
Clinical and Molecular Diagnosis of Beckwith-Wiedemann Syndrome with Single- or Multi-Locus Imprinting Disturbance

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Genetic and Epigenetic Alterations in BWS
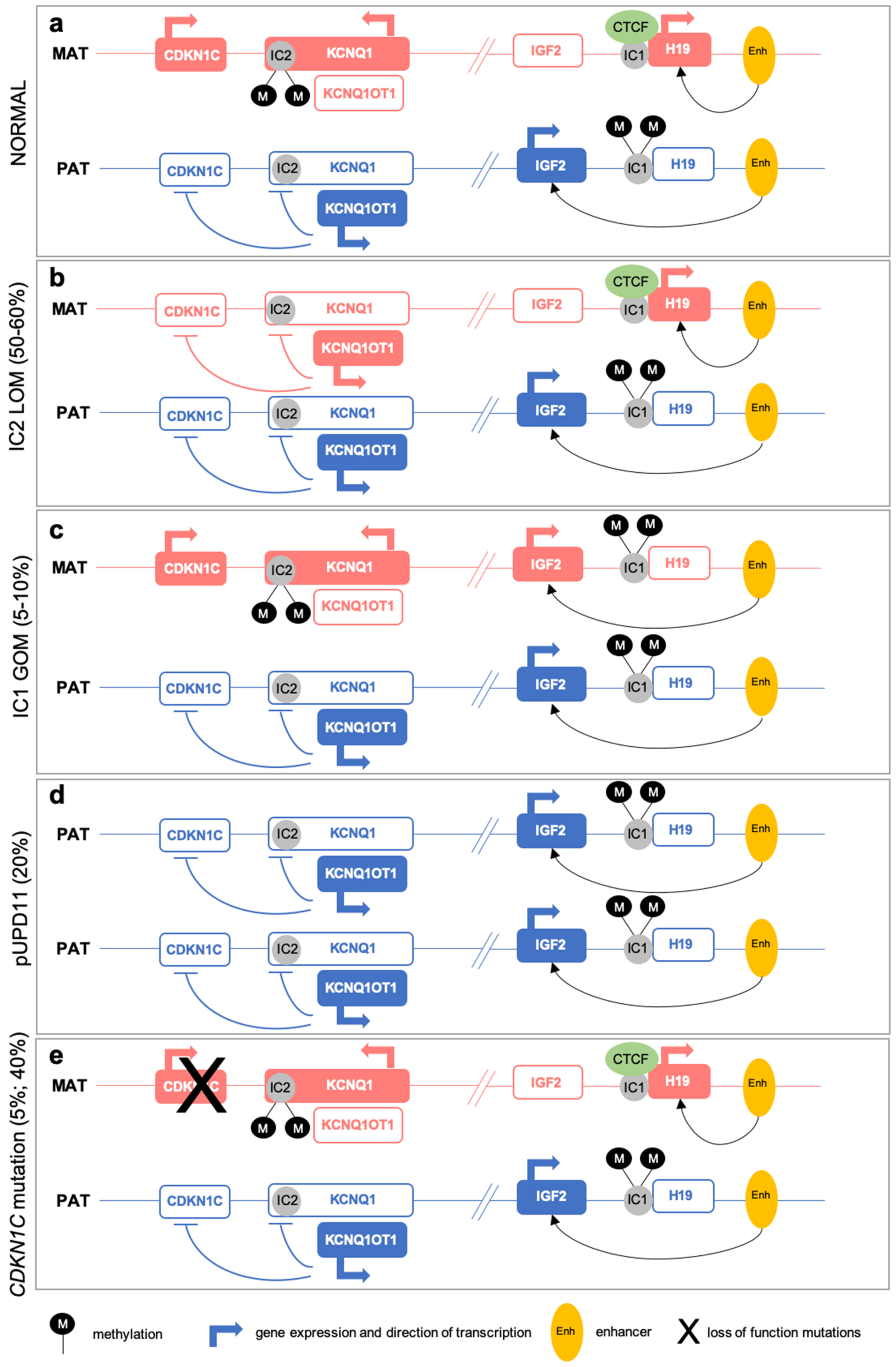
2.1. (Epi)Genetic Defects at 11p15
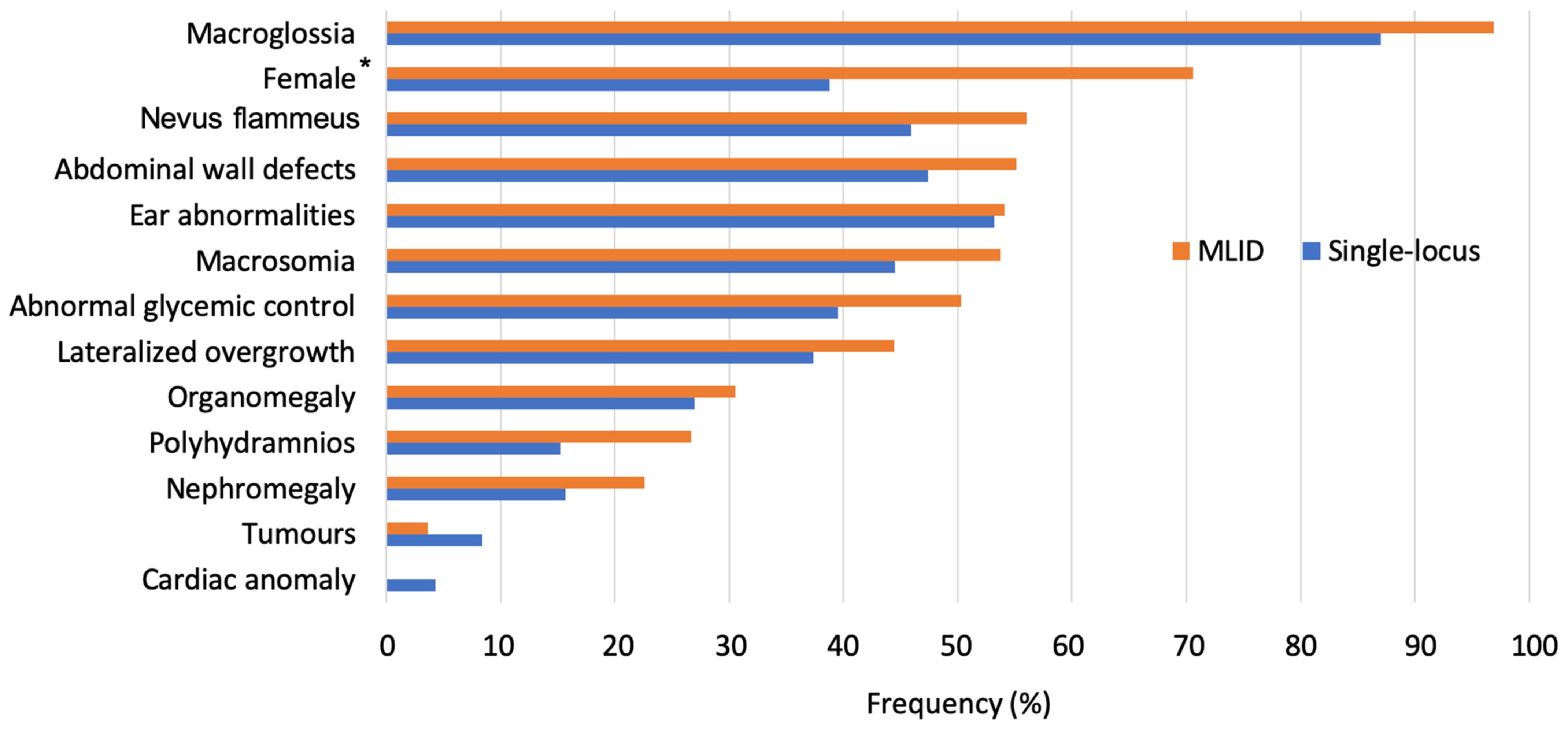
2.2. Multi-Locus Imprinting Disturbance (MLID)
3. Clinical Features and Diagnosis
3.1. Prenatal Clinical Diagnosis
3.2. Postnatal Clinical Presentation
3.3. Differential Diagnosis
3.4. Clinical Management and Follow-up
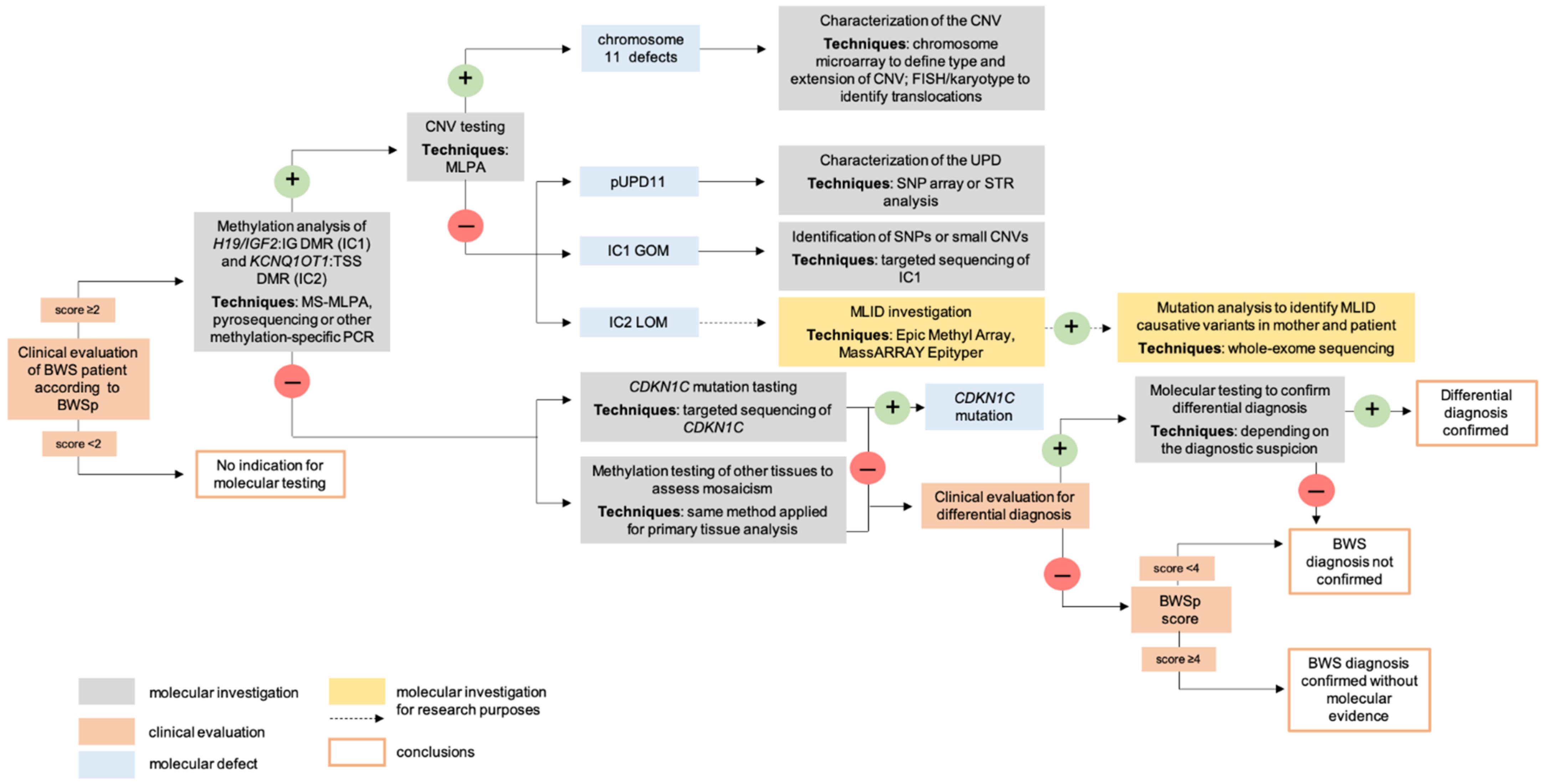
4. Molecular Diagnosis
4.1. Postnatal Testing
4.2. Prenatal Testing
5. (Epi)Genotype-Phenotype Correlations
5.1. Prenatal and Neonatal Correlations
5.2. Postnatal Correlations in Single- and Multi-Locus Patients
6. Deciphering the Timing of Epigenetic Errors Establishment
6.1. Discordant Monozygotic Twins and X-Chromosome Inactivation
6.2. Assisted Reproductive Technologies (ART)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mussa, A.; Russo, S.; De Crescenzo, A.; Chiesa, N.; Molinatto, C.; Selicorni, A.; Richiardi, L.; Larizza, L.; Silengo, M.C.; Riccio, A.; et al. Prevalence of Beckwith-Wiedemann Syndrome in North West of Italy. Am. J. Med. Genet. Part A 2013, 161, 2481–2486. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski, J.; Shuman, C.; Choufani, S.; Ray, P.; Stavropoulos, D.J.; Basran, R.; Steele, L.; Parkinson, N.; Grant, R.; Thorner, P.; et al. Wilms Tumour in Beckwith—Wiedemann Syndrome and Loss of Methylation at Imprinting Centre 2: Revisiting Tumour Surveillance Guidelines. Eur. J. Hum. Genet. 2017, 25, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Shuman, C.; Beckwith, J.B. Beckwith—Wiedemann Syndrome. Eur. J. Hum. Genet. 2009, 18, 8–14. [Google Scholar] [CrossRef]
- Azzi, S.; Steunou, V.; Tost, J.; Rossignol, S.; Thibaud, N.; Das Neves, C.; Le Jule, M.; Habib, W.A.; Blaise, A.; Koudou, Y.; et al. Exhaustive Methylation Analysis Revealed Uneven Profiles of Methylation at IGF2/ICR1/H19 11p15 Loci in Russell Silver Syndrome. J. Med. Genet. 2014, 52, 53–60. [Google Scholar] [CrossRef]
- Azzi, S.; Rossignol, S.; Steunou, V.; Sas, T.; Thibaud, N.; Danton, F.; Le Jule, M.; Heinrichs, C.; Cabrol, S.; Gicquel, C.; et al. Multilocus Methylation Analysis in a Large Cohort of 11p15-Related Foetal Growth Disorders (Russell Silver and Beckwith Wiedemann Syndromes) Reveals Simultaneous Loss of Methylation at Paternal and Maternal Imprinted Loci. Hum. Mol. Genet. 2009, 18, 4724–4733. [Google Scholar] [CrossRef] [PubMed]
- Bens, S.; Kolarova, J.; Beygo, J.; Buiting, K.; Caliebe, A.; Eggermann, T.; Gillessen-Kaesbach, G.; Prawitt, D.; Thiele-Schmitz, S.; Begemann, M.; et al. Phenotypic Spectrum and Extent of DNA Methylation Defects Associated with Multilocus Imprinting Disturbances. Epigenomics 2016, 8, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.; I Rezwan, F.; Beygo, J.; E Docherty, L.; Kolarova, J.; Schroeder, C.; Buiting, K.; Chokkalingam, K.; Degenhardt, F.; Wakeling, E.L.; et al. Maternal Variants in NLRP and Other Maternal Effect Proteins Are Associated with Multilocus Imprinting Disturbance in Offspring. J. Med. Genet. 2018, 55, 497–504. [Google Scholar] [CrossRef]
- Fontana, L.; Bedeschi, M.F.; Maitz, S.; Cereda, A.; Faré, C.; Motta, S.; Seresini, A.; D’Ursi, P.; Orro, A.; Pecile, V.; et al. Characterization of Multi-Locus Imprinting Disturbances and Underlying Genetic Defects in Patients with Chromosome 11p15.5 Related Imprinting Disorders. Epigenetics 2018, 13, 897–909. [Google Scholar] [CrossRef]
- Olney, A.H.; Buehler, B.A.; Waziri, M.; Optiz, J.M.; Reynolds, J.F. Wiedemann-Beckwith Syndrome in Apparently Discordant Monozygotic Twins. Am. J. Med. Genet. 1988, 29, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Shuman, C.; Caluseriu, O.; Smith, A.C.; Fei, Y.-L.; Nishikawa, J.; Stockley, T.L.; Best, L.; Chitayat, D.; Olney, A.; et al. Discordant KCNQ1OT1 Imprinting in Sets of Monozygotic Twins Discordant for Beckwith-Wiedemann Syndrome. Hum. Mol. Genet. 2002, 11, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Bedeschi, M.F.; Cagnoli, G.A.; Costanza, J.; Persico, N.; Gangi, S.; Porro, M.; Ajmone, P.F.; Colapietro, P.; Santaniello, C.; et al. (Epi)Genetic Profiling of Extraembryonic and Postnatal Tissues from Female Monozygotic Twins Discordant for Beckwith—Wiedemann Syndrome. Mol. Genet. Genom. Med. 2020, 8, e1386. [Google Scholar] [CrossRef] [PubMed]
