
The Role of Rigid Residues in Modulating TEM-1 β-Lactamase Function and Thermostability

 ,
,  , and
, and
Abstract
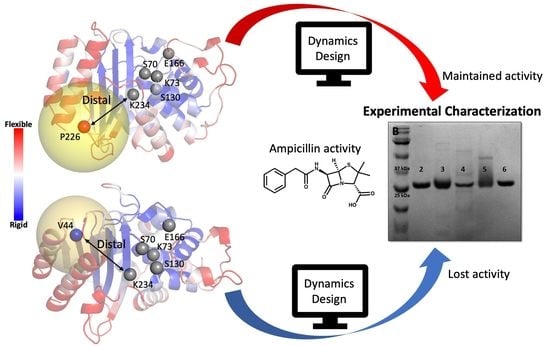
:
1. Introduction
2. Results and Discussion
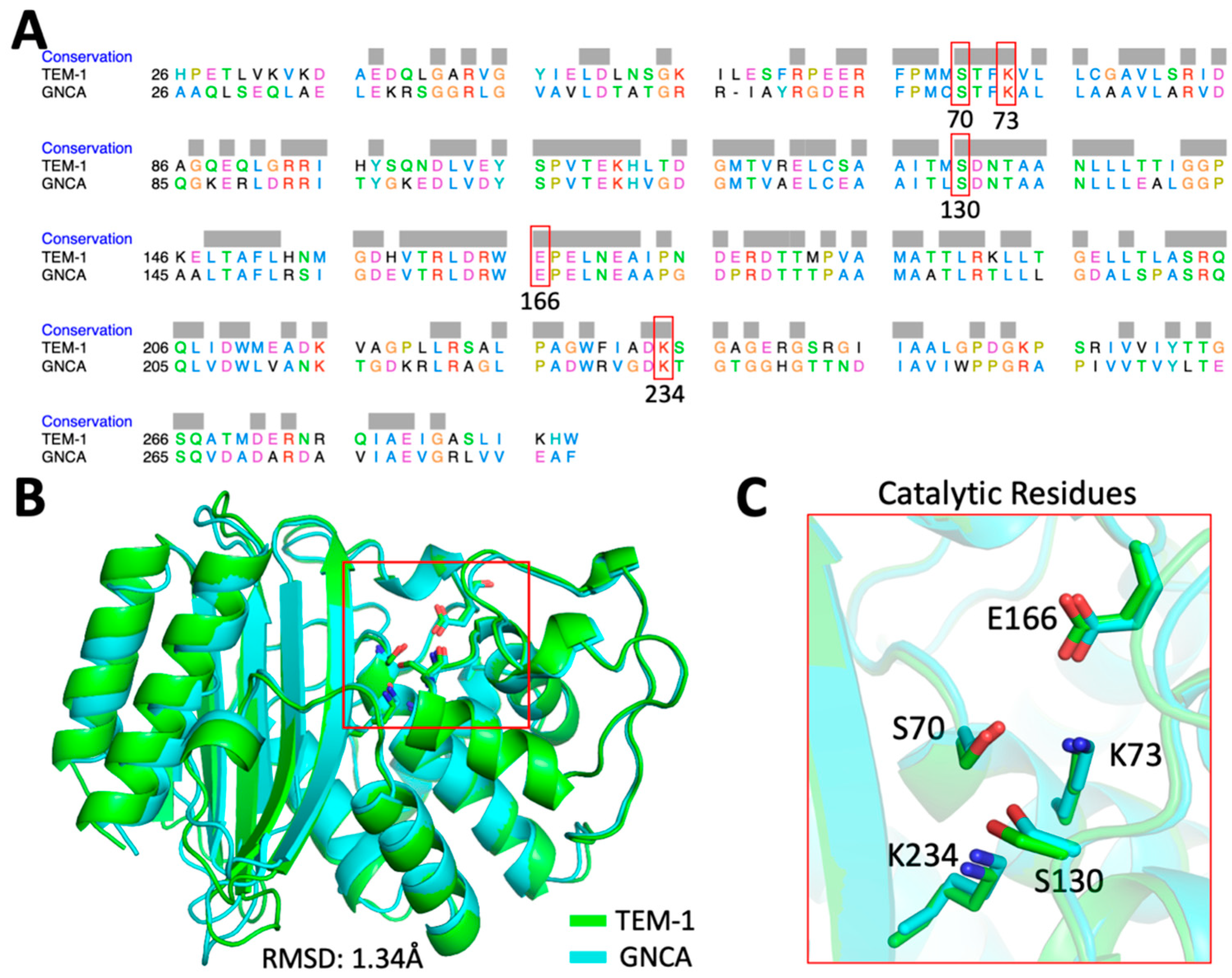
