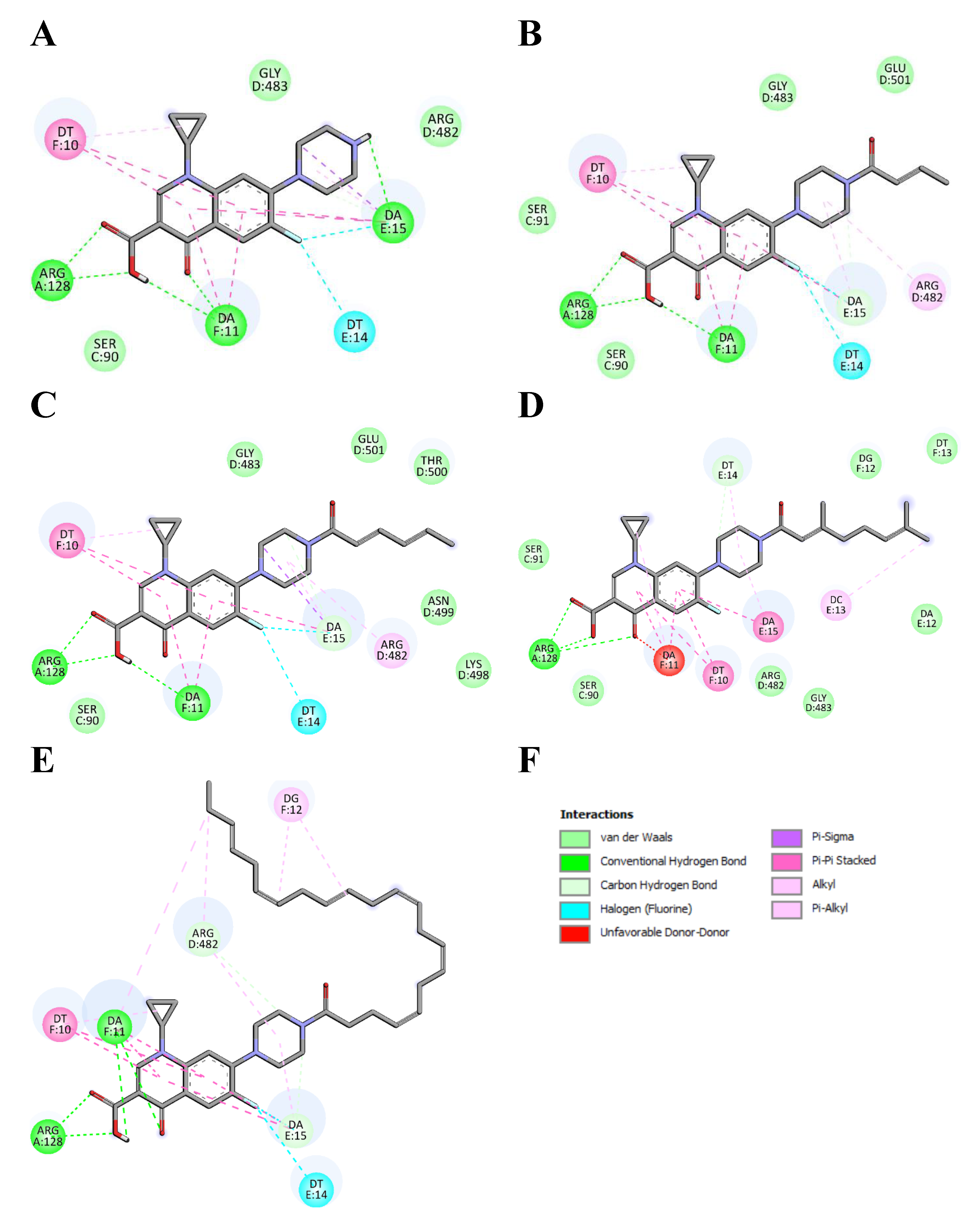
2.6. Molecular Docking Studies
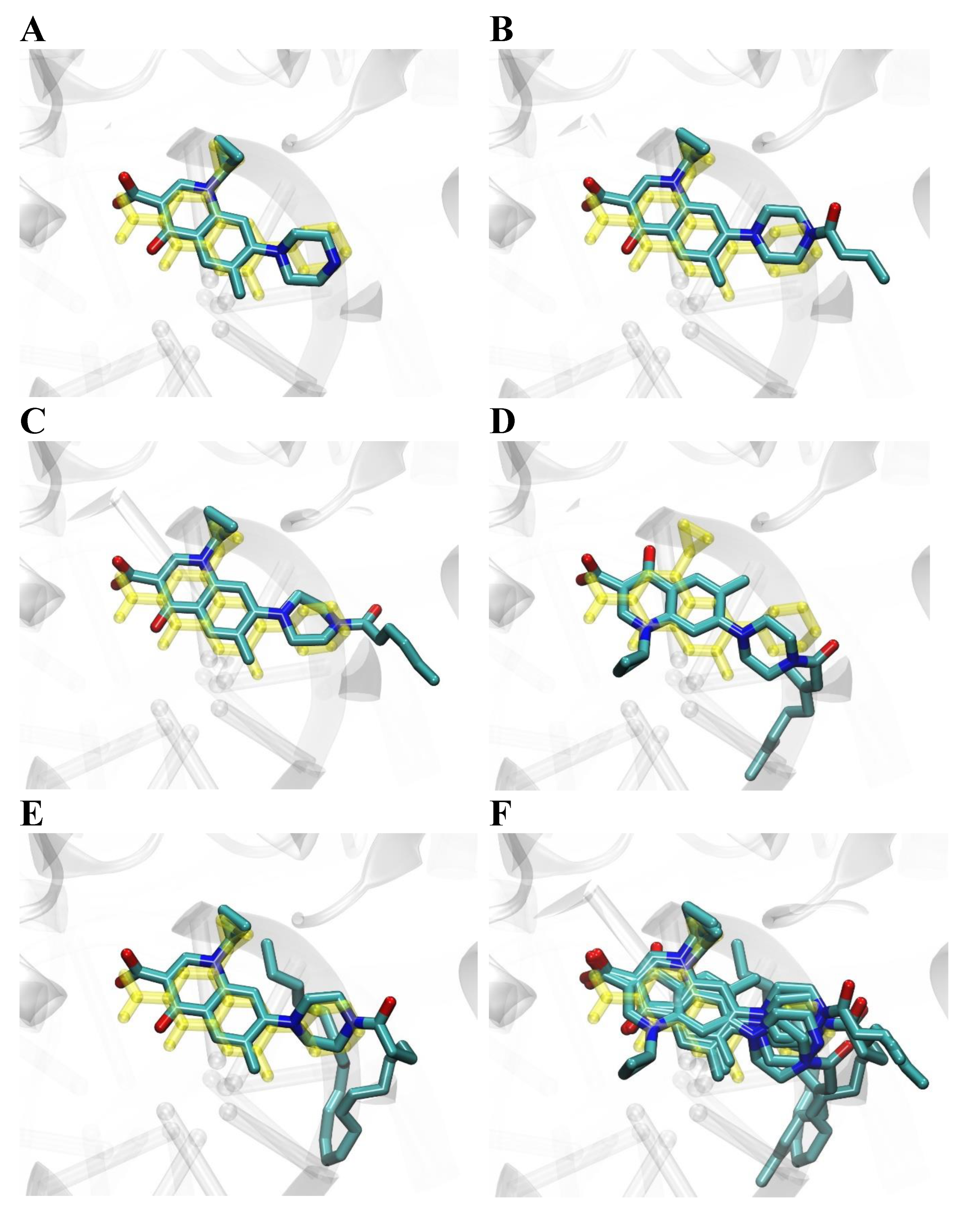
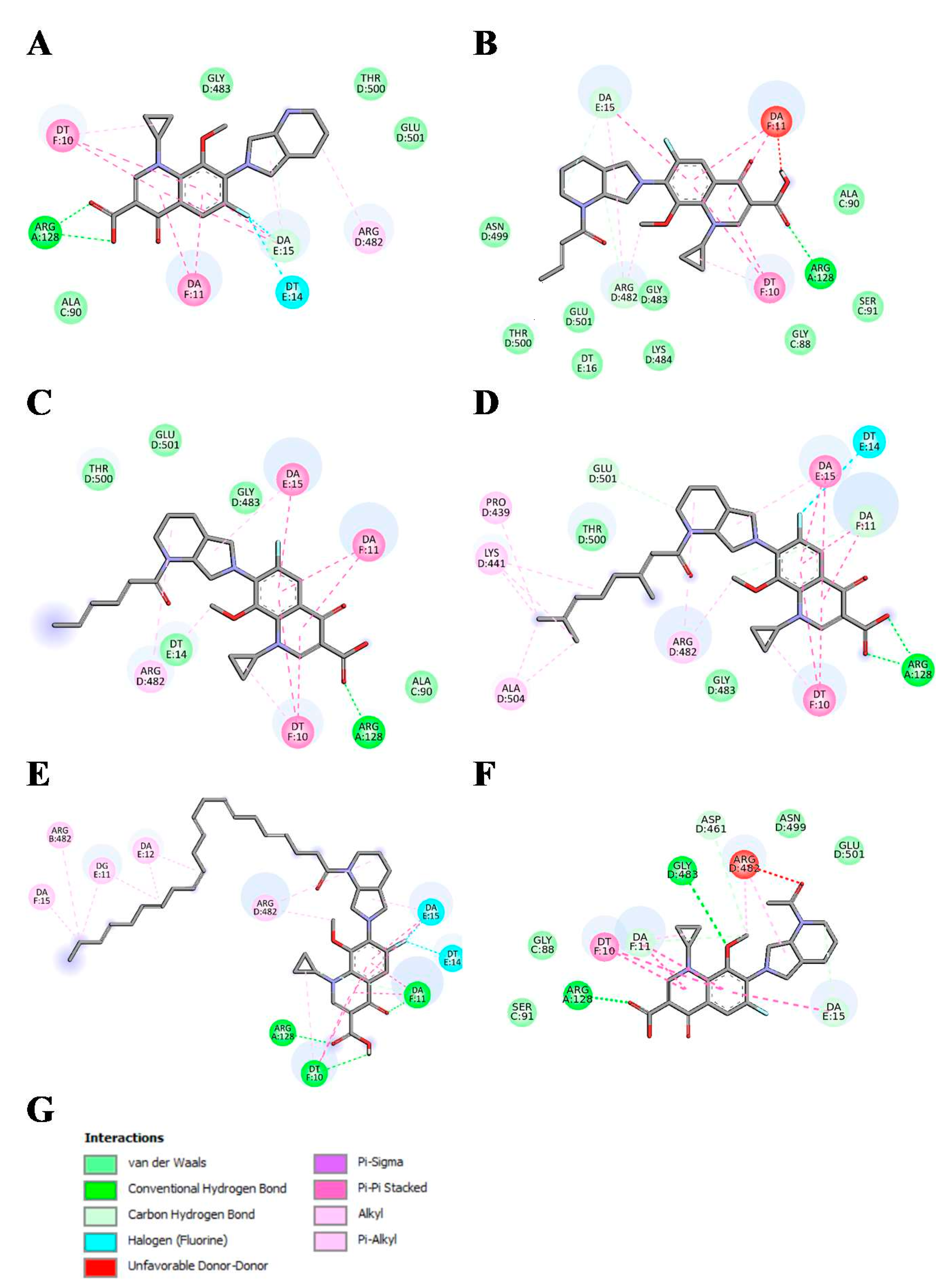
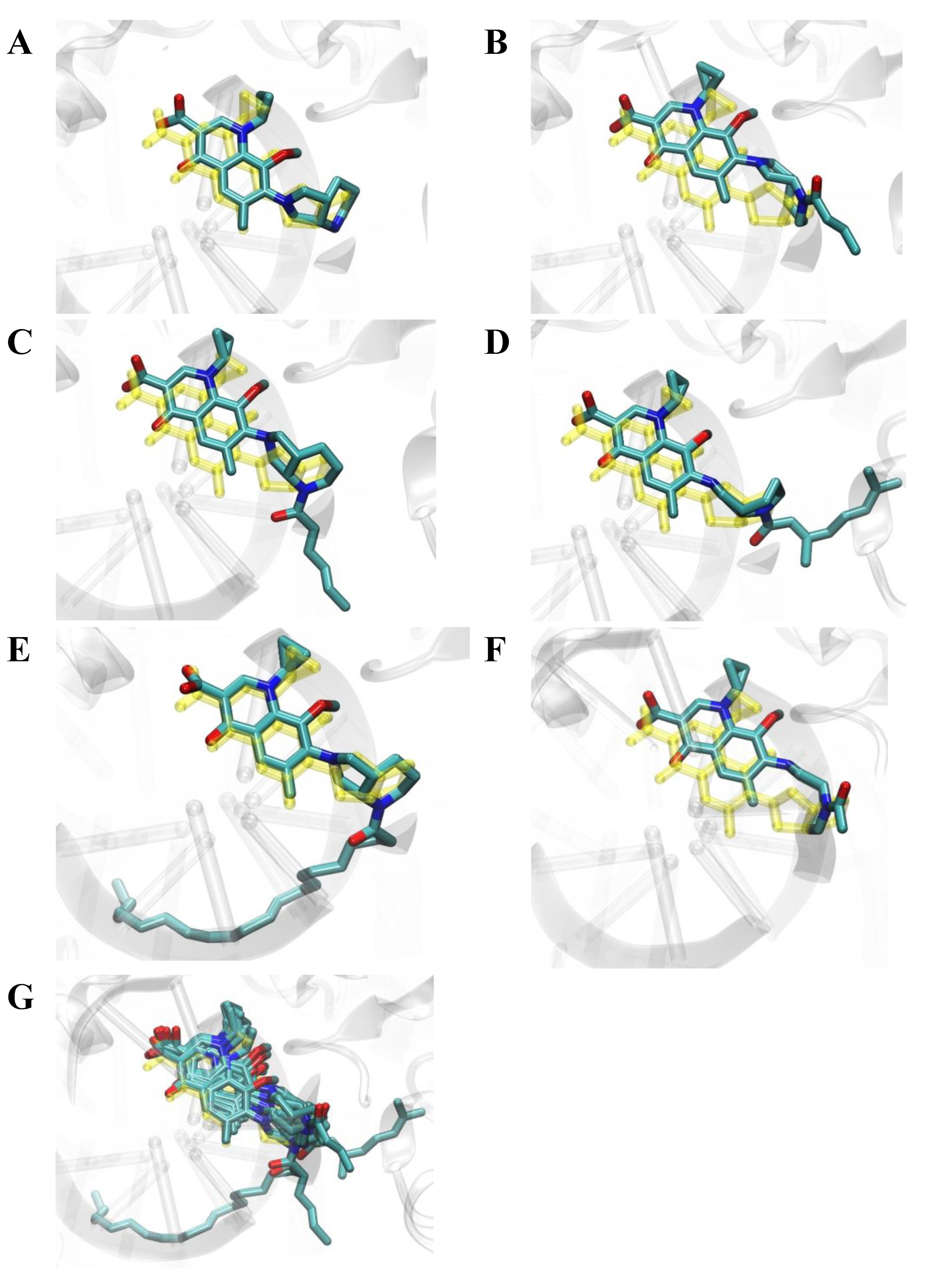
2.7. Conclusions
3. Materials and Methods
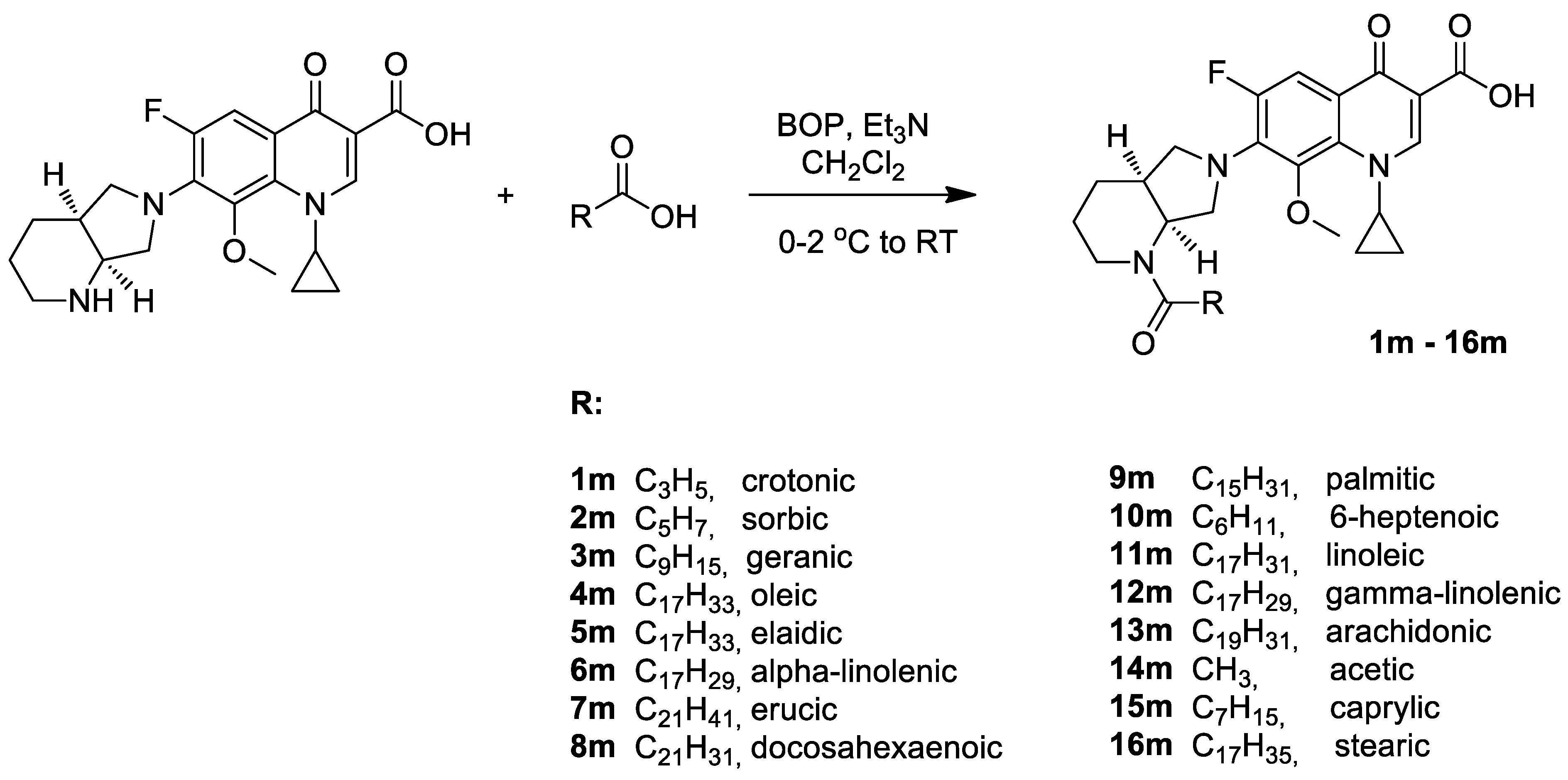
3.1. Chemistry
- 7-{1-(E)-But-2-enoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (1m)
- 1-Cyclopropyl-6-fluoro-7-{1-(2E,4E)-hexa-2,4-dienoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (2m)
- 1-Cyclopropyl-7-{1-(2E)-(3,7-dimethyl-octa-2,6-dienoyl)-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (contaminated with (Z)-isomer) (3m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-{1-(Z)-octadec-9-enoyl-octahydro-pyrrolo[3,4-b] pyridin-6-yl}-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (4m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-{1-(E)-octadec-9-enoyl-octahydro-pyrrolo[3,4-b] pyridin-6-yl}-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (5m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-{1-(9Z,12Z,15Z)-octadeca-9,12,15-trienoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (6m)
- 1-Cyclopropyl-7-{1-(Z)-docos-13-enoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (7m)
- 1-Cyclopropyl-7-{1-(4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (8m)
- 1-Cyclopropyl-6-fluoro-7-(1-hexadecanoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl)-8-methox-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (9m)
- 1-Cyclopropyl-6-fluoro-7-(1-hept-6-enoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl)-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (10m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-{1-(9Z,12Z)-octadeca-9,12-dienoyl-octahydropyrrolo-[3,4-b]pyridin-6-yl)-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (11m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-{1-(6Z,9Z,12Z)-octadeca-6,9,12-trienoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (12m)
- 1-Cyclopropyl-6-fluoro-7-{1-(5Z,8Z,11Z,14Z)-icosa-5,8,11,14-tetraenoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl}-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (13m)
