
The ‘Jekyll and Hyde’ of Gluconeogenesis: Early Life Adversity, Later Life Stress, and Metabolic Disturbances



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Physiological Response to External Psychosocial Stressors
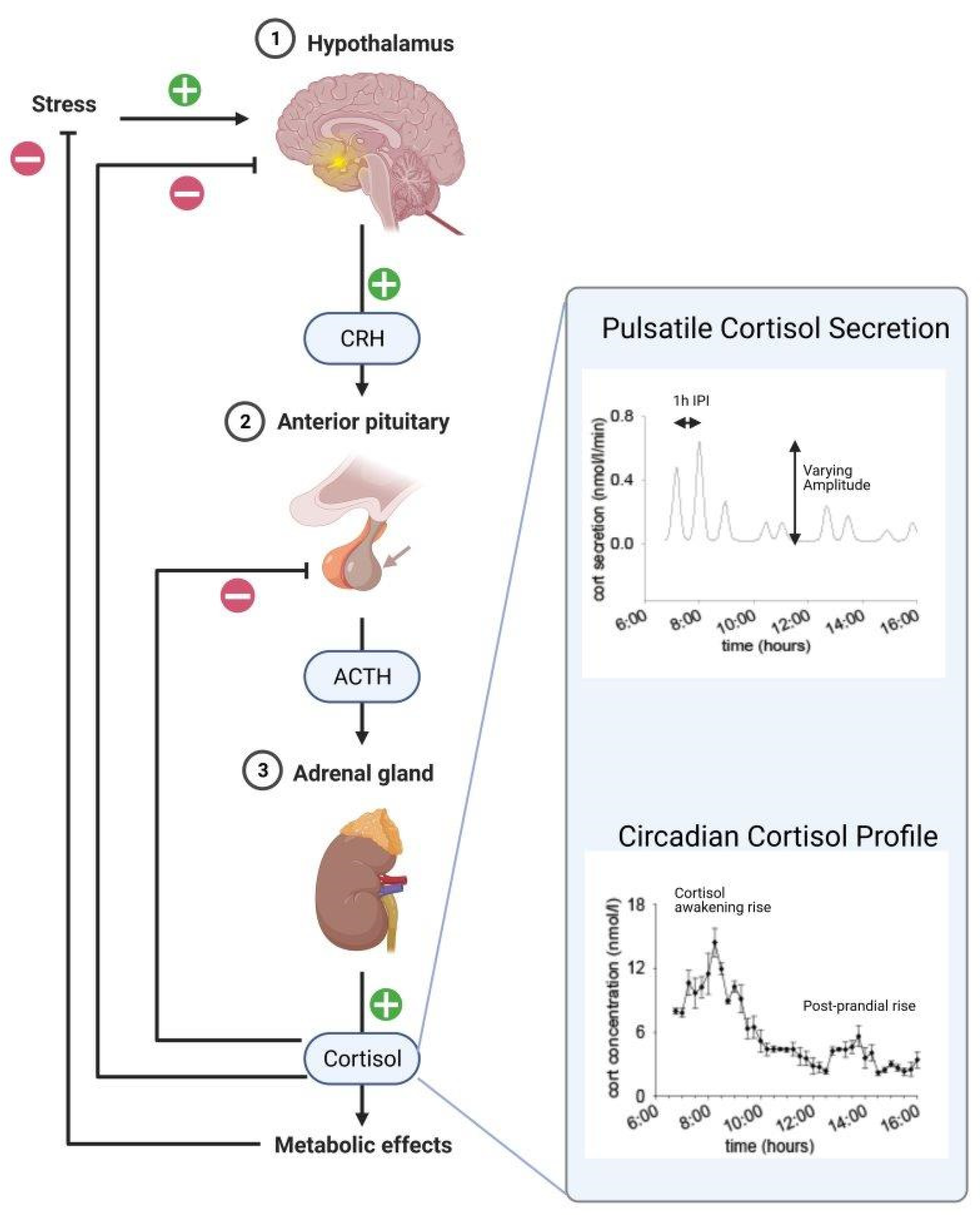
2.1. The ANS and the HPA Axis Stress Response
2.2. Metabolic Adaption
2.3. Metabolism and Acute Stress Interactions
2.4. Metabolism and Chronic Stress
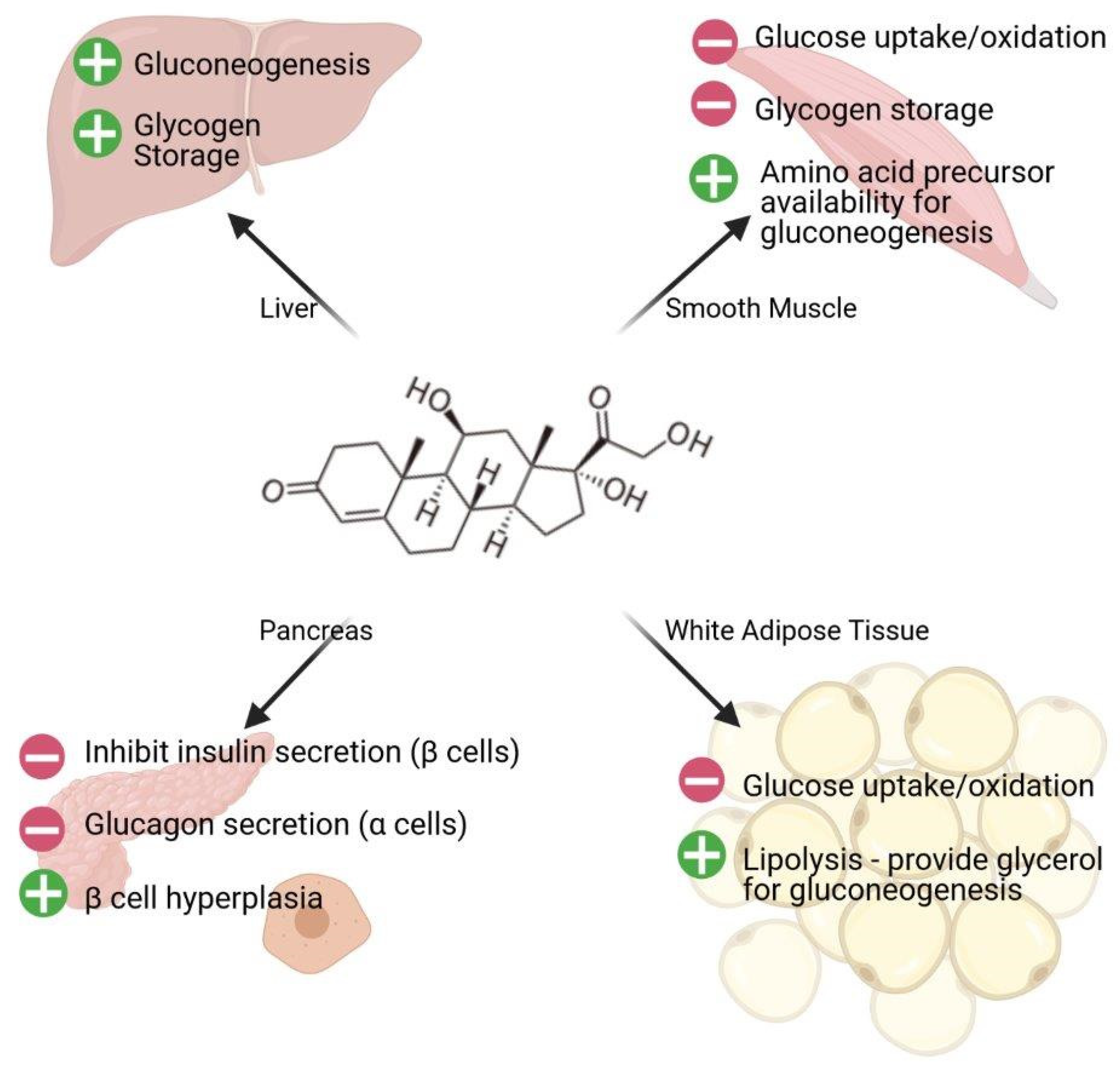
2.5. Glucocorticoids and Diabetes
3. Early Life Adversity
3.1. Early Life Adversity and Changes in the HPA Axis
3.2. Early Life Adversity, Diabetes and the Metabolic Syndrome
3.3. Glucose Metabolism, Allostasis and Allostatic Load
4. Gluconeogenesis at the Crossroads between Adversity and Metabolism
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandi, C.; Haller, J. Stress and the social brain: Behavioural effects and neurobiological mechanisms. Nat. Rev. Neurosci. 2015, 16, 290–304. [Google Scholar] [CrossRef]
- Herzig, S. Liver: A target of late diabetic complications. Exp. Clin. Endocrinol. Diabetes 2012, 120, 202–204. [Google Scholar] [CrossRef]
- de Guia, R.M.; Rose, A.J.; Herzig, S. Glucocorticoid hormones and energy homeostasis. Horm. Mol. Biol. Clin. Investig. 2014, 19, 117–128. [Google Scholar] [CrossRef]
- Nuttall, F.Q.; Ngo, A.; Gannon, M.C. Regulation of hepatic glucose production and the role of gluconeogenesis in humans: Is the rate of gluconeogenesis constant? Diabetes Metab. Res. Rev. 2008, 24, 438–458. [Google Scholar] [CrossRef]
- Konig, M.; Bulik, S.; Holzhutter, H.G. Quantifying the contribution of the liver to glucose homeostasis: A detailed kinetic model of human hepatic glucose metabolism. PLoS Comput. Biol. 2012, 8, e1002577. [Google Scholar] [CrossRef]
- ter Horst, G.J.; Luiten, P.G. The projections of the dorsomedial hypothalamic nucleus in the rat. Brain Res. Bull. 1986, 16, 231–248. [Google Scholar] [CrossRef]
- Cui, A.; Fan, H.; Zhang, Y.; Niu, D.; Liu, S.; Liu, Q.; Ma, W.; Shen, Z.; Shen, L.; Liu, Y.; et al. Dexamethasone-induced Kruppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J. Clin. Investig. 2019, 129, 2266–2278. [Google Scholar] [CrossRef]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C.; National Heart, Lung and Blood Institiute; American Heart Association. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Delpierre, C.; Fantin, R.; Barboza-Solis, C.; Lepage, B.; Darnaudéry, M.; Kelly-Irving, M. The early life nutritional environment and early life stress as potential pathways towards the metabolic syndrome in mid-life? A lifecourse analysis using the 1958 British Birth cohort. BMC Public Health 2016, 16, 815. [Google Scholar]
- Turner, J.D. Childhood adversity from conception onwards: Are our tools unnecessarily hindering us? J. Behav. Med. 2018, 41, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Suglia, S.F.; Koenen, K.C.; Boynton-Jarrett, R.; Chan, P.S.; Clark, C.J.; Danese, A.; Faith, M.S.; Goldstein, B.I.; Hayman, L.L.; Isasi, C.R.; et al. Childhood and adolescent adversity and cardiometabolic outcomes: A scientific statement from the american heart association. Circulation 2018, 137, e15–e28. [Google Scholar] [CrossRef]
- Jaaskelainen, P.; Magnussen, C.G.; Pahkala, K.; Mikkila, V.; Kakohen, M.; Sabin, M.A.; Fogelholm, M.; Hutri-Kakohen, N.; Taittonen, L.; Telama, R.; et al. Childhood nutrition in predicting metabolic syndrome in adults: The cardiovascular risk in Young Finns Study. Diabetes Care 2012, 35, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Gariepy, G.; Gavin, A.R.; Rowhani-Rahbar, A.; Siscovick, D.S.; Enquobahrie, D.A. Maternal education in early life and risk of metabolic syndrome in young adult american females and males: Disentangling life course processes through causal models. Epidemiology 2019, 30, S28–S36. [Google Scholar] [CrossRef]
- Afifi, T.O.; MacMillan, H.L.; Boyle, M.; Cheung, K.; Taillieu, T.; Turner, S.; Sareen, J. Child abuse and physical health in adulthood. Health Rep. 2016, 27, 10–18. [Google Scholar] [PubMed]