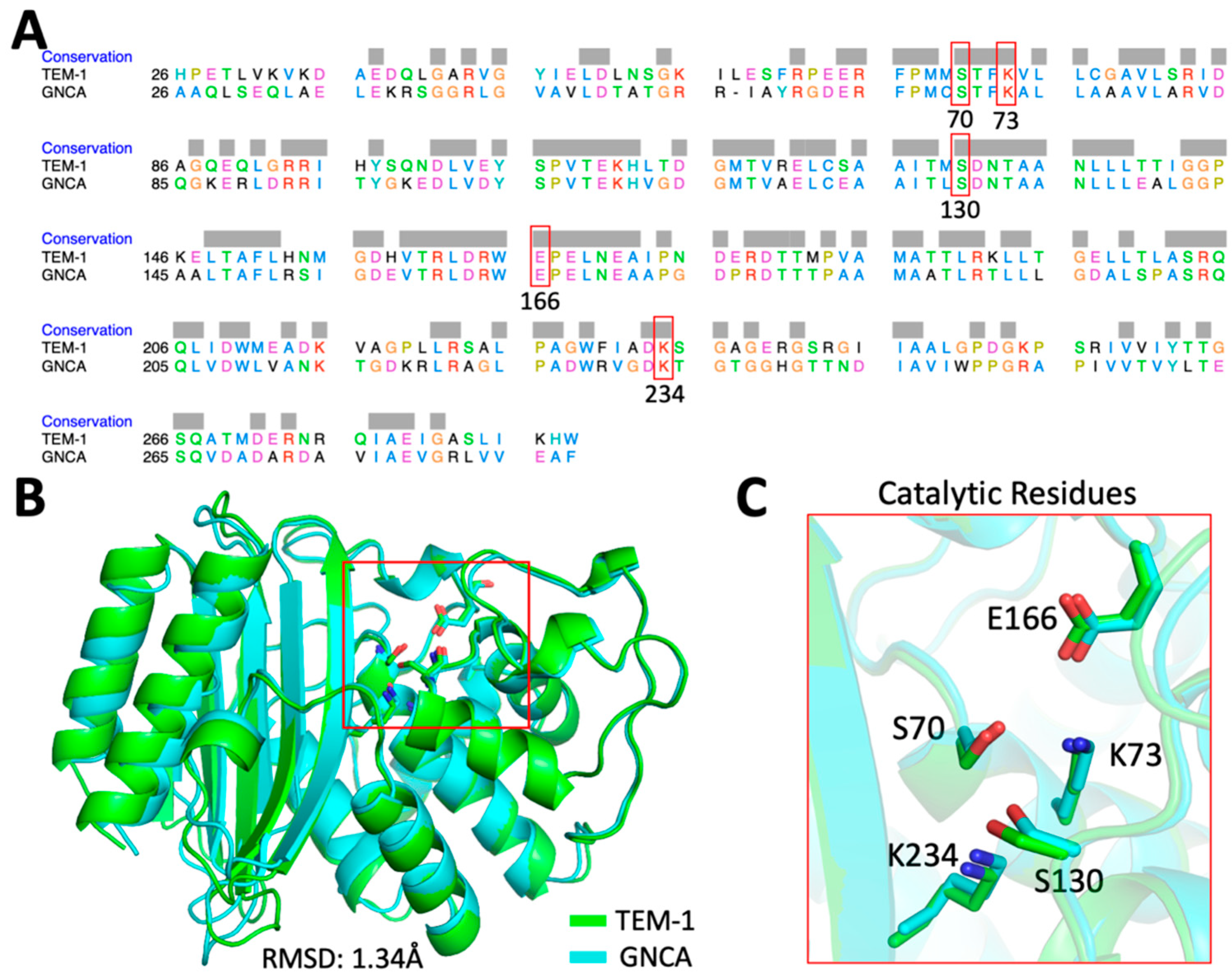
2.1. Computational Analysis Using dfi and dci
