
Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors †


 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Design and Synthesis
2.2. Probability-Guided Pharmacophore Mapping
2.3. Similarity-Driven Property Assesment
2.4. Docking and Molecular Dynamics Simulations
2.5. In Vitro Cell Viability Assay
3. Materials and Methods
3.1. General Methods
3.2. Synthesis
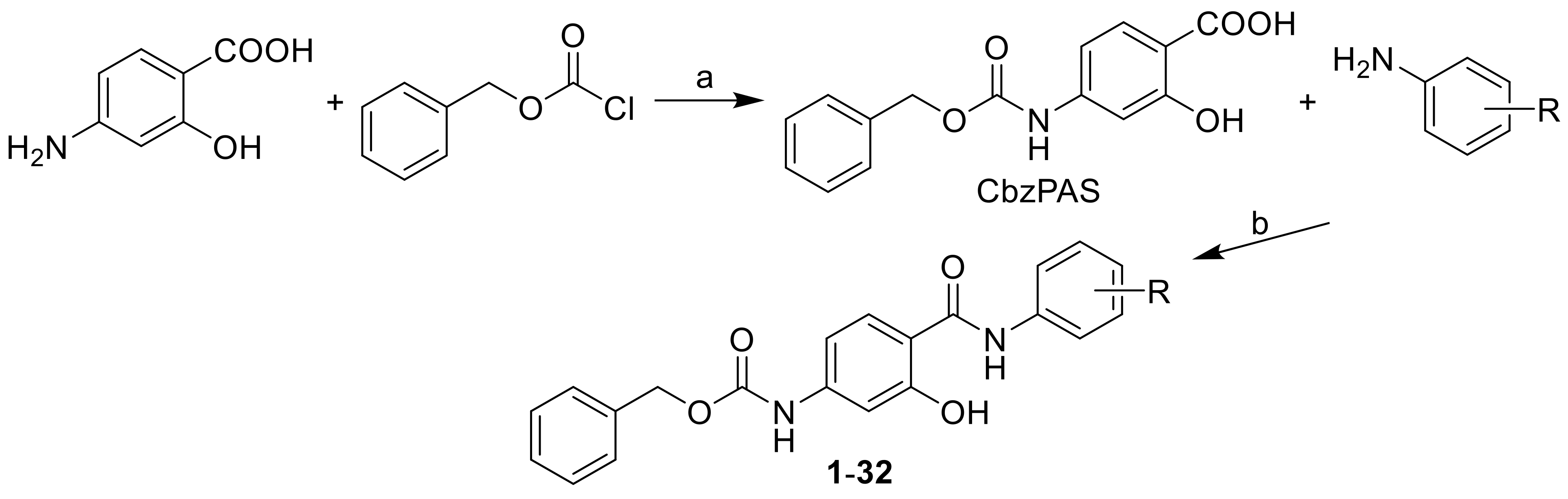
3.2.1. General Procedure Used to Synthesize the Carbamates 1–32
(1) Benzyl[3-hydroxy-4-(phenylcarbamoyl)phenyl]carbamate (1)
(2) Benzyl{3-hydroxy-4-[(2-methoxyphenyl)carbamoyl]phenyl}carbamate (2)
(3) Benzyl{3-hydroxy-4-[(3-methoxyphenyl)carbamoyl]phenyl}carbamate (3)
(4) Benzyl{3-hydroxy-4-[(4-methoxyphenyl)carbamoyl]phenyl}carbamate (4)
(5) Benzyl{3-hydroxy-4-[(3-methylphenyl)carbamoyl]phenyl}carbamate (5)
(6) Benzyl{4-[(2-fluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (6)
(7) Benzyl{4-[(3-fluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (7)
(8) Benzyl{4-[(4-fluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (8)
(9) Benzyl{4-[(2-chlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (9)
(10) Benzyl{4-[(3-chlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (10)
(11) Benzyl{4-[(4-chlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (11)
(12) Benzyl(3-hydroxy-4-{[2-(trifluoromethyl)phenyl]carbamoyl}phenyl)carbamate (12)
(13) Benzyl(3-hydroxy-4-{[3-(trifluoromethyl)phenyl]carbamoyl}phenyl)carbamate (13)
(14) Benzyl(3-hydroxy-4-{[4-(trifluoromethyl)phenyl]carbamoyl}phenyl)carbamate (14)
(15) Benzyl(3-hydroxy-4-{[2-(trifluoromethoxy)phenyl]carbamoyl}phenyl)carbamate (15)
(16) Benzyl{4-[(2,3-difluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (16)
(17) Benzyl{4-[(2,4-difluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (17)
(18) Benzyl{4-[(2,5-difluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (18)
(19) Benzyl{4-[(2,6-difluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (19)
(20) Benzyl{4-[(3,5-difluorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (20)
(21) Benzyl{4-[(2,3-dichlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (21)
(22) Benzyl{4-[(2,5-dichlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (22)
