
A Degeneration Gradient of Poplar Trees Contributes to the Taxonomic, Functional, and Resistome Diversity of Bacterial Communities in Rhizosphere Soils

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
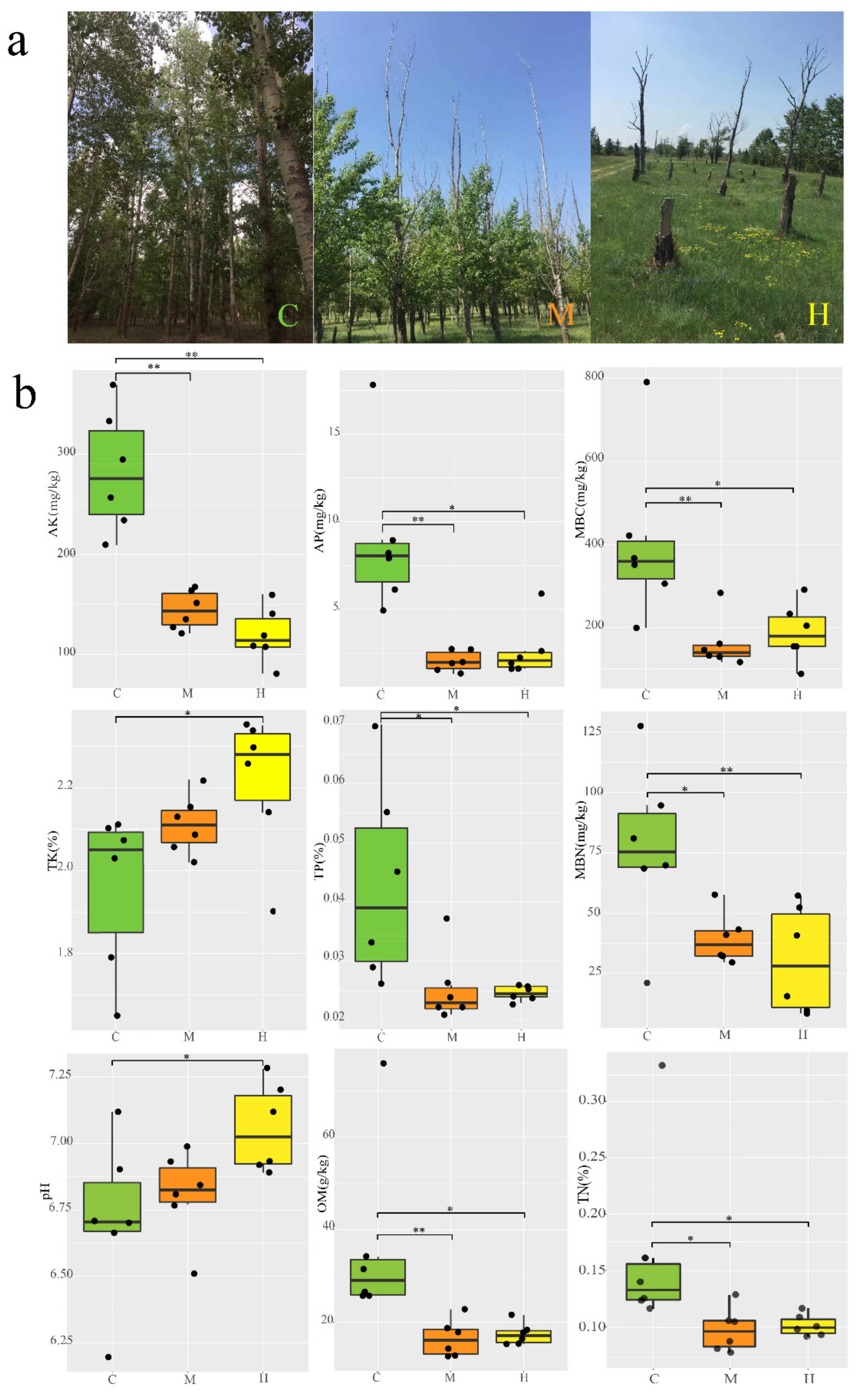
2.1. Populus and Rhizosphere Soil Characterization
2.2. Sequencing Results
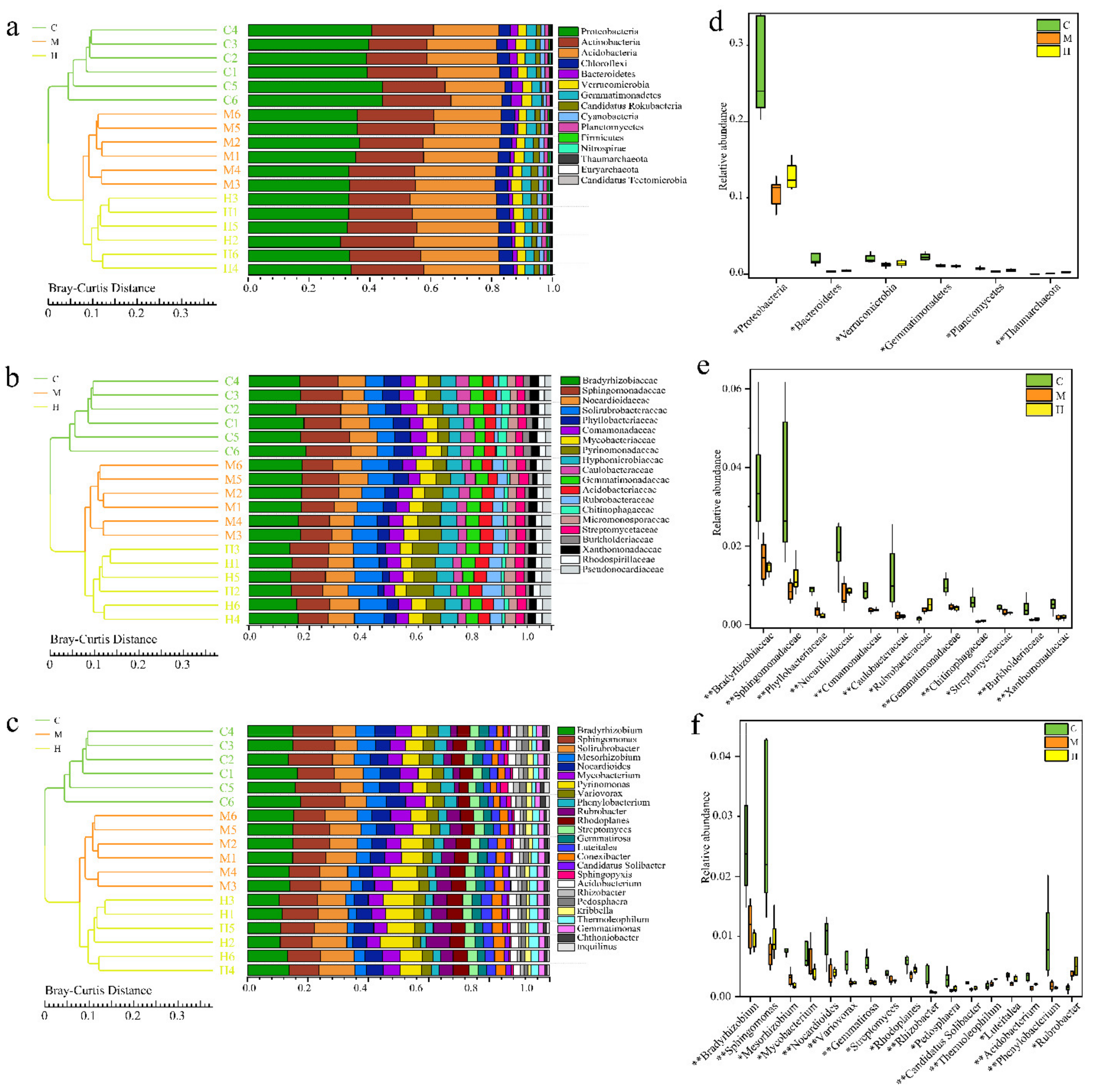
2.3. Comparative Analysis of Microbial Communities
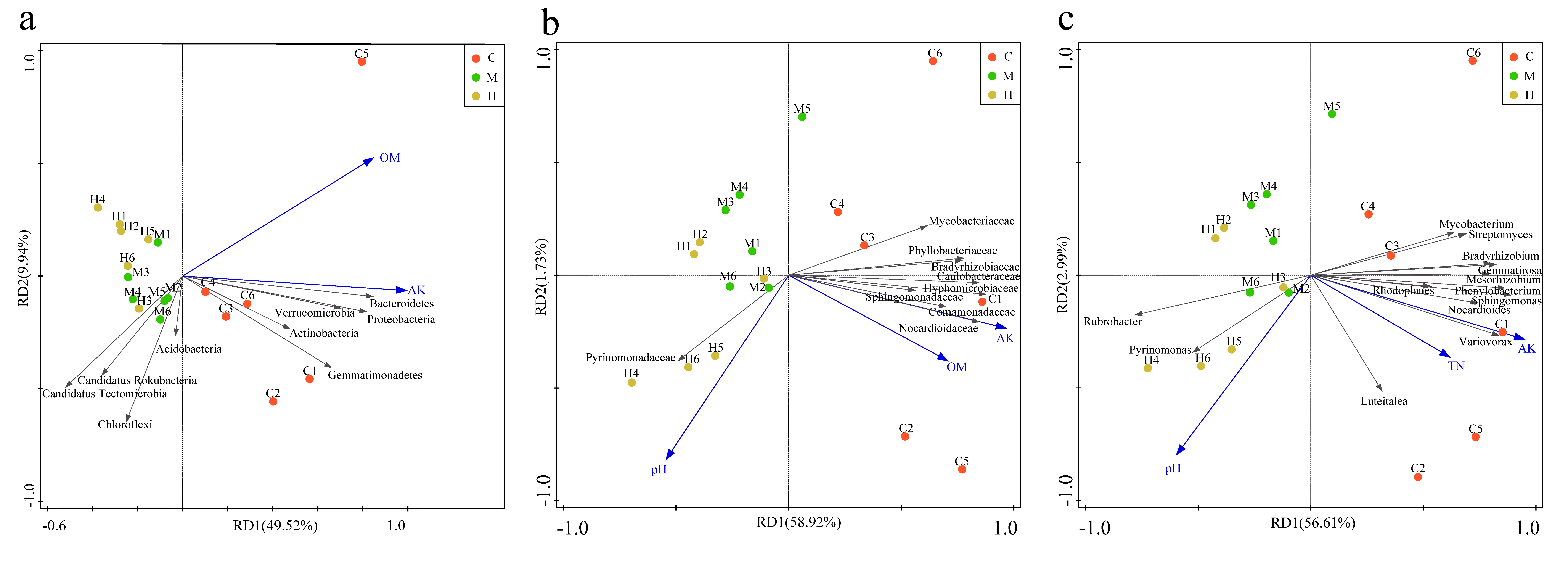
2.4. Correlation Analysis of Soil Characterization and Microbial Communities
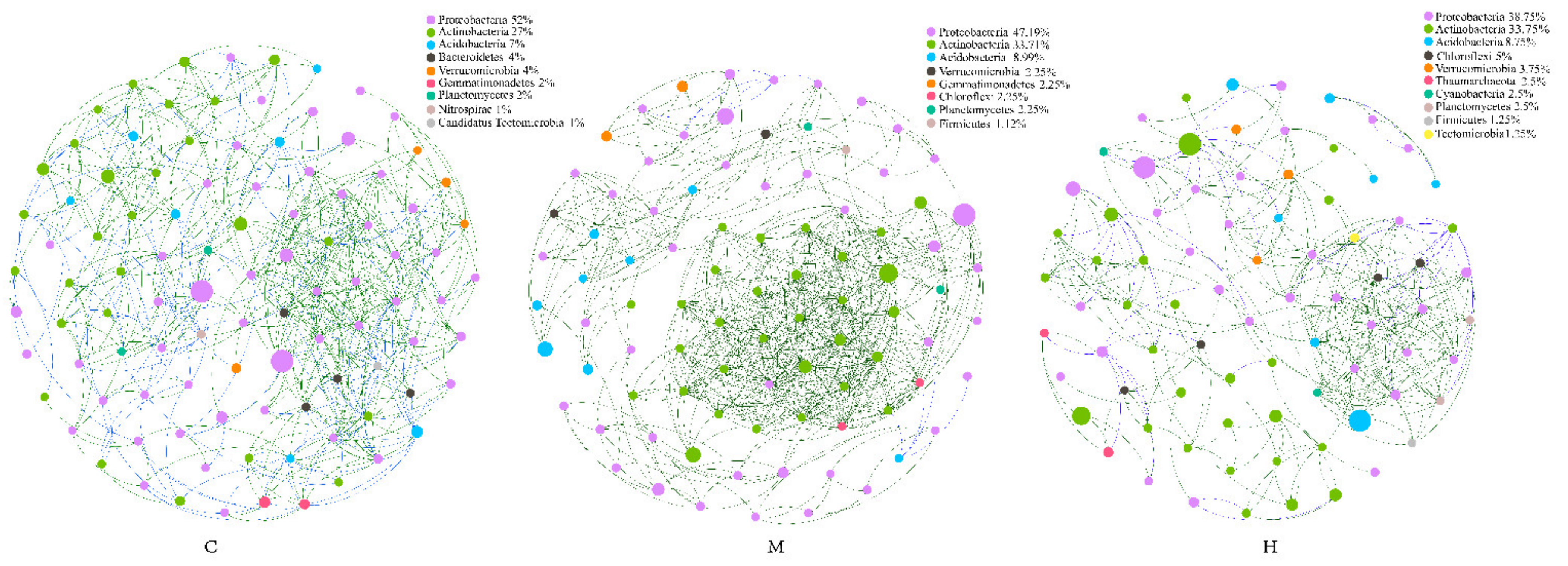
2.5. Comparison of Microbial Interaction Networks
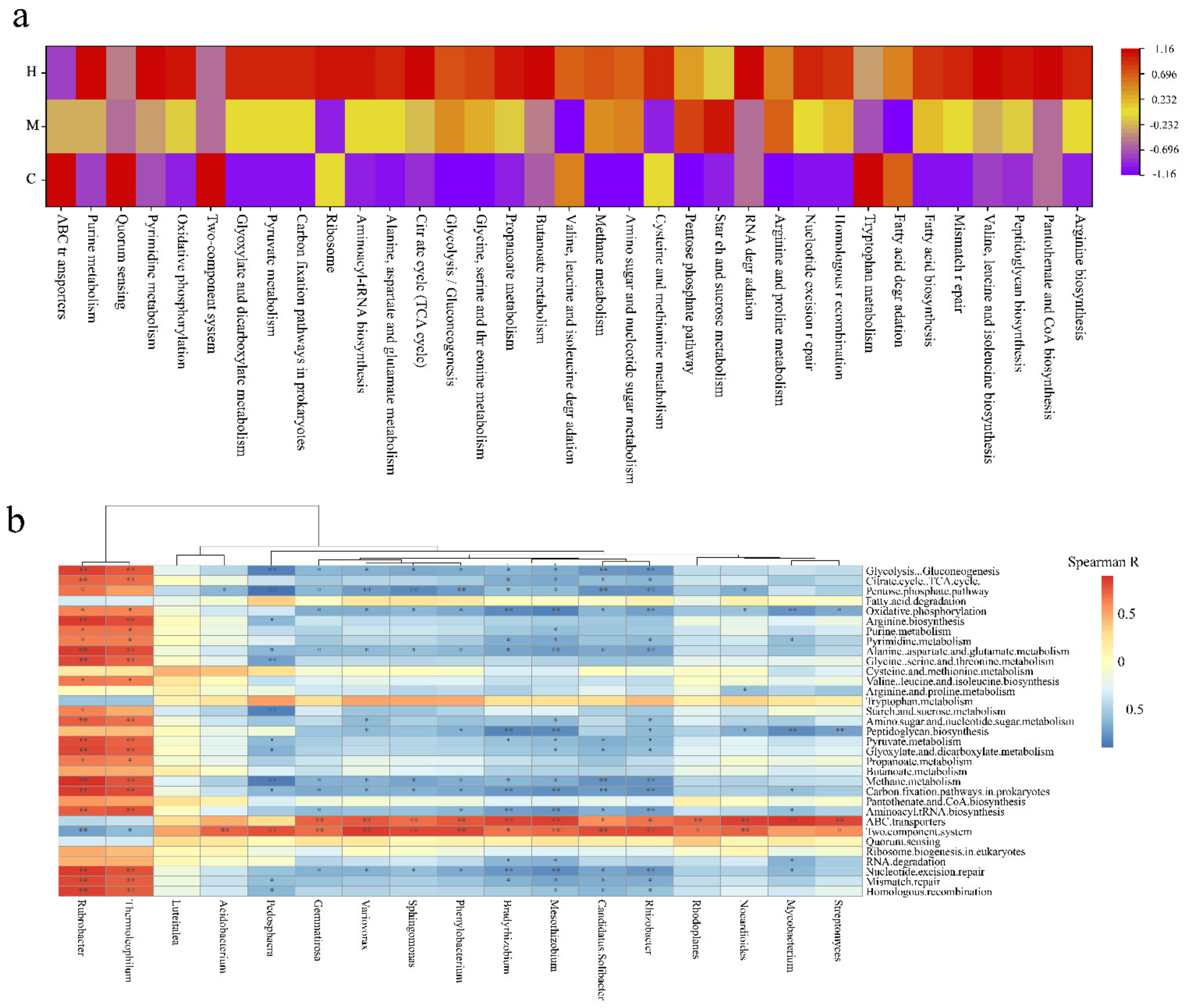
2.6. Function Potential Analysis of Microbes
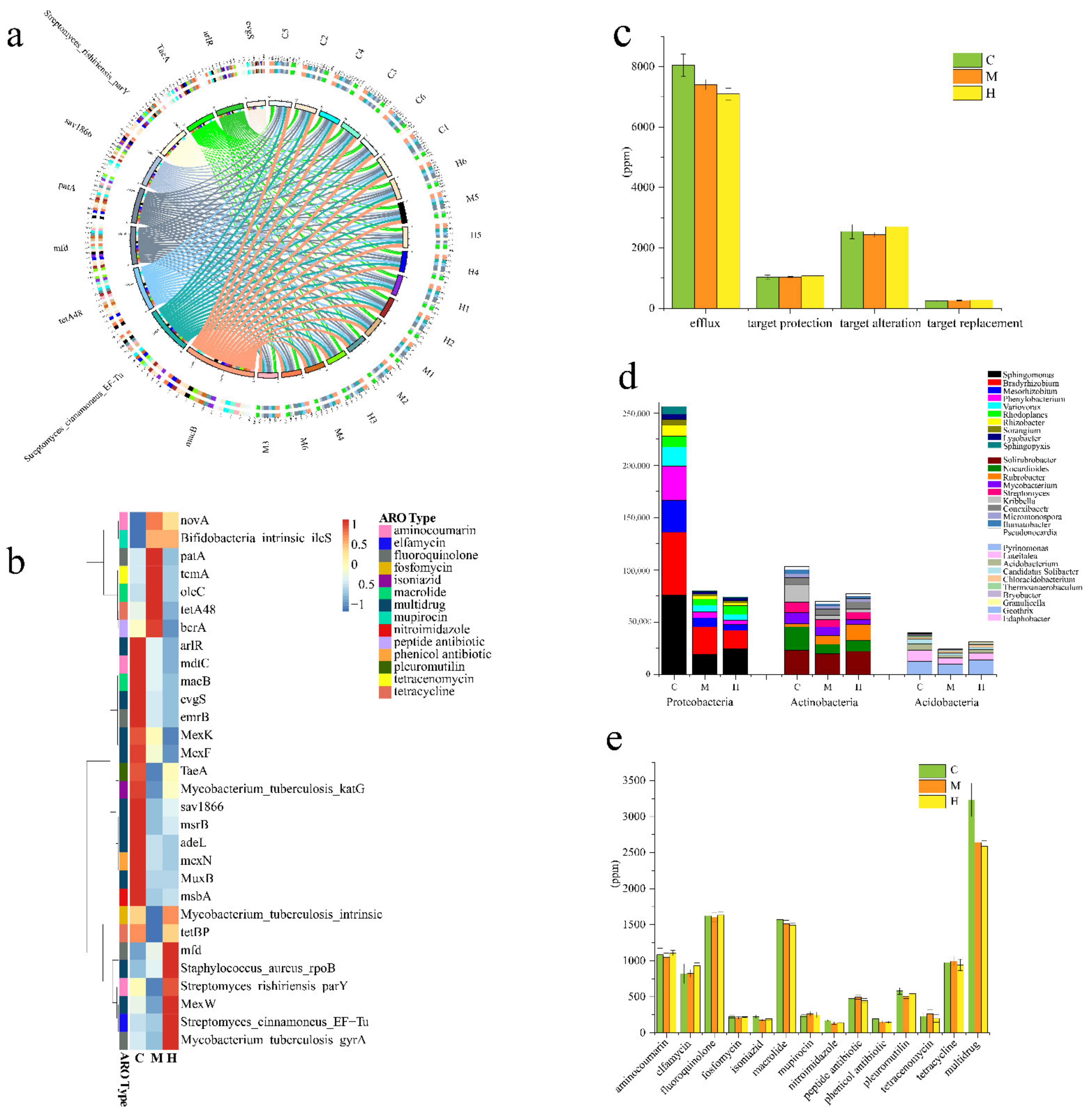
2.7. Resistance Genome
3. Materials and Methods
3.1. Sampling Sites and Sample Collection
3.2. Degeneration Data Determination and Soil Characterization
3.3. DNA Extraction, Shotgun Sequencing, and Metagenome Assembly
3.4. Taxonomic Profiling
3.5. Network Construction
3.6. Function Annotations
3.7. Resistance Gene Annotation
3.8. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Egamberdieva, D.; Kamilova, F.; Validov, S.; Gafurova, L.; Kucharova, Z.; Lugtenberg, B. High incidence of plant growth-stimulating bacteria associated with the rhizosphere of wheat grown on salinated soil in Uzbekistan. Environ. Microbiol. 2008, 10, 1–9. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.A.; Santos-Medellin, C.M.; Liechty, Z.S.; Bao, N.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.G.; Schlatter, D.C.; Otto-Hanson, L.; Kinkel, L.L. Diffuse symbioses: Roles of plant-plant, plant-microbe and microbe-microbe interactions in structuring the soil microbiome. Mol. Ecol. 2014, 23, 1571–1583. [Google Scholar] [CrossRef]
- Rolli, E.; Marasco, R.; Vigani, G.; Ettoumi, B.; Mapelli, F.; Deangelis, M.L.; Gandolfi, C.; Casati, E.; Previtali, F.; Gerbino, R.; et al. Improved plant resistance to drought is promoted by the root-associated microbiome as a water stress-dependent trait. Environ. Microbiol. 2015, 17, 316–331. [Google Scholar] [CrossRef]
- Castrillo, G.; Teixeira, P.J.P.L.; Paredes, S.H.; Law, T.F.; de Lorenzo, L.; Feltcher, M.E.; Finkel, O.M.; Breakfield, N.W.; Mieczkowski, P.; Jones, C.D.; et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 2017, 543, 513–518. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 733. [Google Scholar] [CrossRef]