- 7-(1-Acetyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl)-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (14m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-(1-octanoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl)-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (15m)
- 1-Cyclopropyl-6-fluoro-8-methoxy-7-(1-octadecanoyl-octahydro-pyrrolo[3,4-b]pyridin-6-yl)-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (16m)
3.2. Biological Studies
3.2.1. Cell Culture
3.2.2. MTT Assay
3.2.3. In Vitro Antibacterial Studies
3.2.4. In Vitro Antimycobacterial Activity
3.2.5. Topoisomerases Inhibition Determination
- S. aureus DNA Gyrase Supercoiling Assay
- S. aureus Topoisomerase IV Decatenation Assay
3.2.6. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Emmerson, A.M.; Jones, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51 (Suppl. 1), 13–20. [Google Scholar] [CrossRef] [Green Version]
- Bush, N.G.; Diez-Santos, I.; Abbott, L.R.; Maxwell, A. Quinolones: Mechanism, Lethality and Their Contributions to Antibiotic Resistance. Molecules 2020, 25, 5662. [Google Scholar] [CrossRef]
- Al-Omar, M.A. Ciprofloxacin: Drug metabolism and pharmacokinetic profile. Profiles Drug Subst. Excip. Relat. Methodol. 2005, 31, 209–214. [Google Scholar]
- Peterson, L.R. Quinolone molecular structure-activity relationships: What we have learned about improving antimicrobial activity. Clin. Infect. Dis. 2001, 33 (Suppl. 3), S180–S186. [Google Scholar] [CrossRef]
- Al Omari, M.M.; Jaafari, D.S.; Al-Sou’od, K.A.; Badwan, A.A. Moxifloxacin hydrochloride. Profiles Drug Subst. Excip. Relat. Methodol. 2014, 39, 299–431. [Google Scholar] [CrossRef]
- Keating, G.M.; Scott, L.J. Moxifloxacin: A review of its use in the management of bacterial infections. Drugs 2004, 64, 2347–2377. [Google Scholar] [CrossRef]
- Yilmaz, F.F.; Erac, B.; Ermertcan, S.; Cavusoglu, C.; Bicmen, C.; Aktogu Ozkan, S.; Hosgor Limoncu, M. In vitro effects of ciprofloxacin, levofloxacin and moxifloxacin on Mycobacterium tuberculosis isolates. Tuberk Toraks 2018, 66, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.E.; Nahid, P.; Kurbatova, E.V.; Phillips, P.P.J.; Bryant, K.; Dooley, K.E.; Engle, M.; Goldberg, S.V.; Phan, H.T.T.; Hakim, J.; et al. Four-Month Rifapentine Regimens with or without Moxifloxacin for Tuberculosis. N. Engl. J. Med. 2021, 384, 1705–1718. [Google Scholar] [CrossRef]