(23) Benzyl{4-[(2,6-dichlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (23)
(24) Benzyl{4-[(3,4-dichlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (24)
(25) Benzyl{4-[(3,5-dichlorophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (25)
(26) Benzyl{4-[(2,4-dibromophenyl)carbamoyl]-3-hydroxyphenyl}carbamate (26)
(27) Benzyl{4-[(3,5-di(trifluoromethyl))carbamoyl]-3-hydroxyphenyl}carbamate (27)
(28) Benzyl{3-hydroxy-4-[(2,4,6-trifluorophenyl)carbamoyl]phenyl}carbamate (28)
(29) Benzyl{3-hydroxy-4-[(3,4,5-trifluorophenyl)carbamoyl]phenyl}carbamate (29)
(30) Benzyl{4-[(2,4,5-trichlorophenyl)carbamoyl]phenyl}carbamate (30)
(31) Benzyl{3-hydroxy-4-[(2,4,6-trichlorophenyl)carbamoyl]phenyl}carbamate (31)
(32) Benzyl{3-hydroxy-4-[(2,4,6-tribromophenyl)carbamoyl]phenyl}carbamate (32)
3.3. Determination of Lipophilicity by HPLC
3.4. Evaluating In Vitro AChE and BChE-Inhibition Potencies
3.5. In Vitro Viability Assay
3.6. Model Building and Molecular Modeling
3.7. Principal Component and Hierarchical Custering Analysis
3.8. Theoretical Lipophilicity Evaluation
3.9. Similarity-Based Activity Landscape Index
3.10. Ligand/Structure-Based Analysis
4. Conclusions
- A library of novel 4-{[(benzyloxy)carbonyl]amino}-2-hydroxybenzoic acid amides was designed and synthesized as potential acetyl- and butyrylcholinesterase inhibitors; hence, the in vitro inhibitory profile and selectivity index were determined. Benzyl(3-hydroxy-4-{[2-(trifluoromethoxy)phenyl]carbamoyl}phenyl)carbamate (15) showed the best AChE inhibition (IC50 = 36.05 µM) and benzyl{3-hydroxy-4-[(2-methoxyphenyl)carbamoyl]phenyl}carbamate (2) demonstrated the best BChE inhibiting activity (IC50 = 22.23 µM) with the highest selectivity for BChE (SI = 2.26).
- The distance-oriented property evaluation was conducted using the Principal Component Analysis (PCA) and Hierarchical Clustering Analysis (HCA), respectively. The dissimilarities of molecules 12–14, 27 with trifluoromethyl substituent(s) (-CF3) and compound 15 with trifluromethoxy group (-OCF3) from the remaining ones was observed. Noticeably, objects 2–4 that are methoxy-based (-OCH3) positional isomers and molecule 5 with methyl substituent (-CH3) were also clustered together.