- Hostinar, C.E.; Ross, K.M.; Chan, E.; Miller, G.E. Early-life socioeconomic disadvantage and metabolic health disparities. Psychosom. Med. 2017, 79, 514–523. [Google Scholar] [CrossRef]
- Tomasdottir, M.O.; Sigurdsson, J.A.; Petursson, H.; Kirkengen, A.L.; Krokstad, S.; McEwen, B.; Hetlevik, I.; Getz, L. Self reported childhood difficulties, adult multimorbidity and allostatic load. A cross-sectional analysis of the Norwegian HUNT study. PLoS ONE 2015, 10, e0130591. [Google Scholar] [CrossRef]
- Danese, A.; Tan, M. Childhood maltreatment and obesity: Systematic review and meta-analysis. Mol. Psychiatry 2014, 19, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yan, P.; Shan, Z.; Li, M.; Luo, C.; Gao, H.; Hao, L.; Liu, L. Adverse childhood experiences and risk of type 2 diabetes: A systematic review and meta-analysis. Metabolism 2015, 64, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Rattanatray, L.; Morrison, J.L.; Nicholas, L.M.; Lie, S.; McMillen, I.C. Maternal obesity and the early origins of childhood obesity: Weighing up the benefits and costs of maternal weight loss in the periconceptional period for the offspring. Exp. Diabetes Res. 2011, 2011, 585749. [Google Scholar] [CrossRef]
- Chrousos, G.P.; Gold, P.W. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 1992, 267, 1244–1252. [Google Scholar] [CrossRef]
- Tsigos, C.; Chrousos, G.P. Physiology of the hypothalamic-pituitary-adrenal axis in health and dysregulation in psychiatric and autoimmune disorders. Endocrinol. Metab. Clin. N. Am. 1994, 23, 451–466. [Google Scholar] [CrossRef]
- Rotenberg, S.; McGrath, J.J. Inter-relation between autonomic and HPA axis activity in children and adolescents. Biol. Psychol. 2016, 117, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Yang, T.Z. The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J. Comp. Neurol. 1985, 242, 56–92. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.I.; Coote, J.H. Excitation and inhibition of rat sympathetic preganglionic neurones by catecholamines. Brain Res. 1990, 530, 229–234. [Google Scholar] [CrossRef]
- Unnerstall, J.R.; Kopajtic, T.A.; Kuhar, M.J. Distribution of alpha 2 agonist binding sites in the rat and human central nervous system: Analysis of some functional, anatomic correlates of the pharmacologic effects of clonidine and related adrenergic agents. Brain Res. 1984, 319, 69–101. [Google Scholar] [CrossRef]
- Reiche, E.M.; Nunes, S.O.; Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Ito, R.; Lee, A.C.H. The role of the hippocampus in approach-avoidance conflict decision-making: Evidence from rodent and human studies. Behav. Brain Res. 2016, 313, 345–357. [Google Scholar] [CrossRef]
- Fee, C.; Prevot, T.; Misquitta, K.; Banasr, M.; Sibille, E. Chronic stress-induced behaviors correlate with exacerbated acute stress-induced cingulate cortex and ventral hippocampus activation. Neuroscience 2020, 440, 113–129. [Google Scholar] [CrossRef]
- Jankord, R.; Herman, J.P. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann. N. Y. Acad. Sci. 2008, 1148, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Nixon, M.; Mackenzie, S.D.; Taylor, A.I.; Homer, N.Z.M.; Livingstone, D.E.; Mouras, R.; Morgan, R.A.; Mole, D.J.; Stimson, R.H.; Reynolds, R.M. ABCC1 confers tissue-specific sensitivity to cortisol versus corticosterone: A rationale for safer glucocorticoid replacement therapy. Sci. Transl. Med. 2016, 8, 352ra109. [Google Scholar] [CrossRef] [PubMed]
- Wang, M. The role of glucocorticoid action in the pathophysiology of the metabolic syndrome. Nutr. Metab. (Lond.) 2005, 2, 3. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef]
- Antoni, F.A. Hypothalamic control of adrenocorticotropin secretion: Advances since the discovery of 41-residue corticotropin-releasing factor. Endocr. Rev. 1986, 7, 351–378. [Google Scholar] [CrossRef]