- Sun, M.; Fan, J. Moxifloxacin is a safe and effective candidate agent for tuberculosis treatment: A meta-analysis of randomized controlled trials. Ann. Palliat. Med. 2021, 10, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Kreuzer, K.N.; Cozzarelli, N.R. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: Effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. J. Bacteriol. 1979, 140, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Khodursky, A.B.; Zechiedrich, E.L.; Cozzarelli, N.R. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 11801–11805. [Google Scholar] [CrossRef] [Green Version]
- Fournier, B.; Zhao, X.; Lu, T.; Drlica, K.; Hooper, D.C. Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: Different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrob. Agents Chemother. 2000, 44, 2160–2165. [Google Scholar] [CrossRef] [Green Version]
- Ince, D.; Zhang, X.; Silver, L.C.; Hooper, D.C. Dual targeting of DNA gyrase and topoisomerase IV: Target interactions of garenoxacin (BMS-284756, T-3811ME), a new desfluoroquinolone. Antimicrob. Agents Chemother. 2002, 46, 3370–3380. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Huang, X.; Wang, X.; Yan, S.; Guo, S.; Abdalla, A.E.; Huang, C.; Xie, J. l-Serine potentiates fluoroquinolone activity against Escherichia coli by enhancing endogenous reactive oxygen species production. J. Antimicrob. Chemother. 2016, 71, 2192–2199. [Google Scholar] [CrossRef] [Green Version]
- Becerra, M.C.; Albesa, I. Oxidative stress induced by ciprofloxacin in Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2002, 297, 1003–1007. [Google Scholar] [CrossRef]
- Kloskowski, T.; Gurtowska, N.; Bajek, A.; Drewa, T. Ciprofloxacin as a prophylactic agent against prostate cancer: A “two hit” hypothesis. Med. Hypotheses 2012, 78, 235–238. [Google Scholar] [CrossRef]
- Yadav, V.; Talwar, P. Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: An underestimated truth. Biomed. Pharmacother. 2019, 111, 934–946. [Google Scholar] [CrossRef]
- Pakiet, A.; Kobiela, J.; Stepnowski, P.; Sledzinski, T.; Mika, A. Changes in lipids composition and metabolism in colorectal cancer: A review. Lipids Health Dis. 2019, 18, 29. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111–120. [Google Scholar]