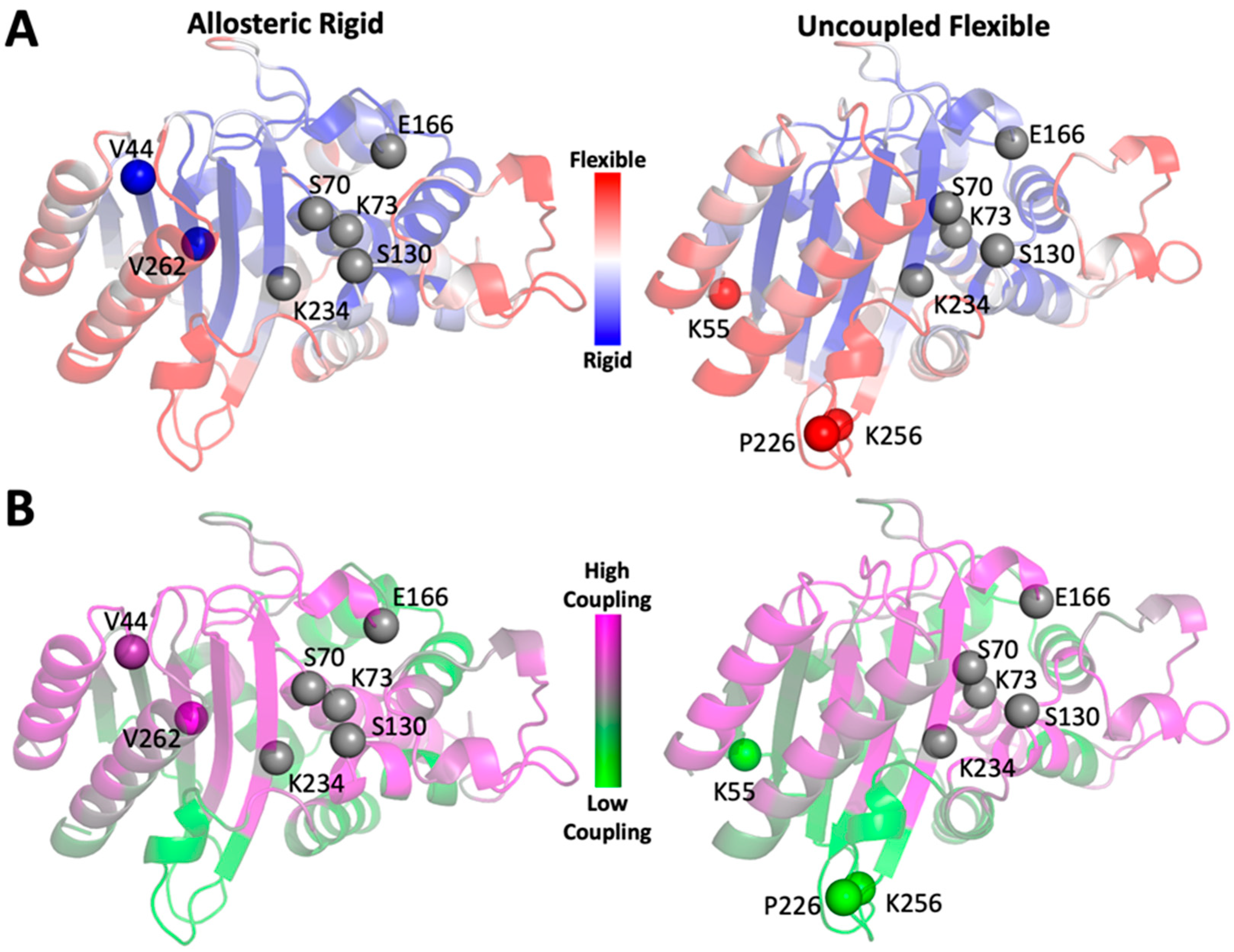
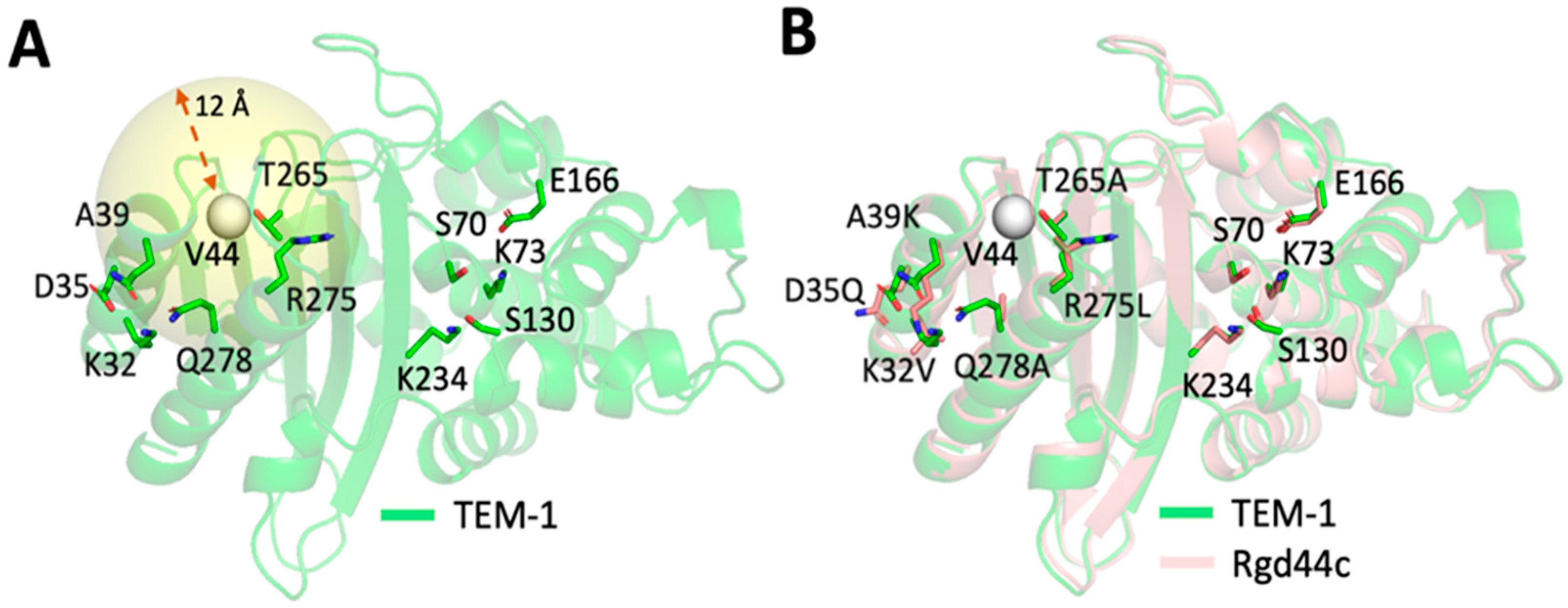