- Tsigos, C.; Chrousos, G.P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Stavreva, D.A.; Wiench, M.; John, S.; Conway-Campbell, B.L.; McKenna, M.A.; Pooley, J.R.; Johnson, T.A.; Lightman, T.C.V.; Hager, G.L. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat. Cell. Biol. 2009, 11, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Trifonova, S.T.; Gantenbein, M.; Turner, J.D.; Muller, C.P. The use of saliva for assessment of cortisol pulsatile secretion by deconvolution analysis. Psychoneuroendocrinology 2013, 38, 1090–1101. [Google Scholar] [CrossRef]
- Lightman, S.L.; Wiles, C.C.; Atkinson, H.C.; Henley, D.E.; Russell, G.M.; Leendertz, J.A.; McKenna, M.A.; Spiga, F.; Wood, S.A.; Conway-Campbell, B.L. The significance of glucocorticoid pulsatility. Eur. J. Pharmacol. 2008, 583, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.J.; Terry, J.R.; Lightman, S.L. Origin of ultradian pulsatility in the hypothalamic-pituitary-adrenal axis. Proc. Biol. Sci. 2010, 277, 1627–1633. [Google Scholar] [CrossRef]
- Sapolsky, R.M.; Meaney, M.J. Maturation of the adrenocortical stress response: Neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res. 1986, 396, 64–76. [Google Scholar] [CrossRef]
- Romeo, R.D. The metamorphosis of adolescent hormonal stress reactivity: A focus on animal models. Front. Neuroendocrinol. 2018, 49, 43–51. [Google Scholar] [CrossRef]
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of glucose homeostasis by glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell. Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Gartner, K.; Buttner, D.; Dohler, K.; Friedel, R.; Lindena, J.; Trautschold, I. Stress response of rats to handling and experimental procedures. Lab. Anim. 1980, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Nirupama, R.; Devaki, M.; Yajurvedi, H.N. Chronic stress and carbohydrate metabolism: Persistent changes and slow return to normalcy in male albino rats. Stress 2012, 15, 262–271. [Google Scholar] [CrossRef]
- Wu, P.; Sato, J.; Zhao, Y.; Jaskiewicz, J.; Popov, K.M.; Harris, R.A. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 1998, 329, 197–201. [Google Scholar] [CrossRef]
- Sato, T.; Yamamoto, H.; Sawada, N.; Nashiki, K.; Tsuji, M.; Muto, K.; Kume, H.; Sasiki, H.; Arai, H.; Nikawa, T.; et al. Restraint stress alters the duodenal expression of genes important for lipid metabolism in rat. Toxicology 2006, 227, 248–261. [Google Scholar] [CrossRef]
- Foley, P.; Kirschbaum, C. Human hypothalamus-pituitary-adrenal axis responses to acute psychosocial stress in laboratory settings. Neurosci. Biobehav. Rev. 2010, 35, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, C.; Bono, E.G.; Rohleder, N.; Gessner, C.; Pirke, K.M.; Salvador, A.; Hellhammer, D.H. Effects of fasting and glucose load on free cortisol responses to stress and nicotine. J. Clin. Endocrinol. Metab. 1997, 82, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bono, E.; Rohleder, N.; Hellhammer, D.H.; Salvador, A.; Kirschbaum, C. Glucose but not protein or fat load amplifies the cortisol response to psychosocial stress. Horm. Behav. 2002, 41, 328–333. [Google Scholar] [CrossRef]
- Choi, S.; Horsley, C.; Aguilla, S.; Dallman, M.F. The hypothalamic ventromedial nuclei couple activity in the hypothalamo-pituitary-adrenal axis to the morning fed or fasted state. J. Neurosci. 1996, 16, 8170–8180. [Google Scholar] [CrossRef]
- Rosmond, R.; Holm, G.; Bjorntorp, P. Food-induced cortisol secretion in relation to anthropometric, metabolic and haemodynamic variables in men. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 416–422. [Google Scholar] [CrossRef]
- Bergendahl, M.; Iranmanesh, A.; Evans, W.S.; Veldhuis, J.D. Short-term fasting selectively suppresses leptin pulse mass and 24-hour rhythmic leptin release in healthy midluteal phase women without disturbing leptin pulse frequency or its entropy control (pattern orderliness). J. Clin. Endocrinol. Metab. 2000, 85, 207–213. [Google Scholar]
- Sherwin, R.S.; Sacca, L. Effect of epinephrine on glucose metabolism in humans: Contribution of the liver. Am. J. Physiol. 1984, 247, E157–E165. [Google Scholar] [CrossRef]
- Burgess, S.C.; He, T.; Yan, Z.; Lindner, J.; Sherry, A.D.; Malloy, C.R.; Browning, J.D.; Magnuson, M.A. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell. Metab. 2007, 5, 313–320. [Google Scholar] [CrossRef]