- Shulse, C.N.; Allen, E.E. Widespread occurrence of secondary lipid biosynthesis potential in microbial lineages. PLoS ONE 2011, 6, e20146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.J.; Yoo, J.S.; Lee, T.G.; Cho, H.Y.; Kim, Y.H.; Kim, W.G. Fatty acid synthesis is a target for antibacterial activity of unsaturated fatty acids. FEBS Lett. 2005, 579, 5157–5162. [Google Scholar] [CrossRef] [Green Version]
- McGaw, L.J.; Jäger, A.K.; van Staden, J. Antibacterial effects of fatty acids and related compounds from plants. S. Afr. J. Bot. 2002, 68, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Churchward, C.P.; Alany, R.G.; Snyder, L.A.S. Alternative antimicrobials: The properties of fatty acids and monoglycerides. Crit. Rev. Microbiol. 2018, 44, 561–570. [Google Scholar] [CrossRef]
- Chrzanowska, A.; Roszkowski, P.; Bielenica, A.; Olejarz, W.; Stepien, K.; Struga, M. Anticancer and antimicrobial effects of novel ciprofloxacin fatty acids conjugates. Eur. J. Med. Chem. 2020, 185, 111810. [Google Scholar] [CrossRef] [PubMed]
- Ptaszynska, N.; Gucwa, K.; Olkiewicz, K.; Heldt, M.; Serocki, M.; Stupak, A.; Martynow, D.; Debowski, D.; Gitlin-Domagalska, A.; Lica, J.; et al. Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 4696. [Google Scholar] [CrossRef]
- Shen, L.L.; Pernet, A.G. Mechanism of inhibition of DNA gyrase by analogues of nalidixic acid: The target of the drugs is DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Tunitskaya, V.L.; Khomutov, A.R.; Kochetkov, S.N.; Kotovskaya, S.K.; Charushin, V.N. Inhibition of DNA gyrase by levofloxacin and related fluorine-containing heterocyclic compounds. Acta Naturae 2011, 3, 94–99. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual Screening with AutoDock: Theory and Practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Wright, H.T.; Reynolds, K.A. Antibacterial targets in fatty acid biosynthesis. Curr. Opin. Microbiol. 2007, 10, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Darvishi, M.; Farahani, S.; Haeri, A. Moxifloxacin-Loaded Lipidic Nanoparticles for Antimicrobial Efficacy. Curr. Pharm. Des. 2021, 27, 135–140. [Google Scholar] [CrossRef]