- Wei, Z.; Gu, Y.; Friman, V.-P.; Kowalchuk, G.A.; Xu, Y.; Shen, Q.; Jousset, A. Initial soil microbiome composition and functioning predetermine future plant health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Nomura, K.; Wang, X.; Sohrabi, R.; Xu, J.; Yao, L.; Paasch, B.C.; Ma, L.; Kremer, J.; Cheng, Y.; et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 2020, 580, 653–657. [Google Scholar] [CrossRef]
- Timm, C.M.; Pelletier, D.A.; Jawdy, S.S.; Gunter, L.E.; Henning, J.A.; Engle, N.; Aufrecht, J.; Gee, E.; Nookaew, I.; Yang, Z.; et al. Two Poplar-Associated Bacterial Isolates Induce Additive Favorable Responses in a Constructed Plant-Microbiome System. Front. Plant Sci. 2016, 7, 497. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.A.; Weston, D.J.; Pelletier, D.A.; Timm, C.M.; Jawdy, S.S.; Classen, A.T. Root bacterial endophytes alter plant phenotype, but not physiology. Peerj 2016, 4, e2606. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, S.; Garafola, C.; Monchy, S.; Newman, L.; Hoffman, A.; Weyens, N.; Barac, T.; Vangronsveld, J.; van der Lelie, D. Genome Survey and Characterization of Endophytic Bacteria Exhibiting a Beneficial Effect on Growth and Development of Poplar Trees. Appl. Environ. Microbiol. 2009, 75, 748–757. [Google Scholar] [CrossRef]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Gottel, N.R.; Castro, H.F.; Kerley, M.; Yang, Z.M.; Pelletier, D.A.; Podar, M.; Karpinets, T.; Uberbacher, E.; Tuskan, G.A.; Vilgalys, R.; et al. Distinct Microbial Communities within the Endosphere and Rhizosphere of Populus deltoides Roots across Contrasting Soil Types. Appl. Environ. Microbiol. 2011, 77, 5934–5944. [Google Scholar] [CrossRef] [PubMed]
- Shakya, M.; Gottel, N.; Castro, H.; Yang, Z.K.; Gunter, L.; Labbe, J.; Muchero, W.; Bonito, G.; Vilgalys, R.; Tuskan, G.; et al. A Multifactor Analysis of Fungal and Bacterial Community Structure in the Root Microbiome of Mature Populus deltoides Trees. PLoS ONE 2013, 8, e76382. [Google Scholar] [CrossRef]
- Wei, W.; Wang, B.; Niu, X. Soil Erosion Reduction by Grain for Green Project in Desertification Areas of Northern China. Forests 2020, 11, 473. [Google Scholar] [CrossRef]
- Deng, L.; Kim, D.-G.; Li, M.; Huang, C.; Liu, Q.; Cheng, M.; Shangguan, Z.; Peng, C. Land-use changes driven by ‘Grain for Green’ program reduced carbon loss induced by soil erosion on the Loess Plateau of China. Glob. Planet. Chang. 2019, 177, 101–115. [Google Scholar] [CrossRef]