- Higashimoto, K.; Jozaki, K.; Kosho, T.; Matsubara, K.; Fuke, T.; Yamada, D.; Yatsuki, H.; Maeda, T.; Ohtsuka, Y.; Nishioka, K.; et al. A Novelde Novopoint Mutation of the OCT-Binding Site in the IGF2/H19-Imprinting Control Region in a Beckwith-Wiedemann Syndrome Patient. Clin. Genet. 2013, 86, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Korostowski, L.; Sedlak, N.; Engel, N. The Kcnq1ot1 Long Non-Coding RNA Affects Chromatin Conformation and Expression of Kcnq1, but Does Not Regulate Its Imprinting in the Developing Heart. PLoS Genet. 2012, 8, e1002956. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, N.; De Crescenzo, A.; Mishra, K.; Perone, L.; Carella, M.; Palumbo, O.; Mussa, A.; Sparago, A.; Cerrato, F.; Russo, S.; et al. The KCNQ1OT1 Imprinting Control Region and Non-Coding RNA: New Properties Derived from the Study of Beckwith—Wiedemann Syndrome and Silver—Russell Syndrome Cases. Hum. Mol. Genet. 2011, 21, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Jacob, K.J.; Robinson, W.P.; Lefebvre, L. Beckwith-Wiedemann and Silver-Russell Syndromes: Opposite Developmental Imbalances in Imprinted Regulators of Placental Function and Embryonic Growth. Clin. Genet. 2013, 84, 326–334. [Google Scholar] [CrossRef]
- Cerrato, F.; Vernucci, M.; Pedone, P.V.; Chiariotti, L.; Sebastio, G.; Bruni, C.B.; Riccio, A. The 5’ End of the KCNQ1OT1 Gene Is Hypomethylated in the Beckwith-Wiedemann Syndrome. Hum. Genet. 2002, 111, 105–107. [Google Scholar] [CrossRef]
- Mancini-DiNardo, D.; Steele, S.J.; Ingram, R.S.; Tilghman, S.M. A Differentially Methylated Region within the Gene Kcnq1 Functions as an Imprinted Promoter and Silencer. Hum. Mol. Genet. 2003, 12, 283–294. [Google Scholar] [CrossRef]
- Mussa, A.; Russo, S.; De Crescenzo, A.; Freschi, A.; Calzari, L.; Maitz, S.; Macchiaiolo, M.; Molinatto, C.; Baldassarre, G.; Mariani, M.; et al. (Epi)Genotype—Phenotype Correlations in Beckwith—Wiedemann Syndrome. Eur. J. Hum. Genet. 2016, 24, 183–190. [Google Scholar] [CrossRef]
- Eggermann, T.; Algar, E.; Lapunzina, P.; Mackay, D.; Maher, E.R.; Mannens, M.; Netchine, I.; Prawitt, D.; Riccio, A.; Temple, I.K.; et al. Clinical Utility Gene Card for: Beckwith—Wiedemann Syndrome. Eur. J. Hum. Genet. 2013, 22, 435. [Google Scholar] [CrossRef]
- Baskin, B.; Choufani, S.; Chen, Y.-A.; Shuman, C.; Parkinson, N.; Lemyre, E.; Innes, A.M.; Stavropoulos, D.J.; Ray, P.N.; Weksberg, R. High Frequency of Copy Number Variations (CNVs) in the Chromosome 11p15 Region in Patients with Beckwith—Wiedemann Syndrome. Hum. Genet. 2014, 133, 321–330. [Google Scholar] [CrossRef]
- Choufani, S.; Shuman, C.; Weksberg, R. Beckwith-Wiedemann Syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154C, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Mackay, D.J.G.; Eggermann, T.; Buiting, K.; Garin, I.; Netchine, I.; Linglart, A.; de Nanclares, G.P. Multilocus Methylation Defects in Imprinting Disorders. Biomol. Concepts 2015, 6, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.L.; Docherty, L.E.; Al Sayegh, A.; Caliebe, A.; Turner, C.; Baple, E.; Wakeling, E.; Harrison, L.; Lehmann, A.; Temple, I.K.; et al. Targeted Methylation Testing of a Patient Cohort Broadens the Epigenetic and Clinical Description of Imprinting Disorders. Am. J. Med. Genet. Part A 2013, 161, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Bliek, J.; Verde, G.; Callaway, J.; Maas, S.M.; De Crescenzo, A.; Sparago, A.; Cerrato, F.; Russo, S.; Ferraiuolo, S.; Rinaldi, M.M.; et al. Hypomethylation at Multiple Maternally Methylated Imprinted Regions Including PLAGL1 and GNAS Loci in Beckwith—Wiedemann Syndrome. Eur. J. Hum. Genet. 2008, 17, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Court, F.; Martin-Trujillo, A.; Romanelli, V.; Garin, I.; Iglesias-Platas, I.; Salafsky, I.; Guitart, M.; de Nanclares, G.P.; Lapunzina, P.; Monk, D. Genome-Wide Allelic Methylation Analysis Reveals Disease-Specific Susceptibility to Multiple Methylation Defects in Imprinting Syndromes. Hum. Mutat. 2013, 34, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Arima, T.; Kamikihara, T.; Hayashida, T.; Kato, K.; Inoue, T.; Shirayoshi, Y.; Oshimura, M.; Soejima, H.; Mukai, T.; Wake, N. ZAC, LIT1 (KCNQ1OT1) and P57KIP2 (CDKN1C) Are in an Imprinted Gene Network That May Play a Role in Beckwith-Wiedemann Syndrome. Nucleic Acids Res. 2005, 33, 2650–2660. [Google Scholar] [CrossRef]