- Millanao, A.R.; Mora, A.Y.; Villagra, N.A.; Bucarey, S.A.; Hidalgo, A.A. Biological Effects of Quinolones: A Family of Broad-Spectrum Antimicrobial Agents. Molecules 2021, 26, 7153. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Williamson, B.H.; Kerns, R.J.; Berger, J.M. Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, 1706–1713. [Google Scholar] [CrossRef] [Green Version]
- Melvin, P. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2019; Volume 38. [Google Scholar]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Reck, F.; Alm, R.; Brassil, P.; Newman, J.; Dejonge, B.; Eyermann, C.J.; Breault, G.; Breen, J.; Comita-Prevoir, J.; Cronin, M.; et al. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: Broad-spectrum antibacterial agents with reduced hERG activity. J. Med. Chem. 2011, 54, 7834–7847. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound a | Cancer Cells | Normal Cells | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SW480 d | SW620 e | PC3 f | HaCaT g | ||||||
| Name of Fatty Acids | Chain Length: Unsaturation | IC50 b | SI c | IC50 | SI | IC50 | SI | IC50 | |
| 1m | crotonic | 4:1 (E2) | 31.6 ± 4.7 | 2.0 | 24.8 ± 3.2 | 2.6 | 23.8 ± 3.4 | 2.7 | 64.1 ± 2.3 |
| 2m | sorbic | 6:2 (E2, E4) | 6.3 ± 1.2 | 15.7 | 3.9 ± 1.1 | 25.4 | 2.7 ± 1.1 | 36.7 | 99.1 ± 1.8 |
| 3m | geranic | 10:2 (E2, E6) | 26.7 ± 1.4 | 3.0 | 38.2 ± 4.1 | 2.1 | 23.5 ± 4.7 | 3.5 | 81.2 ± 2.6 |
| 4m | oleic | 18:1 (Z9) | 4.6 ± 1.4 | 20.1 | 3.4 ± 0.7 | 27.1 | 1.3 ± 0.1 | 71.0 | 92.3 ± 6.1 |
| 5m | elaidic | 18:1 (E9) | 6.7 ± 0.6 | 13.7 | 8.7 ± 1.2 | 10.5 | 5.3 ± 1.3 | 17.3 | 91.6 ± 4.6 |
| 6m | α-linolenic | 18:3 (Z9, Z12, Z15) | 16.7 ± 3.1 | 5.8 | 15.6 ± 2.6 | 6.2 | 14.4 ± 2.4 | 6.7 | 96.6 ± 5.5 |
| 7m | erucic | 22:1 (Z13) | 48.6 ± 2.6 | 1.8 | 51.8 ± 4.3 | 1.6 | 36.5 ± 2.3 | 2.3 | 85.2 ± 3.3 |
| 8m | DHA | 22:6 (Z4, Z7, Z10, Z13, Z16, Z19) | 6.2 ± 1.3 | 13.1 | 9.4 ± 3.7 | 8.7 | 5.6 ± 1.9 | 14.5 | 81.4 ± 3.2 |
| 9m | palmitic | 16:0 | 9.4 ± 2.3 | 10.3 | 10.4 ± 1.4 | 9.3 | 8.1 ± 1.5 | 11.9 | 96.7 ± 2.6 |
| 10m | 6-heptenoic | 7:1 | 5.8 ± 0.8 | 15.1 | 6.2 ± 1.1 | 14.1 | 4.6 ± 1.1 | 19.0 | 87.6 ± 4.5 |
| 11m | linoleic | 18:2 (Z9, Z12) | 4.8 ± 1.3 | 19.0 | 7.3 ± 0.9 | 12.5 | 3.6 ± 1.1 | 25.4 | 91.4 ± 5.3 |
| 12m | γ-linolenic | 18:3 (Z6, Z9, Z12) | 16.8 ± 2.3 | 5.3 | 18.3 ± 3.2 | 4.8 | 14.7 ± 2.3 | 6.0 | 88.7 ± 3.3 |
| 13m | arachidonic | 20:4 (Z5, Z8, Z11, Z14) | 32.3 ± 4.1 | 2.9 | 39.4 ± 4.1 | 2.4 | 28.3 ± 2.8 | 3.3 | 93.3 ± 2.8 |