- Opherk, C.; Tronche, F.; Kellendonk, C.; Kohlmuller, D.; Schulze, A.; Schimd, W.; Schutz, G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol. Endocrinol. 2004, 18, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E685–E692. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nakagawa, Y.; Wang, Y.; Sakurai, R.; Tripathi, P.V.; Lutfy, K.; Friedman, T.C. Increased glucocorticoid receptor and 11{Betancur, #1833}-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to the phenotype of type 2 diabetes in db/db mice. Diabetes 2005, 54, 32–40. [Google Scholar] [PubMed]
- Lee, D.; Le Lay, J.; Kaestner, K.H. The transcription factor CREB has no non-redundant functions in hepatic glucose metabolism in mice. Diabetologia 2014, 57, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Basu, A.K.; Mandal, B.; Mukhopadhyay, P.; Maity, A.; Chakraborty, S.; Devrabhai, P.K. 11beta Hydroxysteroid dehydrogenase-1 activity in type 2 diabetes mellitus: A comparative study. BMC Endocr. Disord. 2019, 19, 15. [Google Scholar] [CrossRef]
- Granner, D.K. In pursuit of genes of glucose metabolism. J. Biol. Chem. 2015, 290, 22312–22324. [Google Scholar] [CrossRef] [PubMed]
- Schacke, H.; Docke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Blondeau, B.; Sahly, I.; Massourides, E.; Singh-Estivalet, A.; Valtat, B.; Dorchene, D.; Jaisser, F.; Breant, B.; Tronche, F. Novel transgenic mice for inducible gene overexpression in pancreatic cells define glucocorticoid receptor-mediated regulations of beta cells. PLoS ONE 2012, 7, e30210. [Google Scholar] [CrossRef]
- Buren, J.; Lai, Y.C.; Lundgren, M.; Eriksson, J.W.; Jensen, J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch. Biochem. Biophys. 2008, 474, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Karnia, M.J.; Myslinska, D.; Dzik, K.P.; Flis, D.J.; Podlacha, M.; Kaczor, J.J. BST stimulation induces atrophy and changes in aerobic energy metabolism in rat skeletal muscles-the biphasic action of endogenous glucocorticoids. Int. J. Mol. Sci. 2020, 21, 2787. [Google Scholar] [CrossRef]
- Chiodini, I.; Adda, G.; Scillitani, A.; Coletti, F.; Morelli, V.; Di Lembo, S.; Epaminonda, P.; Masserini, B.; Beck-Peccoz, P.; Orsi, E.; et al. Cortisol secretion in patients with type 2 diabetes: Relationship with chronic complications. Diabetes Care 2007, 30, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Nouwen, A.; Winkley, K.; Twisk, J.; Lloyd, C.E.; Peyrot, M.; Ismail, K.; Pouwer, F.; European Depression in Diabetes (EDID) Research Consortium. Type 2 diabetes mellitus as a risk factor for the onset of depression: A systematic review and meta-analysis. Diabetologia 2010, 53, 2480–2486. [Google Scholar] [CrossRef]
- Mosili, P.; Mchize, B.C.; Ngubane, P.; Sibiya, N.; Khathi, A. The dysregulation of the hypothalamic-pituitary-adrenal axis in diet-induced prediabetic male Sprague Dawley rats. Nutr. Metab. (Lond.) 2020, 17, 104. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar]
- Swierczynska, M.M.; Mateska, I.; Peitzsch, M.; Bornstein, S.R.; Chavakis, T.; Eisenhofer, G.; Lamounier-Zepter, V.; Eaton, S. Changes in morphology and function of adrenal cortex in mice fed a high-fat diet. Int. J. Obes. (Lond.) 2015, 39, 321–330. [Google Scholar] [CrossRef]
- Shu, H.J.; Isenberg, K.; Cormier, R.J.; Benz, A.; Zorumski, C.F. Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience 2006, 140, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.S.; Burgado, J.; Kelly, S.D.; Johnson, Z.P.; Neigh, G.N. High-fructose diet during periadolescent development increases depressive-like behavior and remodels the hypothalamic transcriptome in male rats. Psychoneuroendocrinology 2015, 62, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease, Control and Prevention (CDC). Adverse childhood experiences reported by adults—Five states, 2009. MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 1609–1613. [Google Scholar]
- van Bodegom, M.; Homberg, J.R.; Henckens, M. Modulation of the hypothalamic-pituitary-adrenal axis by early life stress exposure. Front. Cell. Neurosci. 2017, 11, 87. [Google Scholar] [CrossRef]