- Gong, G.; Liu, J.; Shao, Q.; Zhai, J. Sand-Fixing Function under the Change of Vegetation Coverage in a Wind Erosion Area in Northern China. J. Resour. Ecol. 2014, 5, 105–114. [Google Scholar] [CrossRef]
- Huang, L.; Wang, B.; Niu, X.; Gao, P.; Song, Q. Changes in ecosystem services and an analysis of driving factors for China’s Natural Forest Conservation Program. Ecol. Evol. 2019, 9, 3700–3716. [Google Scholar] [CrossRef]
- Cao, S.; Suo, X.; Xia, C. Payoff from afforestation under the Three-North Shelter Forest Program. J. Clean. Prod. 2020, 256. [Google Scholar] [CrossRef]
- Qiu, B.; Chen, G.; Tang, Z.; Lu, D.; Wang, Z.; Chen, C. Assessing the Three-North Shelter Forest Program in China by a novel framework for characterizing vegetation changes. ISPRS J. Photogramm. Remote Sens. 2017, 133, 75–88. [Google Scholar] [CrossRef]
- Wang, C.; Chu, X.; Zhan, J.; Wang, P.; Zhang, F.; Xin, Z. Factors Contributing to Efficient Forest Production in the Region of the Three-North Shelter Forest Program, China. Sustainability 2020, 12, 302. [Google Scholar] [CrossRef]
- Chunya, Z.; Zhongqi, X.; Changming, M.; Shoujia, S.; Tengfei, Y. The Factors Influencing the Poplar Shelterbelt Degradation in the Bashang Plateau of Northwest Hebei Province. For. Resour. Manag. 2018, 1, 9–16. (In Chinese) [Google Scholar]
- Huan, Z.; Jun, C.; Hua-bing, W.; Bo, S.; Guo-dong, J.; Zi-qiang, L.; Xin-xiao, Y.; Jia, Z. Water utilization characteristics of the degraded poplar shelterbelts in Zhangbei, Hebei, China. Chin. J. Appl. Ecol. 2018, 29, 1381–1388. (In Chinese) [Google Scholar]
- Jump, A.S.; Ruiz-Benito, P.; Greenwood, S.; Allen, C.D.; Kitzberger, T.; Fensham, R.; Martinez-Vilalta, J.; Lloret, F. Structural overshoot of tree growth with climate variability and the global spectrum of drought-induced forest dieback. Glob. Chang. Biol. 2017, 23, 3742–3757. [Google Scholar] [CrossRef]
- Sanchez-Salguero, R.; Julio Camarero, J. Greater sensitivity to hotter droughts underlies juniper dieback and mortality in Mediterranean shrublands. Sci. Total Environ. 2020, 721, 137599. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zhou, G.; Li, Z.; Wang, S.; Zhou, H.; Song, X. Triggers of widespread dieback and mortality of poplar (Populus spp.) plantations across northern China. J. Arid Environ. 2020, 174, 104076. [Google Scholar] [CrossRef]