| 14m | acetic | 2:0 | 14.3 ± 3.2 | 6.0 | 18.4 ± 3.2 | 4.7 | 12.5 ± 1.6 | 6.9 | 86.5 ± 2.1 |
| 15m | caprylic | 8:0 | 3.2 ± 0.8 | 27.3 | 4.7 ± 1.5 | 18.6 | 3.1 ± 0.9 | 28.1 | 87.2 ± 3.6 |
| 16m | stearic | 18:0 | 2.7 ± 0.7 | 30.2 | 6.5 ± 1.2 | 12.5 | 2.4 ± 0.6 | 34.0 | 81.5 ± 4.2 |
| MXF h | - | - | 58.4 ± 3.1 | 2.1 | 54.5 ± 3.8 | 2.2 | 46.4 ± 3.2 | 2.6 | 121.2 ± 6.8 |
| CP i | - | - | 160.4 ± 6.7 | 1.4 | 200.4 ± 4.9 | 1.1 | 101.4 ± 3.6 | 2.2 | 222.1 ± 5.2 |
| Cisplatin j | - | - | 10.4 ± 0.9 | 0.6 | 6.7 ± 1.1 | 0.9 | 13.2 ± 2.1 | 0.5 | 6.3 ± 0.7 |
| Doxorubicin k | - | - | 0.75 ± 0.1 | 0.4 | 0.26 ± 0.1 | 1.1 | 0.31 ± 0.1 | 0.9 | 0.29 ± 0.1 |
| Compound | S. aureus NCTC 4163 | S. aureus ATCC 25923 | S. aureus ATCC 6538 | S. aureus ATCC 29213 | S. epidermidis ATCC 12228 | S. epidermidis ATCC 35984 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 |
|---|---|---|---|---|---|---|---|---|
| 1m | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 0.5 | 16 | >512 |
| 2m | 1 | 1 | 1 | 1 | 2 | 1 | 64 | >512 |
| 3m | 8 | 8 | 8 | 8 | 16 | 8 | 128 | >64 |
| 4m | 128 | 256 | 128 | 512 | >512 | 256 | 512 | 128 |
| 5m | >512 | >512 | >512 | >512 | >512 | >512 | >512 | 128 |
| 6m | 16 | 32 | 16 | 16 | 32 | 32 | 256 | >512 |
| 7m | 256 | 512 | 128 | 512 | 512 | 256 | 256 | >512 |
| 8m | 2 | 2 | 2 | 4 | 4 | 2 | 2 | >512 |
| 9m | >512 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| 10m | 2 | 2 | 2 | 2 | 4 | 2 | 32 | >512 |
| 11m | >512 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| 12m | 64 | 64 | 64 | 64 | 64 | 32 | 128 | >512 |
| 13m | 16 | 16 | 16 | 16 | 16 | 16 | 16 | >512 |
| 14m | 0.25 | 0.25 | 0.25 | 0.5 | 0.5 | 0.5 | 8 | >512 |
| 15m | 4 | 4 | 4 | 4 | 4 | 8 | 128 | 512 |
| 16m | 64 | 64 | 64 | 64 | 64 | 64 | 128 | >512 |
| MXF | 0.06 | <0.03 | 0.06 | 0.06 | 0.06 | 0.06 | <0.03 | >512 |
| CP | 0.25 | 0.5 | 0.5 | 0.5 | 0.25 | 0.125 | 0.031 | 2 |
| Compound | KR 4047 825/19 | KR 4243 829/19 | KR 4313 834/19 | KR 4358/2 840/19 | T 5253 845/19 | T 5399 848/19 | T 5501 851/19 |
|---|---|---|---|---|---|---|---|
| 1 | 256 | 1 | 2 | 16 | 512 | 2 | 8 |
| 2 | 16 | 0.25 | 0.25 | 0.5 | 64 | 0.25 | 0.5 |
| 3 | 64 | 1 | 1 | 1 | 32 | 1 | 1 |
| 4 | 128 | 128 | 128 | 128 | 256 | 128 | 128 |