- E Docherty, L.; I Rezwan, F.; Poole, R.L.; Jagoe, H.; Lake, H.; A Lockett, G.; Arshad, H.; I Wilson, D.; Holloway, J.W.; Temple, I.K.; et al. Genome-Wide DNA Methylation Analysis of Patients with Imprinting Disorders Identifies Differentially Methylated Regions Associated with Novel Candidate Imprinted Genes. J. Med. Genet. 2014, 51, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, S.; Steunou, V.; Chalas, C.; Kerjean, A.; Rigolet, M.; Viegas-Pequignot, E.; Jouannet, P.; Le Bouc, Y.; Gicquel, C. The Epigenetic Imprinting Defect of Patients with Beckwith—Wiedemann Syndrome Born after Assisted Reproductive Technology Is Not Restricted to the 11p15 Region. J. Med. Genet. 2006, 43, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Bowdin, S.C.; Tee, L.; Kirby, G.A.; Blair, E.; Fryer, A.; Lam, W.; Oley, C.; Cole, T.; Brueton, L.A.; et al. Clinical and Molecular Genetic Features of Beckwith—Wiedemann Syndrome Associated with Assisted Reproductive Technologies. Hum. Reprod. 2008, 24, 741–747. [Google Scholar] [CrossRef]
- Hiura, H.; Okae, H.; Miyauchi, N.; Sato, F.; Sato, A.; Van De Pette, M.; John, R.M.; Kagami, M.; Nakai, K.; Soejima, H.; et al. Characterization of DNA Methylation Errors in Patients with Imprinting Disorders Conceived by Assisted Reproduction Technologies. Hum. Reprod. 2012, 27, 2541–2548. [Google Scholar] [CrossRef]
- Boonen, S.E.; Pörksen, S.; Mackay, D.J.G.; Oestergaard, E.; Olsen, B.; Bröndum-Nielsen, K.; Temple, I.K.; Hahnemann, J.M.D. Clinical Characterisation of the Multiple Maternal Hypomethylation Syndrome in Siblings. Eur. J. Hum. Genet. 2008, 16, 453–461. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eggermann, T.; Brück, J.; Knopp, C.; Fekete, G.; Kratz, C.; Tasic, V.; Kurth, I.; Elbracht, M.; Eggermann, K.; Begemann, M. Need for a Precise Molecular Diagnosis in Beckwith-Wiedemann and Silver-Russell Syndrome: What Has to Be Considered and Why It Is Important. J. Mol. Med. 2020, 98, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Higashimoto, K.; Jozaki, K.; Yatsuki, H.; Nakabayashi, K.; Makita, Y.; Tonoki, H.; Okamoto, N.; Takada, F.; Ohashi, H.; et al. Comprehensive and Quantitative Multilocus Methylation Analysis Reveals the Susceptibility of Specific Imprinted Differentially Methylated Regions to Aberrant Methylation in Beckwith—Wiedemann Syndrome with Epimutations. Genet. Med. 2014, 16, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Krzyzewska, I.M.; Alders, M.; Maas, S.M.; Bliek, J.; Venema, A.; Henneman, P.; Rezwan, F.I.; Lip, K.V.D.; Mul, A.N.; Mackay, D.J.G.; et al. Genome-Wide Methylation Profiling of Beckwith-Wiedemann Syndrome Patients without Molecular Confirmation after Routine Diagnostics. Clin. Epigenetics 2019, 11, 53. [Google Scholar] [CrossRef]
- Meyer, E.; Lim, D.; Pasha, S.; Tee, L.J.; Rahman, F.; Yates, J.R.W.; Woods, C.G.; Reik, W.; Maher, E.R. Germline Mutation in NLRP2 (NALP2) in a Familial Imprinting Disorder (Beckwith-Wiedemann Syndrome). PLoS Genet. 2009, 5, e1000423. [Google Scholar] [CrossRef]
- Docherty, L.E.; Rezwan, F.I.; Poole, R.L.; Turner, C.L.S.; Kivuva, E.; Maher, E.R.; Smithson, S.F.; Hamilton-Shield, J.P.; Patalan, M.; Gizewska, M.; et al. Mutations in NLRP5 Are Associated with Reproductive Wastage and Multilocus Imprinting Disorders in Humans. Nat. Commun. 2015, 6, 8086. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Albertini, D.F. The Road to Maturation: Somatic Cell Interaction and Self-Organization of the Mammalian Oocyte. Nat. Rev. Mol. Cell Biol. 2013, 14, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Monk, D.; Sanchez-Delgado, M.; Fisher, R. NLRPs, the Subcortical Maternal Complex and Genomic Imprinting. Reproduction 2017, 154, R161–R170. [Google Scholar] [CrossRef]