- Shimizu, H. An overview of the “Three-North” Shelterbelt project in China. J. For. Stud. China 2012, 14, 70–79. [Google Scholar] [CrossRef]
- Zhang, F.; Xing, Z.; Rees, H.W.; Dong, Y.; Li, S.; Meng, F. Assessment of effects of two runoff control engineering practices on soil water and plant growth for afforestation in a semi-arid area after 10 years. Ecol. Eng. 2014, 64, 430–442. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, C.; Li, W.; Tian, L.; Zhu, Q.; Chen, H.; Fang, X.; Zhang, G.; Liu, G.; Mu, X.; et al. Multiple afforestation programs accelerate the greenness in the ‘Three North’ region of China from 1982 to 2013. Ecol. Indic. 2016, 61, 404–412. [Google Scholar] [CrossRef]
- Liu, J.; Dang, P.; Gao, Y.; Zhu, H.; Zhu, H.; Zhao, F.; Zhao, Z. Effects of tree species and soil properties on the composition and diversity of the soil bacterial community following afforestation. For. Ecol. Manag. 2018, 427, 342–349. [Google Scholar] [CrossRef]
- Kaldorf, M.; Fladung, M.; Muhs, H.J.; Buscot, F. Mycorrhizal colonization of transgenic aspen in a field trial. Planta 2002, 214, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Stefani, F.O.P.; Moncalvo, J.-M.; Seguin, A.; Berube, J.A.; Hamelin, R.C. Impact of an 8-Year-Old Transgenic Poplar Plantation on the Ectomycorrhizal Fungal Community. Appl. Environ. Microbiol. 2009, 75, 7527–7536. [Google Scholar] [CrossRef]
- Danielsen, L.; Thuermer, A.; Meinicke, P.; Buee, M.; Morin, E.; Martin, F.; Pilate, G.; Daniel, R.; Polle, A.; Reich, M. Fungal soil communities in a young transgenic poplar plantation form a rich reservoir for fungal root communities. Ecol. Evol. 2012, 2, 1935–1948. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.-C.; Song, S.-M.; Mu, J.; Hu, W.-L.; Xiao, P.-G. Unearthing microbial diversity of Taxus rhizosphere via MiSeq high-throughput amplicon sequencing and isolate characterization. Sci. Rep. 2016, 6, 22006. [Google Scholar] [CrossRef]
- Dai, Z.; Su, W.; Chen, H.; Barberan, A.; Zhao, H.; Yu, M.; Yu, L.; Brookes, P.C.; Schadt, C.W.; Chang, S.X.; et al. Long-term nitrogen fertilization decreases bacterial diversity and favors the growth of Actinobacteria and Proteobacteria in agro-ecosystems across the globe. Glob. Chang. Biol. 2018, 24, 3452–3461. [Google Scholar] [CrossRef] [PubMed]