- The distribution of the Tanimoto coefficients was analyzed for the triangular T32×32 matrix. Interestingly, the potent molecules 2 and 15 are marked as fairly similar (T = 0.93). In fact, molecule 15 is characterized by low values of SALI indexes with the remaining compounds in the dataset, while the most potent molecule 2 is accompanied by gray pots of the inactive molecules 3, 4 (positional isomers), and 1 (unsubstituted analogue), respectively. Noticeably, three pairs of fairy similar fluorine/bromine-containing compounds (17 vs. 19 and 28, 26 vs. 32) can potentially form the activity cliff that is manifested formally by high SALI numerical values.
- The preferential selection of the active molecules (2 and 6) and inactive ones (3, 13, 23, and 26) that are basically ortho- and meta-substituted analogues resulted in the generation of the robust models with the acceptable predictive power for the test set.
- The molecular docking approach was engaged for the most potent AChE/BChE inhibitors in order to get comprehensive knowledge of the binding mode. The hydrophobic interactions were overwhelmingly generated with Gln119, Asp70, Pro285, Thr120, and Trp82 aminoacid residues, while the HB-donor ones were dominated with Thr120. π-stacking interactions were specified with Trp82 aminoacid residue of chain A as well. 6. The stability of some fused ligand–enzyme systems were evaluated using the molecular dynamic simulations. The trifluoromethoxy substituent of molecule 15 is characterized by lower spatial flexibility compared to methoxy group of compound 2. Obviously, –OCF3 is considerably heavier and can generate greater steric hindrance in comparison to –OCH3, as the most electronegative element fluorine can interact with the surrounding aminoacid residues, which stiffens the ligand–enzyme system.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bak, A.; Kozik, V.; Kozakiewicz, D.; Gajcy, K.; Strub, D.J.; Swietlicka, A.; Stepankova, S.; Imramovsky, A.; Polanski, J.; Smolinski, A.; et al. Novel benzene-based carbamates for AChE/BChE inhibition: Synthesis and ligand/structure-oriented SAR study. Int. J. Mol. Sci. 2019, 20, 1524. [Google Scholar] [CrossRef]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bak, A.; Kauerova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-based carbamates as cholinesterase inhibitors. Molecules 2017, 22, 1969. [Google Scholar] [CrossRef]
- Moss, D.E.; Perez, R.G.; Kobayashi, H. Cholinesterase inhibitor therapy in Alzheimer’s disease: The limits and tolerability of irreversible CNS-selective acetylcholinesterase inhibition in primates. J. Alzheimers Dis. 2017, 55, 1285–1294. [Google Scholar] [CrossRef]
- Bajic, V.; Milovanovic, E.S.; Spremo-Potparevic, B.; Zivkovic, L.; Miliccivc, Z.; Stanimirovic, J.; Bogdanovic, N.; Isenovic, E.R. Treatment of Alzheimer’s disease: Classical therapeutic approach. Curr. Pharm. Anal. 2016, 12, 82–90. [Google Scholar] [CrossRef]