- Gutteling, B.M.; de Weerth, C.; Buitelaar, J.K. Prenatal stress and children’s cortisol reaction to the first day of school. Psychoneuroendocrinology 2005, 30, 541–549. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.G.; Ben-Shlomo, Y.; Heron, J.; Golding, J.; Adams, D.; Glover, V. Prenatal anxiety predicts individual differences in cortisol in pre-adolescent children. Biol. Psychiatry 2005, 58, 211–217. [Google Scholar] [CrossRef]
- Davies, P.T.; Sturge-Apple, M.L.; Cicchetti, D.; Manning, L.G.; Zale, E. Children’s patterns of emotional reactivity to conflict as explanatory mechanisms in links between interpartner aggression and child physiological functioning. J. Child Psychol. Psychiatry 2009, 50, 1384–1391. [Google Scholar] [CrossRef]
- Bugental, D.B.; Martorell, G.A.; Barraza, V. The hormonal costs of subtle forms of infant maltreatment. Horm. Behav. 2003, 43, 237–244. [Google Scholar] [CrossRef]
- Carlson, M.; Earls, F. Psychological and neuroendocrinological sequelae of early social deprivation in institutionalized children in Romania. Ann. N. Y. Acad. Sci. 1997, 807, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Zwerling, J.; Dozier, M. Effects of early adversity on young children’s diurnal cortisol rhythms and externalizing behavior. Dev. Psychobiol. 2015, 57, 935–947. [Google Scholar] [CrossRef]
- Hengesch, X.; Elwenspoek, M.M.C.; Schaan, V.K.; Larra, M.F.; Finke, J.B.; Zhang, X.; Bachmann, P.; Turner, J.D.; Vogele, C.; Muller, C.P.; et al. Blunted endocrine response to a combined physical-cognitive stressor in adults with early life adversity. Child Abuse Negl. 2018, 85, 137–144. [Google Scholar] [CrossRef]
- Pesonen, A.K.; Raikkonen, K.; Feldt, K.; Heinnonen, K.; Osmond, C.; Philips, D.I.W.; Barker, D.J.P.; Eriksson, J.G.; Kajantie, E. Childhood separation experience predicts HPA axis hormonal responses in late adulthood: A natural experiment of World War II. Psychoneuroendocrinology 2010, 35, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Fries, E.; Hesse, J.; Hellhammer, J.; Hellhammer, D.H. A new view on hypocortisolism. Psychoneuroendocrinology 2005, 30, 1010–1016. [Google Scholar] [CrossRef]
- Yehuda, R.; Yang, R.-K.; Buchsbaum, M.S.; Golier, J.A. Alterations in cortisol negative feedback inhibition as examined using the ACTH response to cortisol administration in PTSD. Psychoneuroendocrinology 2006, 31, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Elwenspoek, M.M.C.; Hengesch, X.; Leenen, F.A.D.; Sias, K.; Fernandes, S.B.; Schaan, V.K.; Meriaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schachinger, H.; et al. Glucocorticoid receptor signaling in leukocytes after early life adversity. Dev. Psychopathol. 2019, 32, 1–11. [Google Scholar] [CrossRef]
- Teicher, M.H.; Tomoda, A.; Andersen, S. Neurobiological consequences of early stress and childhood maltreatment: Are results from human and animal studies comparable? Ann. N. Y. Acad. Sci. 2006, 1071, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Tomada, A.; Vincow, E.S.; Valente, E.; Polcari, A.; Teicher, M.H. Preliminary evidence for sensitive periods in the effect of childhood sexual abuse on regional brain development. J. Neuropsychiatry Clin. Neurosci. 2008, 20, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Maercker, A.; Michael, T.; Fehm, L.; Becker, E.S.; Margraf, J. Age of traumatisation as a predictor of post-traumatic stress disorder or major depression in young women. Br. J. Psychiatry 2004, 184, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Granberg, R.; Tseng, K.Y. Mechanisms contributing to prefrontal cortex maturation during adolescence. Neurosci. Biobehav. Rev. 2016, 70, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Starr, L.R.; Dienes, K.; Stroud, C.B.; Shaw, Z.A.; Li, Y.I.; Mlawer, F.; Huang, M. Childhood adversity moderates the influence of proximal episodic stress on the cortisol awakening response and depressive symptoms in adolescents. Dev. Psychopathol. 2017, 29, 1877–1893. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, N.; Canli, T. Early life stress and cortisol: A meta-analysis. Horm. Behav. 2018, 98, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Bunea, I.M.; Szentagotai-Tatar, A.; Miu, A.C. Early-life adversity and cortisol response to social stress: A meta-analysis. Transl. Psychiatry 2017, 7, 1274. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.; Okoloise, M.; Williams, K.; Stern, M.P.; Haffner, S.M.; San Antonio Heart Study. The metabolic syndrome as predictor of type 2 diabetes: The San Antonio heart study. Diabetes Care 2003, 26, 3153–3159. [Google Scholar] [CrossRef]
- Rich-Edwards, J.W.; Spiegelman, D.; Lividoti Hibert, E.N.; Jun, H.-J.; Todd, T.J.; Kawachi, I.; Wright, R.J. Abuse in childhood and adolescence as a predictor of type 2 diabetes in adult women. Am. J. Prev. Med. 2010, 39, 529–536. [Google Scholar] [CrossRef]
- Boynton-Jarrett, R.; Rosenberg, L.; Palmer, J.R.; Boggs, D.A.; Wise, L.A. Child and adolescent abuse in relation to obesity in adulthood: The black women’s health study. Pediatrics 2012, 130, 245–253. [Google Scholar] [CrossRef]
- Elwenspoek, M.M.C.; Hengesch, X.; Leenen, F.A.D.; Schritz, A.; Sias, K.; Schaan, V.K.; Meriaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schachinger, H.; et al. Proinflammatory T cell status associated with early life adversity. J. Immunol. 2017, 199, 4046–4055. [Google Scholar] [CrossRef]
- Horner, E.M.; Strombotne, K.; Huang, A.; Lapham, S. Investigating the early life determinants of type-II diabetes using a project talent-medicare linked data-set. SSM Popul. Health 2018, 4, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Elwenspoek, M.M.C.; Kuehn, A.; Muller, C.P.; Turner, J.D. The effects of early life adversity on the immune system. Psychoneuroendocrinology 2017, 82, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Elwenspoek, M.M.C.; Sias, K.; Hengesch, X.; Schaan, V.K.; Leenen, F.A.D.; Adam, P.; Meriaux, S.B.; Schmitz, S.; Bonnemberger, F.; Ewen, A.; et al. T cell immunosenescence after early life adversity: Association with Cytomegalovirus infection. Front. Immunol. 2017, 8, 1263. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Coe, C.L.; Doyle, C.M.; Sheerar, D.; Slukvina, A.; Donzella, B.; Gunnar, M.R. Persistent skewing of the T-cell profile in adolescents adopted internationally from institutional care. Brain Behav. Immun. 2019, 77, 168–177. [Google Scholar] [CrossRef]
- Slopen, N.; Lewis, T.T.; Gruenewald, T.L.; Mujahid, M.S.; Ryff, C.D.; Albert, M.A.; Williams, D.R. Early life adversity and inflammation in African Americans and whites in the midlife in the United States survey. Psychosom. Med. 2010, 72, 694–701. [Google Scholar] [CrossRef]
- Hostinar, C.E.; Lachman, M.E.; Mroczek, D.K.; Seeman, T.E.; Miller, G.E. Additive contributions of childhood adversity and recent stressors to inflammation at midlife: Findings from the MIDUS study. Dev. Psychol. 2015, 51, 1630–1644. [Google Scholar] [CrossRef]
- Klassen, S.A.; Chirico, D.; O’Leary, D.D.; Cairney, J.; Wade, T.J. Linking systemic arterial stiffness among adolescents to adverse childhood experiences. Child Abuse Negl. 2016, 56, 1–10. [Google Scholar] [CrossRef]
- Su, S.; Wang, X.; Pollock, J.S.; Treiber, F.A.; Xu, X.; Snieder, H.; McCall, W.V.; Stefanek, M.; Harshfield, G.A. Adverse childhood experiences and blood pressure trajectories from childhood to young adulthood: The Georgia stress and Heart study. Circulation 2015, 131, 1674–1681. [Google Scholar] [CrossRef]
- Chandan, J.S.; Okoth, K.; Gokhale, K.M.; Bandyopadhyay, S.; Taylor, J.; Nirantharakumar, K. Increased cardiometabolic and mortality risk following childhood maltreatment in the United Kingdom. J. Am. Heart Assoc. 2020, 9, e015855. [Google Scholar] [CrossRef] [PubMed]
- Holuka, C.; Merz, M.P.; Fernandes, S.B.; Charalambous, E.G.; Seal, S.V.; Grova, N.; Turner, J.D. The COVID-19 pandemic: Does Our early life environment, life trajectory and socioeconomic status determine disease susceptibility and severity? Int. J. Mol. Sci. 2020, 21, 5094. [Google Scholar] [CrossRef]