- Menendez, E.; Perez-Yepez, J.; Hernandez, M.; Rodriguez-Perez, A.; Velazquez, E.; Leon-Barrios, M. Plant Growth Promotion Abilities of Phylogenetically Diverse Mesorhizobium Strains: Effect in the Root Colonization and Development of Tomato Seedlings. Microorganisms 2020, 8, 412. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, F.; Huang, Y.; Zhou, M.; Gao, J.; Yan, T.; Sheng, H.; An, L. Sphingomonas sp. Cra20 Increases Plant Growth Rate and Alters Rhizosphere Microbial Community Structure of Arabidopsis thaliana Under Drought Stress. Front. Microbiol. 2019, 10, 1221. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ge, C.; Xu, S.A.; Wu, Y.; Sahito, Z.A.; Ma, L.; Pan, F.; Zhou, Q.; Huang, L.; Feng, Y.; et al. The endophytic bacterium Sphingomonas SaMR12 alleviates Cd stress in oilseed rape through regulation of the GSH-AsA cycle and antioxidative enzymes. BMC Plant Biol. 2020, 20, 63. [Google Scholar] [CrossRef]
- Talia, P.; Sede, S.M.; Campos, E.; Rorig, M.; Principi, D.; Tosto, D.; Hopp, H.E.; Grasso, D.; Cataldi, A. Biodiversity characterization of cellulolytic bacteria present on native Chaco soil by comparison of ribosomal RNA genes. Res. Microbiol. 2012, 163, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Natsagdorj, O.; Sakamoto, H.; Santiago, D.M.O.; Santiago, C.D.; Orikasa, Y.; Okazaki, K.; Ikeda, S.; Ohwada, T. Variovorax sp. Has an Optimum Cell Density to Fully Function as a Plant Growth Promoter. Microorganisms 2019, 7, 82. [Google Scholar] [CrossRef]
- Sun, S.-L.; Yang, W.-L.; Fang, W.-W.; Zhao, Y.-X.; Guo, L.; Dai, Y.-J. The Plant Growth-Promoting Rhizobacterium Variovorax boronicumulans CGMCC 4969 Regulates the Level of Indole-3-Acetic Acid Synthesized from Indole-3-Acetonitrile. Appl. Environ. Microbiol. 2018, 84, e00298-18. [Google Scholar] [CrossRef]
- Buenger, W.; Jiang, X.; Mueller, J.; Hurek, T.; Reinhold-Hurek, B. Novel cultivated endophytic Verrucomicrobia reveal deep-rooting traits of bacteria to associate with plants. Sci. Rep. 2020, 10, 8692. [Google Scholar] [CrossRef]
- Bergmann, G.T.; Bates, S.T.; Eilers, K.G.; Lauber, C.L.; Caporaso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 2011, 43, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; del Rio, T.G.; Jones, C.D.; Tringe, S.G.; et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef]