- Hussein, W.; Saglik, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Ozkay, Y.; Kaplancikli, Z.A. Synthesis and biological evaluation of new cholinesterase inhibitors for Alzheimer’s disease. Molecules 2018, 23, 2033. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, Q.; Gu, K.; Zhu, J.; Jiang, X.; Chen, Y.; Sun, H. Therapeutic agents in Alzheimer’s disease through a multi-target directed ligands strategy: Recent progress based on tacrine core. Curr. Top. Med. Chem. 2017, 17, 3000–3016. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Dai, Y.C.; Li, N.G.; Dong, Z.X.; Gu, T.; Shi, Z.H.; Xue, X.; Tang, Y.P.; Duan, J.A. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2017, 32, 572–587. [Google Scholar] [CrossRef]
- Jampilek, J.; Kralova, K.; Novak, P.; Novak, M. Nanobiotechnology in neurodegenerative diseases. In Nanobiotechnology in Neurodegenerative Diseases; Rai, M., Yadav, A., Eds.; Springer: Cham, Switzerland, 2019; pp. 65–138. [Google Scholar]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging. 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Pejchal, V.; Stepankova, S.; Padelkova, Z.; Imramovsky, A.; Jampilek, J. 1,3-Substituted imidazolidine-2,4,5-triones: Synthesis and inhibition of cholinergic enzymes. Molecules 2011, 16, 7565–7582. [Google Scholar] [CrossRef]
- Imramovsky, A.; Stepankova, S.; Vanco, J.; Pauk, K.; Monreal-Ferriz, J.; Vinsova, J.; Jampilek, J. Acetylcholinesterase-inhibiting activity of salicylanilide N-alkylcarbamates and their molecular docking. Molecules 2012, 17, 10142–10158. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pejchal, V.; Stepankova, S.; Vorcakova, K.; Jampilek, J.; Vanco, J.; Simunek, P.; Kralovec, K.; Bruckova, L.; Mandikova, J.; et al. Synthesis and in vitro evaluation of new derivatives of 2-substituted-6-fluorobenzo[d]thiazoles as cholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 1735–1748. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]benzamides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef] [PubMed]
- Lemke, T.L.; Williams, D.A. Foye’s Principles of Medicinal Chemistry, 7th ed.; Lippincott Williams & Wilkins and Wolters Kluwer: Baltimore, MD, USA, 2013. [Google Scholar]
- Bak, A.; Pizova, H.; Kozik, V.; Vorcakova, K.; Kos, J.; Treml, J.; Odehnalova, K.; Oravec, M.; Imramovsky, A.; Bobal, P.; et al. SAR-mediated similarity assessment of the property profile for new, silicon-based AChE/BChE inhibitors. Int. J. Mol. Sci. 2019, 20, 5385. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Devillers, J. Methods for building QSARs. Methods Mol. Biol. 2013, 930, 3–27. [Google Scholar]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.; Oprea, T. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Walczak, M.; Fraczyk, J.; Kaminski, Z.; Kolesinska, B.; Smolinski, A.; Jampilek, J. Towards intelligent drug design system: Application of artificial dipeptide receptor library in QSAR-oriented studies. Molecules 2018, 23, 1964. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Peltason, L.; Bajorath, J. Systematic computational analysis of structure-activity relationships: Concepts, challenges and recent advances. Future Med Chem. 2009, 1, 451–466. [Google Scholar] [CrossRef]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Structure—activity landscape index: Identifying and quantifying activity cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Peltason, L.; Wawer, M.; Guha, R.; Lajiness, M.S.; Van Drie, J.H. Navigating structure–activity landscapes. Drug Discov. Today 2009, 14, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Irwin, J.J. Docking screens: Right for the right reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef]