- Patten, M.M.; Cowley, M.; Oakey, R.J.; Feil, R. Regulatory Links between Imprinted Genes: Evolutionary Predictions and Consequences. Proc. R. Soc. B Boil. Sci. 2016, 283, 20152760. [Google Scholar] [CrossRef]
- Cai, X.; Cullen, B.R. The Imprinted H19 Noncoding RNA Is a Primary MicroRNA Precursor. RNA 2007, 13, 313–316. [Google Scholar] [CrossRef]
- Rovina, D.; La Vecchia, M.; Cortesi, A.; Fontana, L.; Pesant, M.; Maitz, S.; Tabano, S.; Bodega, B.; Miozzo, M.; Sirchia, S.M. Profound Alterations of the Chromatin Architecture at Chromosome 11p15.5 in Cells from Beckwith-Wiedemann and Silver-Russell Syndromes Patients. Sci. Rep. 2020, 10, 8275. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.H.; Gauthier, D.W.; Maizels, M. Prenatal Diagnosis of Beckwith-Wiedemann Syndrome. Prenat. Diagn. 2005, 25, 879–884. [Google Scholar] [CrossRef]
- Chen, Z.; Hagen, D.E.; Elsik, C.G.; Ji, T.; Morris, C.J.; Moon, L.E.; Rivera, R.M. Characterization of Global Loss of Imprinting in Fetal Overgrowth Syndrome Induced by Assisted Reproduction. Proc. Natl. Acad. Sci. USA 2015, 112, 4618–4623. [Google Scholar] [CrossRef]
- Paganini, L.; Carlessi, N.; Fontana, L.; Silipigni, R.; Motta, S.; Fiori, S.; Guerneri, S.; Lalatta, F.; Cereda, A.; Sirchia, S.M.; et al. Beckwith—Wiedemann Syndrome Prenatal Diagnosis by Methylation Analysis in Chorionic Villi. Epigenetics 2015, 10, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Katorza, E.; Achiron, R. Early Pregnancy Scanning for Fetal Anomalies—the New Standard? Clin. Obstet. Gynecol. 2012, 55, 199–216. [Google Scholar] [CrossRef]
- Shuman, C.; Beckwith, J.B.; Weksberg, R. Beckwith—Wiedemann Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Petry, C.J.; Ong, K.K.; Dunger, D.B. Does the Fetal Genotype Affect Maternal Physiology during Pregnancy? Trends Mol. Med. 2007, 13, 414–421. [Google Scholar] [CrossRef]
- Wangler, M.F.; Chang, A.S.; Moley, K.H.; Feinberg, A.P.; Debaun, M.R. Factors Associated with Preterm Delivery in Mothers of Children with Beckwith-Wiedemann Syndrome: A Case Cohort Study from the BWS Registry. Am. J. Med. Genet. Part A 2005, 134, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Shieh, H.F.; Estroff, J.A.; Barnewolt, C.E.; Zurakowski, D.; Tan, W.; Buchmiller, T.L. Prenatal Imaging throughout Gestation in Beckwith—Wiedemann Syndrome. Prenat. Diagn. 2019, 39, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Lapunzina Badía, P.; del Campo Casanelles, M.; Delicado Navarro, A.; Fernández-Toral, J.; García-Alix, A.; García-Guereta, L.; Pérez Jurado, L.A.; Ramos Fuentes, F.J.; Sánchez Díaz, A.; Urioste Azcorra, M. Clinical guide to the management of patients with Beckwith-Wiedemann syndrome. Anales de Pediatría 2006, 64, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Clinical and Molecular Diagnosis, Screening and Management of Beckwith—Wiedemann Syndrome: An International Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Duffy, K.A.; Cielo, C.M.; Cohen, J.L.; Gonzalez-Gandolfi, C.X.; Griff, J.R.; Hathaway, E.R.; Kupa, J.; Taylor, J.A.; Wang, K.H.; Ganguly, A.; et al. Characterization of the Beckwith—Wiedemann Spectrum: Diagnosis and Management. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 693–708. [Google Scholar] [CrossRef]
- Wang, K.H.; Kupa, J.; Duffy, K.A.; Kalish, J.M. Diagnosis and Management of Beckwith-Wiedemann Syndrome. Front. Pediatr. 2020, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Sajorda, B.J.; Gonzalez-Gandolfi, C.X.; Hathaway, E.R.; Kalish, J.M. Simpson-Golabi-Behmel Syndrome Type 1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Waterson, J.; Stockley, T.L.; Segal, S.; Golabi, M. Novel Duplication in Glypican-4 as an Apparent Cause of Simpson-Golabi-Behmel Syndrome. Am. J. Med. Genet. Part A 2010, 152, 3179–3181. [Google Scholar] [CrossRef] [PubMed]