- Utturkar, S.M.; Cude, W.N.; Robeson, M.S., Jr.; Yang, Z.K.; Klingeman, D.M.; Land, M.L.; Allman, S.L.; Lu, T.-Y.S.; Brown, S.D.; Schadt, C.W.; et al. Enrichment of Root Endophytic Bacteria from Populus deltoides and Single-Cell-Genomics Analysis. Appl. Environ. Microbiol. 2016, 82, 5698–5708. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-von-Wobeser, E.; Rocha-Estrada, J.; Shapiro, L.R.; de la Torre, M. Enrichment of Verrucomicrobia, Actinobacteria and Burkholderiales drives selection of bacterial community from soil by maize roots in a traditional milpa agroecosystem. PLoS ONE 2018, 13, e0208852. [Google Scholar] [CrossRef] [PubMed]
- Sessitsch, A.; Hardoim, P.; Doering, J.; Weilharter, A.; Krause, A.; Woyke, T.; Mitter, B.; Hauberg-Lotte, L.; Friedrich, F.; Rahalkar, M.; et al. Functional Characteristics of an Endophyte Community Colonizing Rice Roots as Revealed by Metagenomic Analysis. Mol. Plant Microbe Interact. 2012, 25, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellin, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef]
- Krings, M.; Hass, H.; Kerp, H.; Taylor, T.N.; Agerer, R.; Dotzler, N. Endophytic cyanobacteria in a 400-million-yr-old land plant: A scenario for the origin of a symbiosis? Rev. Palaeobot. Palynol. 2009, 153, 62–69. [Google Scholar] [CrossRef]
- He, S.; Stevens, S.L.R.; Chan, L.-K.; Bertilsson, S.; del Rio, T.G.; Tringe, S.G.; Malmstrom, R.R.; McMahon, K.D. Ecophysiology of Freshwater Verrucomicrobia Inferred from Metagenome-Assembled Genomes. mSphere 2017, 2, e00277-17. [Google Scholar] [CrossRef]
- Pold, G.; Conlon, E.M.; Huntemann, M.; Pillay, M.; Mikhailova, N.; Stamatis, D.; Reddy, T.B.K.; Daum, C.; Shapiro, N.; Kyrpides, N.; et al. Genome Sequence of Verrucomicrobium sp. Strain GAS474, a Novel Bacterium Isolated from Soil. Genome Announc. 2018, 6, e01451-17. [Google Scholar] [CrossRef] [PubMed]
- Noraini, C.H.C.; Morad, N.; Norli, I.; Teng, T.T.; Ogugbue, C.J. Methylene Blue Degradation by Sphingomonas paucimobilis under Aerobic Conditions. Water Air Soil Pollut. 2012, 223, 5131–5142. [Google Scholar] [CrossRef]
- Ravintheran, S.K.; Sivaprakasam, S.; Loke, S.; Lee, S.Y.; Manickam, R.; Yahya, A.; Croft, L.; Millard, A.; Parimannan, S.; Rajandas, H. Complete genome sequence of Sphingomonas paucimobilis AIMST S2, a xenobiotic-degrading bacterium. Sci. Data 2019, 6, 280. [Google Scholar] [CrossRef]