- Colquhoun, D. The quantitative analysis of drug–receptor interactions: A short history. Trends Pharmacol. Sci. 2006, 27, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of docking. Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Assessing how well a modeling protocol captures a structure—Activity landscape. J. Chem. Inf. Model. 2008, 48, 1716–1728. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Environ. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef]
- Smolinski, A.; Drobek, L.; Dombek, V.; Bak, A. Modeling of experimental data on trace elements and organic content in industrial waste dumps. Chemosphere 2016, 162, 189–198. [Google Scholar] [CrossRef]
- Smolinski, A.; Howaniec, N.; Bak, A. Utilization of energy crops and sewage sludge in the proces of co-gasificiation for sustainable hydrogen production. Energies 2018, 11, 809. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef]
- Rozas, I.; Du, Q.; Arteca, G.A. Interrelation between electrostatic and lipophilicity potentials on molecular surfaces. J. Mol. Graph. 1995, 13, 98–108. [Google Scholar] [CrossRef]
- Chrobak, E.; Marciniec, K.; Dąbrowska, A.; Pęcak, P.; Bębenek, E.; Kadela-Tomanek, M.; Bak, A.; Jastrzębska, M.; Boryczka, S. New phosphorus analogs of bevirimat: Synthesis, evaluation of anti-HIV-1 activity and molecular docking study. Int. J. Mol. Sci. 2019, 20, 5209. [Google Scholar] [CrossRef] [PubMed]
- Voltz, K.; Trylska, J.; Tozzini, V.; Kurkal-Siebert, V.; Langovski, J.; Smith, J. Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comp. Chem. 2008, 29, 1429–1439. [Google Scholar] [CrossRef]
- Tozzini, V.; Trylska, J.; Chang, C.E.; McCammon, J.A. Flap opening dynamics in HIV-1 protease explored with a coarse-grained model. J. Struct. Biol. 2007, 157, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Leonarski, F.; Trylska, J. RedMDStream: Parameterization and simulation toolbox for coarse-grained molecular dynamics models. Biophys. J. 2015, 108, 1843–1847. [Google Scholar] [CrossRef]
- Pospisilova, S.; Malik, I.; Curillova, J.; Michnova, H.; Cerna, L.; Padrtova, T.; Hosek, J.; Pecher, D.; Cizek, A.; Jampilek, J. Insight into antimicrobial activity of substituted phenylcarbamoyloxypiperazinylpropanols. Bioorg. Chem. 2020, 102, 104060. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Gupte, A.; Boshoff, H.I.; Wilson, D.J.; Bennett, E.M.; Somu, R.V.; Barry, C.E., III; Aldrich, C.C. 5′-O-[(N-Acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: An adenylation enzyme required for siderophore biosynthesis of the Mycobactins. J. Med. Chem. 2007, 50, 6080–6094. [Google Scholar] [CrossRef] [PubMed]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Nat. C 2004, 59, 293–296. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ou, S.; Kwok, K.C.; Wang, Y.; Bao, H. An improved method to determine SH and –S–S– group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | R | log k | IC50 (μM) | SI 1 | |
|---|---|---|---|---|---|
| AChE | BChE | ||||
| 1 | H | 0.1160 | 65.17 ± 2.24 | 175.18 ± 5.85 | 0.37 |
| 2 | 2-OCH3 | 0.3368 | 50.14 ± 1.57 | 22.23 ± 0.53 | 2.26 |