- Schirwani, S.; Novelli, A.; Digilio, M.C.; Bourn, D.; Wilson, V.; Roberts, C.; Dallapiccola, B.; Hobson, E. Duplications of GPC3 and GPC4 Genes in Symptomatic Female Carriers of Simpson-Golabi-Behmel Syndrome Type 1. Eur. J. Med. Genet. 2019, 62, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Cole, T.R.; Rahman, N. Sotos Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Baujat, G.; Rio, M.; Rossignol, S.; Sanlaville, D.; Lyonnet, S.; Le Merrer, M.; Munnich, A.; Gicquel, C.; Cormier-Daire, V.; Colleaux, L. Paradoxical NSD1 Mutations in Beckwith-Wiedemann Syndrome and 11p15 Anomalies in Sotos Syndrome. Am. J. Hum. Genet. 2004, 74, 715–720. [Google Scholar] [CrossRef]
- Neylon, O.M.; Werther, G.A.; Sabin, M.A. Overgrowth Syndromes. Curr. Opin. Pediatr. 2012, 24, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ferianec, V.; Bartova, M. Beckwith—Wiedemann Syndrome with Overlapping Perlman Syndrome Manifestation. J. Matern Neonatal Med. 2013, 27, 1607–1609. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Rios, J.J.; Parker, V.E.; Semple, R.K.; Lindhurst, M.J.; Sapp, J.C.; Alomari, A.; Ezaki, M.; Dobyns, W.; Biesecker, L.G. PIK3CA-Related Overgrowth Spectrum (PROS): Diagnostic and Testing Eligibility Criteria, Differential Diagnosis, and Evaluation. Am. J. Med. Genet. Part A 2015, 167, 287–295. [Google Scholar] [CrossRef]
- Weksberg, R.; Shuman, C.; Smith, A.C. Beckwith-Wiedemann Syndrome. Am. J. Med. Genet. Part C Semin. Med Genet. 2005, 137, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Molinatto, C.; Cerrato, F.; Palumbo, O.; Carella, M.; Baldassarre, G.; Carli, D.; Peris, C.; Riccio, A.; Ferrero, G.B. Assisted Reproductive Techniques and Risk of Beckwith-Wiedemann Syndrome. Pediatrics 2017, 140, e20164311. [Google Scholar] [CrossRef]
- Cielo, C.M.; Duffy, K.A.; Vyas, A.; Taylor, J.A.; Kalish, J.M. Obstructive Sleep Apnoea and the Role of Tongue Reduction Surgery in Children with Beckwith-Wiedemann Syndrome. Paediatr. Respir. Rev. 2018, 25, 58–63. [Google Scholar] [CrossRef]
- Maas, S.M.; Kadouch, D.J.; Masselink, A.-C.C.; Van Der Horst, C.M. Taste and Speech Following Surgical Tongue Reduction in Children with Beckwith—Wiedemann Syndrome. J. Cranio-Maxillofac. Surg. 2016, 44, 659–663. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Baldassarre, G.; Riberi, E.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J. Pediatr. 2016, 176, 142–149.e1. [Google Scholar] [CrossRef] [PubMed]
- Eliseörstavik, R.; Tommerup, N.; Eiklid, K.; Heleneörstavik, K. Non-Random X Chromosome Inactivation in an Affected Twin in a Monozygotic Twin Pair Discordant for Wiedemann-Beckwith Syndrome. Am. J. Med. Genet. 1995, 56, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L.; Duffy, K.A.; Sajorda, B.J.; Hathaway, E.R.; Gonzalez-Gandolfi, C.X.; Richards-Yutz, J.; Gunter, A.T.; Ganguly, A.; Kaplan, J.; Deardorff, M.A.; et al. Diagnosis and management of the phenotypic spectrum of twins with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. Part A 2019, 179, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Calzari, L.; Mussa, A.; Mainini, E.; Cassina, M.; Di Candia, S.; Clementi, M.; Guzzetti, S.; Tabano, S.; Miozzo, M.; et al. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver–Russell and Beckwith–Wiedemann syndromes. Clin. Epigenetics 2016, 8, 23. [Google Scholar] [CrossRef]
- Tabano, S.M.; Bonaparte, E.; Miozzo, M. Detection of Loss of Imprinting by Pyrosequencing®. Adv. Struct. Saf. Stud. 2015, 1315, 241–258. [Google Scholar] [CrossRef]
- Eggermann, K.; Bliek, J.; Brioude, F.; Algar, E.; Buiting, K.; Russo, S.; Tümer, Z.; Monk, D.; Moore, G.; Antoniadi, T.; et al. EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver–Russell and Beckwith–Wiedemann syndrome. Eur. J. Hum. Genet. 2016, 24, 1377–1387. [Google Scholar] [CrossRef]
- Sparago, A.; Cerrato, F.; Vernucci, M.; Ferrero, G.B.; Silengo, M.C.; Riccio, A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith—Wiedemann syndrome. Nat. Genet. 2004, 36, 958–960. [Google Scholar] [CrossRef]
- Demars, J.; Shmela, M.E.; Rossignol, S.; Okabe, J.; Netchine, I.; Azzi, S.; Cabrol, S.; Le Caignec, C.; David, A.; Le Bouc, Y.; et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum. Mol. Genet. 2009, 19, 803–814. [Google Scholar] [CrossRef]