- Joung, K.E.; Park, K.-H.; Zaichenko, L.; Sahin-Efe, A.; Thakkar, B.; Brinkotter, M.; Usher, N.; Warner, D.; David, C.R.; Crowell, J.A.; et al. Early life adversity is associated with elevated levels of circulating leptin, irisin, and decreased levels of adiponectin in midlife adults. J. Clin. Endocrinol. Metab. 2014, 99, E1055–E1060. [Google Scholar] [CrossRef] [PubMed]
- Mantzoros, C.S.; Magkos, F.; Brinkoetter, M.; Sienkiewicz, E.; Dardeno, T.A.; Kim, S.-Y.; Hamnvik, O.-P.R.; Koniaris, A. Leptin in human physiology and pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E567–E584. [Google Scholar] [CrossRef]
- Yang, W.S.; Lee, W.J.; Funahashi, T.; Tanaka, S.; Matsuzawa, Y.; Chao, C.L.; Chen, C.L.; Tai, T.Y.; Chuang, L.M. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J. Clin. Endocrinol. Metab. 2001, 86, 3815–3819. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Mohlig, M.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F.H. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003, 361, 226–228. [Google Scholar] [CrossRef]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Naumova, O.Y.; Rychkov, S.Y.; Kornilov, S.A.; Odintsova, V.V.; Anikina, V.O.; Solodunova, M.Y.; Arintcina, I.A.; Zhukova, M.A.; Ovchinnikova, I.V.; Burenkova, O.V.; et al. Effects of early social deprivation on epigenetic statuses and adaptive behavior of young children: A study based on a cohort of institutionalized infants and toddlers. PLoS ONE 2019, 14, e0214285. [Google Scholar] [CrossRef]
- Suderman, M.; Borghol, N.; Pappas, J.J.; Pereira, S.M.P.; Pembrey, M.; Hertzman, C.; Power, C.; Szyf, M. Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med. Genom. 2014, 7, 13. [Google Scholar] [CrossRef]
- Needham, B.L.; Smith, J.A.; Zhao, W.; Wang, X.; Mukherjee, B.; Kardia, S.L.R.; Shively, C.A.; Seeman, T.E.; Liu, Y.; Roux, A.V.D. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: The multi-ethnic study of atherosclerosis. Epigenetics 2015, 10, 958–969. [Google Scholar] [CrossRef]
- Wu, L.H.; Huang, C.C.; Adhikarahunnathu, S.; Mateo, L.R.S.; Duffy, K.E.; Rafferty, P.; Bugelski, P.; Raymond, H.; Deutsch, H.; Picha, K.; et al. Loss of toll-like receptor 3 function improves glucose tolerance and reduces liver steatosis in obese mice. Metabolism 2012, 61, 1633–1645. [Google Scholar] [CrossRef]
- Truax, A.D.; Chen, L.; Tam, J.W.; Cheng, N.; Guo, H.; Koblansky, A.A.; Chou, W.-C.; Wilson, J.E.; Brickey, W.J.; Petrucelli, A.; et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell. Host Microbe 2018, 24, 364–378. [Google Scholar] [CrossRef]
- Jackson, M.; Marks, L.; May, G.H.W.; Wilson, J.B. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723. [Google Scholar] [CrossRef]
- Long, S.A.; Thorpe, J.; DeBerg, H.A.; Gersuk, V.; Eddy, J.; Harris, K.M.; Ehlers, M.; Herold, K.C.; Nepom, G.T.; Linsley, P.S. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol. 2016, 1, 7793. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, R.; Zhu, S.; Fu, S.; Chen, Z.; Zhou, R.; Tian, Z.; Bai, L. M2-specific reduction of CD1d switches NKT cell-mediated immune responses and triggers metaflammation in adipose tissue. Cell. Mol. Immunol. 2018, 15, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.T.; Uimari, P.; Aalto, J.-M.; Pirskanen, M.; Kaikkonen, J.; Todorova, B.; Hypponen, J.; Korhonen, V.-P.; Asikainen, J.; Devine, C.; et al. Type 2 diabetes whole-genome association study in four populations: The DiaGen consortium. Am. J. Hum. Genet. 2007, 81, 338–345. [Google Scholar] [CrossRef]
- Sidibeh, C.O.; Pereira, M.J.; Abalo, X.M.; Boersma, G.J.; Skrtic, S.; Lundkvist, P.; Katsogiannos, P.; Hausch, F.; Cartillejo-Lopez, C.; Errikson, J.W. FKBP5 expression in human adipose tissue: Potential role in glucose and lipid metabolism, adipogenesis and type 2 diabetes. Endocrine 2018, 62, 116–128. [Google Scholar] [CrossRef]
- Carroll, H.A.; James, L.J. Hydration, arginine vasopressin, and glucoregulatory health in humans: A critical perspective. Nutrients 2019, 11, 1201. [Google Scholar] [CrossRef]