| 5 | 128 | 128 | 128 | 128 | 256 | 128 | 128 |
| 6 | 32 | 2 | 2 | 2 | 128 | 2 | 1 |
| 7 | 512 | 256 | 256 | 256 | 512 | 256 | 256 |
| 8 | 128 | 4 | 4 | 4 | 256 | 4 | 4 |
| 9 | 128 | 512 | 128 | 512 | 512 | 256 | 128 |
| 1m | 32 | 0.5 | 1 | 1 | 64 | 1 | 1 |
| 2m | 32 | 1 | 1 | 1 | 32 | 0.5 | 2 |
| 3m | 16 | 8 | 16 | 8 | 8 | 8 | 16 |
| 4m | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| 5m | >512 | >512 | 256 | 512 | 512 | 512 | 512 |
| 6m | 256 | 64 | 64 | 64 | 256 | 32 | 128 |
| 7m | 512 | 256 | 256 | 256 | >512 | 512 | 256 |
| 8m | 512 | 4 | 4 | 4 | 512 | 4 | 4 |
| 9m | 512 | 512 | 512 | >512 | 512 | 512 | 512 |
| 10m | 16 | 2 | 2 | 4 | 16 | 2 | 4 |
| 11m | 512 | 256 | 64 | 128 | 512 | 512 | 512 |
| 12m | 256 | 32 | 256 | 512 | 512 | 32 | 32 |
| 13m | 256 | 4 | 8 | 8 | 512 | 4 | 8 |
| 14m | 32 | 0.5 | 0.5 | 0.5 | 32 | 0.5 | 0.5 |
| 15m | 8 | 8 | 8 | 8 | 8 | 4 | 8 |
| 16m | 256 | 64 | 128 | 512 | 512 | 512 | 512 |
| MXF | 2 | 0.1 | 0.1 | 0.1 | 4 | 0.1 | 0.1 |
| CP | 2 | 0.25 | 0.03 | 0.125 | 64 | 0.25 | 0.03 |
| Compound | M. tuberculosis H37Rv | M. tuberculosis Spec. 210 (Multidrug-Resistant) | M. tuberculosis Spec. 192 (Sensitive to Tuberculostatics) |
|---|---|---|---|
| 1 | 256 | 128 | 64 |
| 2 | 4 | 2 | 1 |
| 3 | 64 | 32 | 32 |
| 4 | >512 | >512 | >512 |
| 5 | >512 | >512 | >512 |
| 6 | 16 | 16 | 16 |
| 7 | >512 | >512 | >512 |
| 8 | 16 | 4 | 8 |
| 9 | >512 | >512 | >512 |
| 1m | 64 | 64 | 64 |
| 2m | 32 | 32 | 32 |
| 3m | 128 | 128 | 128 |
| 4m | >512 | >512 | >512 |
| 5m | 512 | 256 | 256 |
| 6m | 256 | 256 | 128 |
| 7m | >512 | >512 | >512 |
| 8m | 8 | 2 | 4 |
| 9m | >512 | >512 | >512 |
| 10m | 32 | 32 | 32 |
| 11m | 512 | 256 | 128 |
| 12m | 64 | 64 | 64 |
| 13m | 4 | 2 | 4 |
| 14m | 64 | 64 | 64 |
| 15m | 256 | 128 | 128 |
| 16m | >512 | >512 | >512 |
| CP | 0.5 | 0.25 | 0.5 |
| MXF | 0.25 | ≤0.0625 | 0.125 |
| Isoniazid (INH) | 0.125 | 16 | 0.125 |
| Rifampicin (RMP) | 1 | 32 | 1 |
| Streptomycin (SM) | 1 | 16 | 1 |
| Ethambutol (EMB) | 2 | 32 | 2 |
| Compound | * IC50 for Gyrase (μg/mL) | * IC50 for Topo IV (μg/mL) |
|---|---|---|
| 1 | >64 | >64 |
| 2 | 34.0 ± 1.0 | 32.0 ± 1.5 |
| 3 | 51.5 ± 2.5 | >64 |
| 8 | >64 | >64 |
| 1m | >64 | 61.0 ± 3.0 |
| 2m | >64 | >64 |
| 3m | >64 | >64 |
| 8m | >64 | >64 |