- Antonazzo, P.; Alvino, G.; Cozzi, V.; Grati, F.; Tabano, S.M.; Sirchia, S.; Miozzo, M.; Cetin, I. Placental IGF2 Expression in Normal and Intrauterine Growth Restricted (IUGR) Pregnancies. Placenta 2008, 29, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Tabano, S.; Colapietro, P.; Cetin, I.; Grati, F.R.; Zanutto, S.; Mandò, C.; Antonazzo, P.; Pileri, P.; Rossella, F.; Larizza, L.; et al. Epigenetic modulation of theIGF2/H19imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics 2010, 5, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, T.; Brioude, F.; Russo, S.; Lombardi, M.P.; Bliek, J.; Maher, E.R.; Larizza, L.; Prawitt, D.; Netchine, I.; Gonzales, M.; et al. Prenatal molecular testing for Beckwith–Wiedemann and Silver–Russell syndromes: A challenge for molecular analysis and genetic counseling. Eur. J. Hum. Genet. 2015, 24, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Calvello, M.; Tabano, S.M.; Colapietro, P.; Maitz, S.; Pansa, A.; Augello, C.; Lalatta, F.; Gentilin, B.; Spreafico, F.; Calzari, L.; et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith—Wiedemann syndrome. Epigenetics 2013, 8, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Bedeschi, M.F.; Calvello, M.; Paganini, L.; Pezzani, L.; Baccarin, M.; Fontana, L.; Sirchia, S.M.; Guerneri, S.; Canazza, L.; Leva, E.; et al. Sequence variants identification at the KCNQ1OT1:TSS differentially Methylated region in isolated omphalocele cases. BMC Med. Genet. 2017, 18, 115. [Google Scholar] [CrossRef]
- Grati, F.R.; Turolla, L.; D’Ajello, P.; Ruggeri, A.; Miozzo, M.; Bracalente, G.; Baldo, D.; Laurino, L.; Boldorini, R.; Frate, E.; et al. Chromosome 11 segmental paternal isodisomy in amniocytes from two fetuses with omphalocoele: New highlights on phenotype-genotype correlations in Beckwith—Wiedemann syndrome. J. Med. Genet. 2007, 44, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Gaillot-Durand, L.; Brioude, F.; Beneteau, C.; Le Breton, F.; Massardier, J.; Michon, L.; Devouassoux-Shisheboran, M.; Allias, F. Placental Pathology in Beckwith–Wiedemann Syndrome According to Genotype/Epigenotype Subgroups. Fetal Pediatr. Pathol. 2018, 37, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.N.; Luharia, A.; Evans, G.A.; Raza, H.; Haire, A.C.; Grundy, R.G.; Bowdin, S.C.; Riccio, A.; Sebastio, G.; Bliek, J.; et al. Molecular subtypes and phenotypic expression of Beckwith–Wiedemann syndrome. Eur. J. Hum. Genet. 2005, 13, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Lacoste, A.; Netchine, I.; Vazquez, M.-P.; Auber, F.; Audry, G.; Gauthier-Villars, M.; Brugieres, L.; Gicquel, C.; Le Bouc, Y.; et al. Beckwith—Wiedemann Syndrome: Growth Pattern and Tumor Risk according to Molecular Mechanism, and Guidelines for Tumor Surveillance. Horm. Res. Paediatr. 2013, 80, 457–465. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Niemitz, E.L.; McNeil, D.E.; Brandenburg, S.A.; Lee, M.P.; Feinberg, A.P. Epigenetic Alterations of H19 and LIT1 Distinguish Patients with Beckwith—Wiedemann Syndrome with Cancer and Birth Defects. Am. J. Hum. Genet. 2002, 70, 604–611. [Google Scholar] [CrossRef]
- Mussa, A.; Peruzzi, L.; Chiesa, N.; De Crescenzo, A.; Russo, S.; Melis, D.; Tarani, L.; Baldassarre, G.; Larizza, L.; Riccio, A.; et al. Nephrological findings and genotype–phenotype correlation in Beckwith–Wiedemann syndrome. Pediatr. Nephrol. 2011, 27, 397–406. [Google Scholar] [CrossRef]
- Romanelli, V.; Belinchón, A.; Benito-Sanz, S.; Martínez-Glez, V.; Gracia-Bouthelier, R.; Heath, K.E.; Campos-Barros, A.; García-Miñaur, S.; Fernandez, L.; Meneses, H.; et al. CDKN1C (p57Kip2) analysis in Beckwith—Wiedemann syndrome (BWS) patients: Genotype-phenotype correlations, novel mutations, and polymorphisms. Am. J. Med. Genet. Part A 2010, 152, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, F.; Zollino, M.; Oliva, A.; Pascali, V.L.; Orteschi, D.; Pietrobono, R.; Camporeale, A.; Vidal, M.C.; Partemi, S.; Brugada, R.; et al. Mild Beckwith—Wiedemann and severe long-QT syndrome due to deletion of the imprinting center 2 on chromosome 11p. Eur. J. Hum. Genet. 2013, 21, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, S.; Capri, Y.; Rossignol, S.; Denjoy, I.; Soudée, S.; Aboura, A.; Baumann, C.; Verloes, A. Beckwith—Wiedemann syndrome and long QT syndrome due to familial-balanced translocation t(11;17)(p15.5;q21.3) involving the KCNQ1 gene. Clin. Genet. 2012, 84, 78–81. [Google Scholar] [CrossRef]