| 3 | 3-OCH3 | 0.1280 | 69.26 ± 2.00 | 361.80 ± 8.12 | 0.19 |
| 4 | 4-OCH3 | 0.1176 | 57.49 ± 0.08 | 153.98 ± 6.09 | 0.37 |
| 5 | 3-CH3 | 0.2725 | 70.23 ± 1.70 | 167.46 ± 10.21 | 0.42 |
| 6 | 2-F | 0.3577 | 50.07 ± 0.06 | 70.84 ± 5.61 | 0.71 |
| 7 | 3-F | 0.2303 | 66.60 ± 0.14 | 197.62 ± 1.36 | 0.34 |
| 8 | 4-F | 0.3924 | 47.37 ± 0.61 | 105.82 ± 0.43 | 0.45 |
| 9 | 2-Cl | 0.5188 | 41.86 ± 0.15 | 140.06 ± 0.35 | 0.30 |
| 10 | 3-Cl | 0.6411 | 61.62 ± 1.74 | 203.54 ± 7.49 | 0.30 |
| 11 | 4-Cl | 0.6399 | 78.20 ± 1.58 | 138.67 ± 6.86 | 0.56 |
| 12 | 2-CF3 | 0.4318 | 56.42 ± 1.26 | 33.59 ± 0.71 | 1.68 |
| 13 | 3-CF3 | 0.4477 | 70.67 ± 0.57 | 309.25 ± 8.06 | 0.23 |
| 14 | 4-CF3 | 0.7408 | 71.42 ± 5.04 | 136.95 ± 21.94 | 0.52 |
| 15 | 2-OCF3 | 0.6227 | 36.05 ± 1.48 | 44.31 ± 0.87 | 0.81 |
| 16 | 2,3-F | 0.4705 | 62.42 ± 1.42 | 199.81 ± 7.00 | 0.31 |
| 17 | 2,4-F | 0.1912 | 79.23 ± 1.27 | 180.31 ± 7.04 | 0.44 |
| 18 | 2,5-F | 0.4012 | 58.31 ± 0.20 | 157.34 ± 3.11 | 0.37 |
| 19 | 2,6-F | -0.1205 | 64.31 ± 4.74 | 42.95 ± 2.13 | 1.50 |
| 20 | 3,5-F | 0.4072 | 67.36 ± 5.24 | 208.34 ± 5.30 | 0.32 |
| 21 | 2,3-Cl | 0.5497 | 88.78 ± 1.42 | 247.56 ± 14.95 | 0.36 |
| 22 | 2,5-Cl | 0.6138 | 99.14 ± 0.20 | 141.20 ± 7.12 | 0.70 |
| 23 | 2,6-Cl | 0.8125 | 76.74 ± 0.74 | 117.57 ± 1.11 | 0.65 |
| 24 | 3,4-Cl | 0.6664 | 57.99 ± 5.83 | 185.06 ± 6.13 | 0.31 |
| 25 | 3,5-Cl | 0.9084 | 50.62 ± 0.38 | 143.08 ± 3.69 | 0.35 |
| 26 | 2,4-Br | 0.7366 | 94.89 ± 0.97 | 261.65 ± 5.10 | 0.36 |
| 27 | 3,5-CF3 | 0.9667 | 59.35 ± 3.38 | 145.68 ± 2.65 | 0.41 |
| 28 | 2,4,6-F | -0.0131 | 49.87 ± 0.09 | 31.03 ± 2.11 | 1.61 |
| 29 | 3,4,5-F | 0.5169 | 62.27 ± 0.75 | 183.71 ± 0.75 | 0.34 |
| 30 | 2,4,5-Cl | 0.9360 | 94.88 ± 1.78 | 174.98 ± 10.47 | 0.54 |
| 31 | 2,4,6-Cl | 0.3679 | 93.09 ± 1.80 | 57.11 ± 1.72 | 1.63 |
| 32 | 2,4,6-Br | 0.4427 | 65.92 ± 2.81 | 56.01 ± 0.58 | 1.18 |
| CbzPAS | – | – | 58.08 ± 1.04 | 136.85 ± 21.80 | 0.42 |
| RIV | – | – | 50.10 ± 3.08 | 19.95 ± 0.31 | 2.51 |
| GLT | – | – | 4.0 ± 0.13 | 7.96 ± 0.59 | 0.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kos, J.; Kozik, V.; Pindjakova, D.; Jankech, T.; Smolinski, A.; Stepankova, S.; Hosek, J.; Oravec, M.; Jampilek, J.; Bak, A. Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors. Int. J. Mol. Sci. 2021, 22, 3444. https://doi.org/10.3390/ijms22073444
Kos J, Kozik V, Pindjakova D, Jankech T, Smolinski A, Stepankova S, Hosek J, Oravec M, Jampilek J, Bak A. Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors. International Journal of Molecular Sciences. 2021; 22(7):3444. https://doi.org/10.3390/ijms22073444
Chicago/Turabian StyleKos, Jiri, Violetta Kozik, Dominika Pindjakova, Timotej Jankech, Adam Smolinski, Sarka Stepankova, Jan Hosek, Michal Oravec, Josef Jampilek, and Andrzej Bak. 2021. "Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors" International Journal of Molecular Sciences 22, no. 7: 3444. https://doi.org/10.3390/ijms22073444
APA StyleKos, J., Kozik, V., Pindjakova, D., Jankech, T., Smolinski, A., Stepankova, S., Hosek, J., Oravec, M., Jampilek, J., & Bak, A. (2021). Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors. International Journal of Molecular Sciences, 22(7), 3444. https://doi.org/10.3390/ijms22073444