- Worsley, S.F.; Newitt, J.; Rassbach, J.; Batey, S.F.D.; Holmes, N.A.; Murrell, J.C.; Wilkinson, B.; Hutchings, M.I. Streptomyces Endophytes Promote Host Health and Enhance Growth across Plant Species. Appl. Environ. Microbiol. 2020, 86, e01053-20. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Nobre, M.F.; Moore, E.; Rainey, F.A.; Battista, J.R.; da Costa, M.S. Characterization and radiation resistance of new isolates of Rubrobacter radiotolerans and Rubrobacter xylanophilus. Extrem. Life Extrem. Cond. 1999, 3, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–94. [Google Scholar] [CrossRef]
- White, P.J.; George, T.S.; Dupuy, L.X.; Karley, A.J.; Valentine, T.A.; Wiesel, L.; Wishart, J. Root traits for infertile soils. Front. Plant Sci. 2013, 4, 193. [Google Scholar] [CrossRef]
- Armengaud, P.; Breitling, R.; Amtmann, A. Coronatine-Insensitive 1 (COI1) Mediates Transcriptional Responses of Arabidopsis thaliana to External Potassium Supply. Mol. Plant 2010, 3, 309–405. [Google Scholar] [CrossRef]
- Troufflard, S.; Mullen, W.; Larson, T.R.; Graham, I.A.; Crozier, A.; Amtmann, A.; Armengaud, P. Potassium deficiency induces the biosynthesis of oxylipins and glucosinolates in Arabidopsis thaliana. BMC Plant Biol. 2010, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Meena, V.S.; Maurya, B.R.; Verma, J.P. Does a rhizospheric microorganism enhance K+ availability in agricultural soils? Microbiol. Res. 2014, 169, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Lv, L.; Wang, W.; Liu, Y.; Yin, C.; Xu, Q.; Yan, H.; Fu, J.; Liu, X. Differences in Distribution of Potassium-Solubilizing Bacteria in Forest and Plantation Soils in Myanmar. Int. J. Environ. Res. Public Health 2019, 16, 700. [Google Scholar] [CrossRef]
- Mark Ibekwe, A.; Ors, S.; Ferreira, J.F.S.; Liu, X.; Suarez, D.L. Seasonal induced changes in spinach rhizosphere microbial community structure with varying salinity and drought. Sci. Total. Environ. 2017, 579, 1485–1495. [Google Scholar] [CrossRef]
- Sun, S.; Gao, H.; Liu, Y.; Jin, L.; Wang, R.; Wang, X.; Wang, Q.; Yin, Y.; Zhang, Y.; Wang, H. Co-existence of a novel plasmid-mediated efflux pump with colistin resistance gene mcr in one plasmid confers transferable multidrug resistance in Klebsiella pneumoniae. Emerg. Microbes Infect. 2020, 9, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, J.; Huang, W.; Wu, H.; Chen, J.; Yang, Y.; Zhang, Z.; Lin, W. Plant-microbe rhizosphere interactions mediated by Rehmannia glutinosa root exudates under consecutive monoculture. Sci. Rep. 2015, 5, 15871. [Google Scholar] [CrossRef]