- Pisto, L.; Vaden, A.; Sillanmaki, L.; Mattila, K. Childhood adversities are associated with diabetes management in working age in Finland. Int. J. Family Med. 2014, 2014, 864572. [Google Scholar] [CrossRef]
- Selye, H. A syndrome produced by diverse nocuous agents. 1936. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S.; Meaney, M.J.; Sapolsky, R.M.; Spencer, R.L. Reflections on Bruce, S. McEwen’s contributions to stress neurobiology and so much more. Stress 2020, 23, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cepero, A.; Rosal, M.C.; Frisard, C.; Person, S.; Ockene, I.; Tucker, K.L. Changes in glycemic load are positively associated with small changes in primary stress markers of allostatic load in Puerto Rican women. J. Nutr. 2020, 150, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Young, J.B.; Landsberg, L. Stimulation of the sympathetic nervous system during sucrose feeding. Nature 1977, 269, 615–617. [Google Scholar] [CrossRef]
- Schmidt, M.V.; Levine, S.; Alam, S.; Sterlemann, V.; Ganea, K.; de Kloet, E.R.; Holsboer, F.; Muller, M.B. Metabolic signals modulate hypothalamic-pituitary-adrenal axis activation during maternal separation of the neonatal mouse. J. Neuroendocrinol. 2006, 18, 865–874. [Google Scholar] [CrossRef] [PubMed]
- von Dawans, B.; Zimmer, P.; Domes, G. Effects of glucose intake on stress reactivity in young, healthy men. Psychoneuroendocrinology 2020, 126, 105062. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Bagot, R.C.; Parker, K.J.; Vinkers, C.H.; de Kloet, E.R. The three-hit concept of vulnerability and resilience: Toward understanding adaptation to early-life adversity outcome. Psychoneuroendocrinology 2013, 38, 1858–1873. [Google Scholar] [CrossRef] [PubMed]
- Grova, N.; Schroeder, H.; Olivier, J.-L.; Turner, J.D. Epigenetic and neurological impairments associated with early life exposure to persistent organic pollutants. Int. J. Genom. 2019, 2019, 2085496. [Google Scholar] [CrossRef]
- de Jong, M.; Lafeber, H.N.; Cranendonk, A.; van Weissenbruch, M.M. Components of the metabolic syndrome in early childhood in very-low-birth-weight infants. Horm. Res. Paediatr. 2014, 81, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Finken, M.J.; van der Voorn, B.; Heijboer, A.C.; de Waard, M.; van Goudoever, J.B.; Rotteveel, J. Glucocorticoid programming in very preterm birth. Horm. Res. Paediatr. 2016, 85, 221–231. [Google Scholar] [CrossRef]
- Vargas, J.; Junco, M.; Gomez, C.; Lajud, N. Early life stress increases metabolic risk, HPA axis reactivity, and depressive-like behavior when combined with postweaning social isolation in rats. PLoS ONE 2016, 11, e0162665. [Google Scholar] [CrossRef]
- Yau, Y.H.; Potenza, M.N. Stress and eating behaviors. Minerva Endocrinol. 2013, 38, 255–267. [Google Scholar] [PubMed]
- Cohen, J.I. Stress and mental health: A biobehavioral perspective. Issues Ment. Health Nurs. 2000, 21, 185–202. [Google Scholar] [CrossRef]
- Serchov, T.; van Calker, D.; Biber, K. Sucrose preference test to measure anhedonic behaviour in mice. Bio Protoc. 2016, 6, e1958. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seal, S.V.; Turner, J.D. The ‘Jekyll and Hyde’ of Gluconeogenesis: Early Life Adversity, Later Life Stress, and Metabolic Disturbances. Int. J. Mol. Sci. 2021, 22, 3344. https://doi.org/10.3390/ijms22073344
Seal SV, Turner JD. The ‘Jekyll and Hyde’ of Gluconeogenesis: Early Life Adversity, Later Life Stress, and Metabolic Disturbances. International Journal of Molecular Sciences. 2021; 22(7):3344. https://doi.org/10.3390/ijms22073344
Chicago/Turabian StyleSeal, Snehaa V., and Jonathan D. Turner. 2021. "The ‘Jekyll and Hyde’ of Gluconeogenesis: Early Life Adversity, Later Life Stress, and Metabolic Disturbances" International Journal of Molecular Sciences 22, no. 7: 3344. https://doi.org/10.3390/ijms22073344
APA StyleSeal, S. V., & Turner, J. D. (2021). The ‘Jekyll and Hyde’ of Gluconeogenesis: Early Life Adversity, Later Life Stress, and Metabolic Disturbances. International Journal of Molecular Sciences, 22(7), 3344. https://doi.org/10.3390/ijms22073344