- Valente, F.M.; Sparago, A.; Freschi, A.; Hill-Harfe, K.; Maas, S.M.; Frints, S.G.M.; Alders, M.; Pignata, L.; Franzese, M.; Angelini, C.; et al. Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith–Wiedemann locus. Genet. Med. 2019, 21, 1808–1820. [Google Scholar] [CrossRef]
- Bliek, J.; Alders, M.; Maas, S.M.; Oostra, R.-J.; Mackay, D.M.; Van Der Lip, K.; Callaway, J.L.; Brooks, A.; van’t Padje, S.; Westerveld, A.; et al. Lessons from BWS twins: Complex maternal and paternal hypomethylation and a common source of haematopoietic stem cells. Eur. J. Hum. Genet. 2009, 17, 1625–1634. [Google Scholar] [CrossRef]
- Hall, J.G. Twinning. Lancet 2003, 362, 735–743. [Google Scholar] [CrossRef]
- D’Antonio, F.; Khalil, A.; Dias, T.; Thilaganathan, B.; Southwest Thames Obstetric Research Collaborative (STORK). Early fetal loss in monochorionic and dichorionic twin pregnancies: Analysis of the Southwest Thames Obstetric Research Collaborative (STORK) multiple pregnancy cohort. Ultrasound Obstet. Gynecol. 2013, 41, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nakamura, A.; Matsubara, K.; Nyuzuki, H.; Nagasaki, K.; Oka, A.; Fukami, M.; Kagami, M. Continuous hypomethylation of theKCNQ1OT1:TSS-DMR in monochorionic twins discordant for Beckwith—Wiedemann syndrome. Am. J. Med. Genet. Part A 2017, 173, 2847–2850. [Google Scholar] [CrossRef]
- Tierling, S.; Souren, N.; Reither, S.; Zang, K.; Meng-Hentschel, J.; Leitner, D.; Oehl-Jaschkowitz, B.; Walter, J. DNA methylation studies on imprinted loci in a male monozygotic twin pair discordant for Beckwith—Wiedemann syndrome. Clin. Genet. 2010, 79, 546–553. [Google Scholar] [CrossRef]
- Lubinsky, M.; Hall, J. Genomic imprinting, monozygous twinning, and X inactivation. Lancet 1991, 337, 1288. [Google Scholar] [CrossRef]
- Bestor, T.H. Imprinting errors and developmental asymmetry. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- McGraw, S.; Oakes, C.C.; Martel, J.; Cirio, M.C.; De Zeeuw, P.; Mak, W.; Plass, C.; Bartolomei, M.S.; Chaillet, J.R.; Trasler, J.M. Loss of DNMT1o Disrupts Imprinted X Chromosome Inactivation and Accentuates Placental Defects in Females. PLoS Genet. 2013, 9, e1003873. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P.; Pasini, S.; Selmi, C.; Miozzo, M.; Podda, M. Skewing of X chromosome inactivation in autoimmunity. Autoimmun 2008, 41, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Salsano, E.; Tabano, S.M.; Sirchia, S.M.; Colapietro, P.; Castellotti, B.; Gellera, C.; Rimoldi, M.; Pensato, V.; Mariotti, C.; Pareyson, D.; et al. Preferential expression of mutant ABCD1 allele is common in adrenoleukodystrophy female carriers but unrelated to clinical symptoms. Orphanet J. Rare Dis. 2012, 7, 10. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of In Vitro Fertilization with Beckwith—Wiedemann Syndrome and Epigenetic Alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Gicquel, C.; Gaston, V.; Mandelbaum, J.; Siffroi, J.-P.; Flahault, A.; Le Bouc, Y. In Vitro Fertilization May Increase the Risk of Beckwith—Wiedemann Syndrome Related to the Abnormal Imprinting of the KCNQ1OT Gene. Am. J. Hum. Genet. 2003, 72, 1338–1341. [Google Scholar] [CrossRef]
- Maher, E.R. Epigenetic risks related to assisted reproductive technologies: Epigenetics, imprinting, ART and icebergs? Hum. Reprod. 2003, 18, 2508–2511. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Major Features | Minor Features |
|---|---|
| Macroglossia | Polyhydramnios |
| Macrosomia | Nephromegaly/kidney dysgenesis |
| Abdominal wall defect | Suprarenal mass suggestive for adrenal cytomegaly |
| Placental mesenchymal dysplasia |
| Clinical Feature | Frequency (%) |
|---|---|
| Major criteria | |
| Macroglossia | 97 |
| Macrosomia | 84 |
| Abdominal wall defects | 80 |
| Hemihypertrophy | 64 |
| Outer ear anomalies | 63 |
| Kidney and ureter anomalies | 28–61 |
| Visceromegaly | 41 |
| Embryonal tumors | ~8 |
| Cleft palate | ~6 |
| Positive family history | - |
| Minor criteria | |
| Nevus flammeus of the forehead | 54 |