- Xia, Z.; Bai, E.; Wang, Q.; Gao, D.; Zhou, J.; Jiang, P.; Wu, J. Biogeographic Distribution Patterns of Bacteria in Typical Chinese Forest Soils. Front. Microbiol. 2016, 7, 1106. [Google Scholar] [CrossRef] [PubMed]
- Delgado Restrepo, O.M.; Menjivar Flores, J.C.; Muñoz Arboleda, F. Influence of management systems on the nitrogen mineralization and fertilization of sugarcane. Rev. Fac. Nac. Agron. Medellín 2016, 69, 7755–7762. [Google Scholar] [CrossRef]
- Inclan, R.; De la Torre, D.; Benito, M.; Rubio, A. Soil CO2 efflux in a mixed pine-oak forest in Valsain (central Spain). Sci. World J. 2007, 7 (Suppl. 1), 166–174. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.D. Soil and Agricultural Chemistry Analysis; Agricultural Press: Beijing, China, 2000; pp. 355–356. [Google Scholar]
- Wang, J.-H.; Lu, J.; Zhang, Y.-X.; Wu, J.; Luo, Y.; Liu, H. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 2018, 253, 235–243. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A.; Progra, N.C.S. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Shi, Y.; Fan, K.K.; Li, Y.T.; Yang, T.; He, J.S.; Chu, H.Y. Archaea Enhance the Robustness of Microbial Co-occurrence Networks in Tibetan Plateau Soils. Soil Sci. Soc. Am. J. 2019, 83, 1093–1099. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; He, X.; Sun, J.; Ma, Y. A Degeneration Gradient of Poplar Trees Contributes to the Taxonomic, Functional, and Resistome Diversity of Bacterial Communities in Rhizosphere Soils. Int. J. Mol. Sci. 2021, 22, 3438. https://doi.org/10.3390/ijms22073438
Liu J, He X, Sun J, Ma Y. A Degeneration Gradient of Poplar Trees Contributes to the Taxonomic, Functional, and Resistome Diversity of Bacterial Communities in Rhizosphere Soils. International Journal of Molecular Sciences. 2021; 22(7):3438. https://doi.org/10.3390/ijms22073438
Chicago/Turabian StyleLiu, Juan, Xiangwei He, Jingya Sun, and Yuchao Ma. 2021. "A Degeneration Gradient of Poplar Trees Contributes to the Taxonomic, Functional, and Resistome Diversity of Bacterial Communities in Rhizosphere Soils" International Journal of Molecular Sciences 22, no. 7: 3438. https://doi.org/10.3390/ijms22073438
APA StyleLiu, J., He, X., Sun, J., & Ma, Y. (2021). A Degeneration Gradient of Poplar Trees Contributes to the Taxonomic, Functional, and Resistome Diversity of Bacterial Communities in Rhizosphere Soils. International Journal of Molecular Sciences, 22(7), 3438. https://doi.org/10.3390/ijms22073438