| 14 m | 59.0 ± 2.5 | 60.0 ± 2.2 |
| CP | 3.0 ± 0.1 | 1.7 ± 0.1 |
| MXF | 2.0 ± 0.5 | 0.9 ± 0.5 |
| Compound | CS | LE (kcal/mol) | BE (kcal/mol) |
|---|---|---|---|
| CP | 740 | −0.310 | −7.45 (±1.23) |
| 1 | 641 | −0.257 | −7.45 (±1.96) |
| 2 | 546 | −0.274 | −8.48 (±2.55) |
| 3 | 152 | −0.245 | −8.56 (±1.62) |
| 8 | 41 | −0.269 | −12.62 (±3.41) |
| Compound | CS | LE (kcal/mol) | BE (kcal/mol) |
|---|---|---|---|
| MXF | 449 | −0.244 | −7.09 (±1.43) |
| 1m | 281 | −0.239 | −8.13 (±2.55) |
| 2m | 316 | −0.224 | −8.05 (±1.76) |
| 3m | 316 | −0.237 | −9.47 (±2.55) |
| 8m | 44 | −0.259 | −13.46 (±3.54) |
| 14m | 339 | −0.227 | −7.27 (±1.35) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrzanowska, A.; Struga, M.; Roszkowski, P.; Koliński, M.; Kmiecik, S.; Jałbrzykowska, K.; Zabost, A.; Stefańska, J.; Augustynowicz-Kopeć, E.; Wrzosek, M.; et al. The Effect of Conjugation of Ciprofloxacin and Moxifloxacin with Fatty Acids on Their Antibacterial and Anticancer Activity. Int. J. Mol. Sci. 2022, 23, 6261. https://doi.org/10.3390/ijms23116261
Chrzanowska A, Struga M, Roszkowski P, Koliński M, Kmiecik S, Jałbrzykowska K, Zabost A, Stefańska J, Augustynowicz-Kopeć E, Wrzosek M, et al. The Effect of Conjugation of Ciprofloxacin and Moxifloxacin with Fatty Acids on Their Antibacterial and Anticancer Activity. International Journal of Molecular Sciences. 2022; 23(11):6261. https://doi.org/10.3390/ijms23116261
Chicago/Turabian StyleChrzanowska, Alicja, Marta Struga, Piotr Roszkowski, Michał Koliński, Sebastian Kmiecik, Karolina Jałbrzykowska, Anna Zabost, Joanna Stefańska, Ewa Augustynowicz-Kopeć, Małgorzata Wrzosek, and et al. 2022. "The Effect of Conjugation of Ciprofloxacin and Moxifloxacin with Fatty Acids on Their Antibacterial and Anticancer Activity" International Journal of Molecular Sciences 23, no. 11: 6261. https://doi.org/10.3390/ijms23116261
APA StyleChrzanowska, A., Struga, M., Roszkowski, P., Koliński, M., Kmiecik, S., Jałbrzykowska, K., Zabost, A., Stefańska, J., Augustynowicz-Kopeć, E., Wrzosek, M., & Bielenica, A. (2022). The Effect of Conjugation of Ciprofloxacin and Moxifloxacin with Fatty Acids on Their Antibacterial and Anticancer Activity. International Journal of Molecular Sciences, 23(11), 6261. https://doi.org/10.3390/ijms23116261