| Neonatal hypoglycemia | >50 |
| Prematurity | 50 |
| Placentomegaly | 50 |
| Polyhydramnios | 50 |
| Diastasis recti | 28 |
| Cardiomegaly/hypertrophic cardiomyopathy | 20 |
| Typical facies | - |
| Polydactyly | - |
| Supernumerary nipples | - |
| Advanced bone age | - |
| Cardinal Features (2 Points for Feature) |
|---|
| Macroglossia |
| Exomphalos |
| Lateralized overgrowth |
| Multifocal and/or bilateral Wilms tumors or nephroblastomatosis |
| Hyperinsulinism (lasting >1 week and requiring medical treatment) |
| Pathology findings: adrenal cortex cytomegaly, placental mesenchymal dysplasia or pancreatic adenomatosis |
| Suggestive Features (1 Point for Feature) |
| Birthweight >2SDS above the mean |
| Facial nevus flammeus |
| Polyhydramnios and/or placentomegaly |
| Ear creases and/or pits |
| Hyperinsulinism (lasting <1 week) |
| Typical tumors: neuroblastoma, rhabdomyosarcoma, unilateral Wilms tumors, hepatoblastoma, adrenocortical carcinoma or pheochromocytoma |
| Nephromegaly and/or hepatomegaly Umbilical hernia and/or diastasis recti |
| BWS | SGB | Sotos | Perlman | RASopathies | PROS | |
|---|---|---|---|---|---|---|
| Macroglossia | ++ | ++ | + | |||
| Abdominal wall defects | ++ | ++ | + | |||
| Segmental/lateralized overgrowth | ++ | + | ++ | |||
| Increased embryonal tumor risk | ++ | ++ | + § | + ^ | + | |
| Neonatal hypoglycemia | ++ | ++ | + | |||
| Birthweight > 2SD/macrosomia | + | ++ | ++ * | ++ | ++ | + |
| Vascular defects | + | + | ++ | |||
| Ear creases and/or pits | + | + | + | |||
| Macrocephaly | ++ | ++ | ++ | ++ | ||
| Developmental delay/intellectual disability | ++ | ++ | ++ | ++ | + ° | |
| Peculiar dysmorphic features | + | ++ | ++ | ++ | +/− | |
| Congenital heart disease | ++ | ++ | + | ++ | + | |
| Nephromegaly and/or hepatomegaly | + | ++ | + | + | + |
| Molecular Alteration | Frequency | Mosaicism | Risk of Recurrence | (Epi)Genotype-Phenotype Correlations |
|---|---|---|---|---|
| IC1 GOM | 5–10% | Yes | <1% without genetic anomalies; 50% depending on the parental origin and if genetic anomalies are present (up to 20% of SNVs) | Bilateral/multifocal Wilms tumors, macroglossia, macrosoma, organomegaly, nephrourological, hypoglycemia, undescended testes |
| IC2 LOM | 50–60% | Yes | <1% without genetic anomalies; 50% depending on the parental origin if genetic anomalies are present | Omphalocele, macroglossia, undescended testes |
| pUPD11 | 20% | Yes | <1% | Lateralized overgrowth, Wilms tumors, hypoglycemia |
| CDKN1C mutations | 5% sporadic cases; 40% familial cases | Rarely | 50% via maternal transmission | Omphalocele |
| MLID | 50% IC2 LOM patients; rare in patients with no molecular diagnosis | Yes | Low without causative mutations | Not clearly defined with an excess of female and a high frequency of some features |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, L.; Tabano, S.; Maitz, S.; Colapietro, P.; Garzia, E.; Gerli, A.G.; Sirchia, S.M.; Miozzo, M. Clinical and Molecular Diagnosis of Beckwith-Wiedemann Syndrome with Single- or Multi-Locus Imprinting Disturbance. Int. J. Mol. Sci. 2021, 22, 3445. https://doi.org/10.3390/ijms22073445
Fontana L, Tabano S, Maitz S, Colapietro P, Garzia E, Gerli AG, Sirchia SM, Miozzo M. Clinical and Molecular Diagnosis of Beckwith-Wiedemann Syndrome with Single- or Multi-Locus Imprinting Disturbance. International Journal of Molecular Sciences. 2021; 22(7):3445. https://doi.org/10.3390/ijms22073445
Chicago/Turabian StyleFontana, Laura, Silvia Tabano, Silvia Maitz, Patrizia Colapietro, Emanuele Garzia, Alberto Giovanni Gerli, Silvia Maria Sirchia, and Monica Miozzo. 2021. "Clinical and Molecular Diagnosis of Beckwith-Wiedemann Syndrome with Single- or Multi-Locus Imprinting Disturbance" International Journal of Molecular Sciences 22, no. 7: 3445. https://doi.org/10.3390/ijms22073445
APA StyleFontana, L., Tabano, S., Maitz, S., Colapietro, P., Garzia, E., Gerli, A. G., Sirchia, S. M., & Miozzo, M. (2021). Clinical and Molecular Diagnosis of Beckwith-Wiedemann Syndrome with Single- or Multi-Locus Imprinting Disturbance. International Journal of Molecular Sciences, 22(7), 3445. https://doi.org/10.3390/ijms22073445