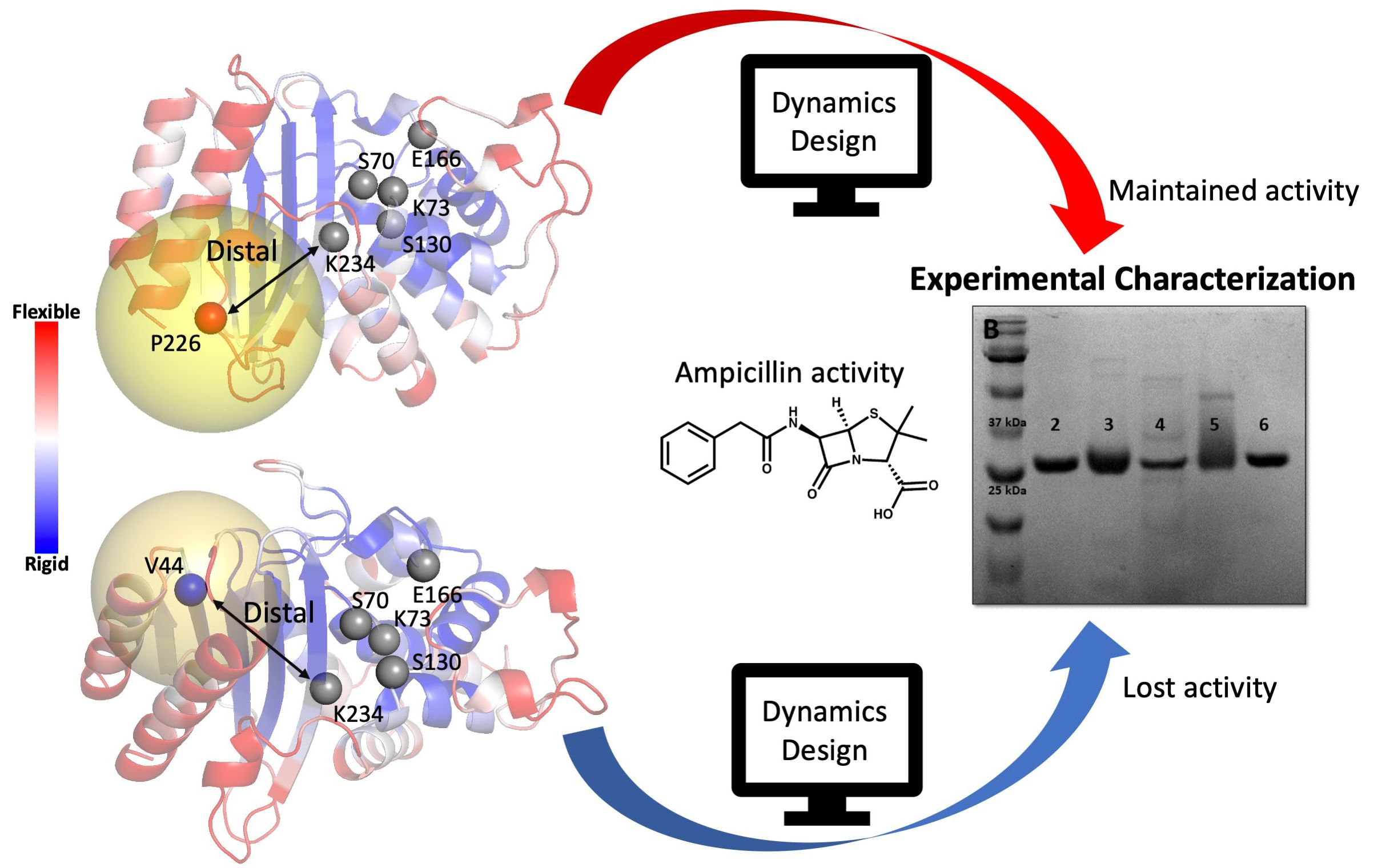
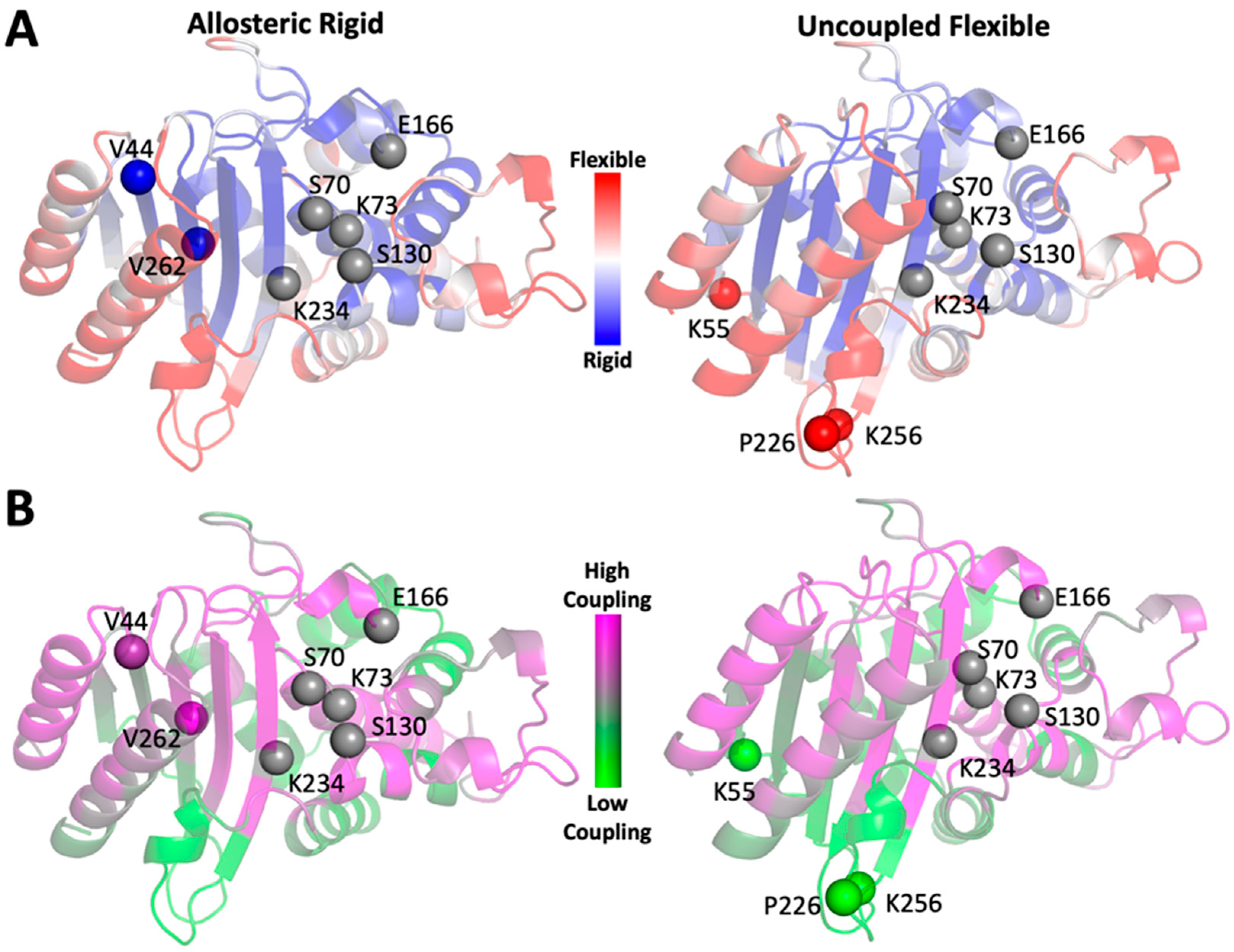
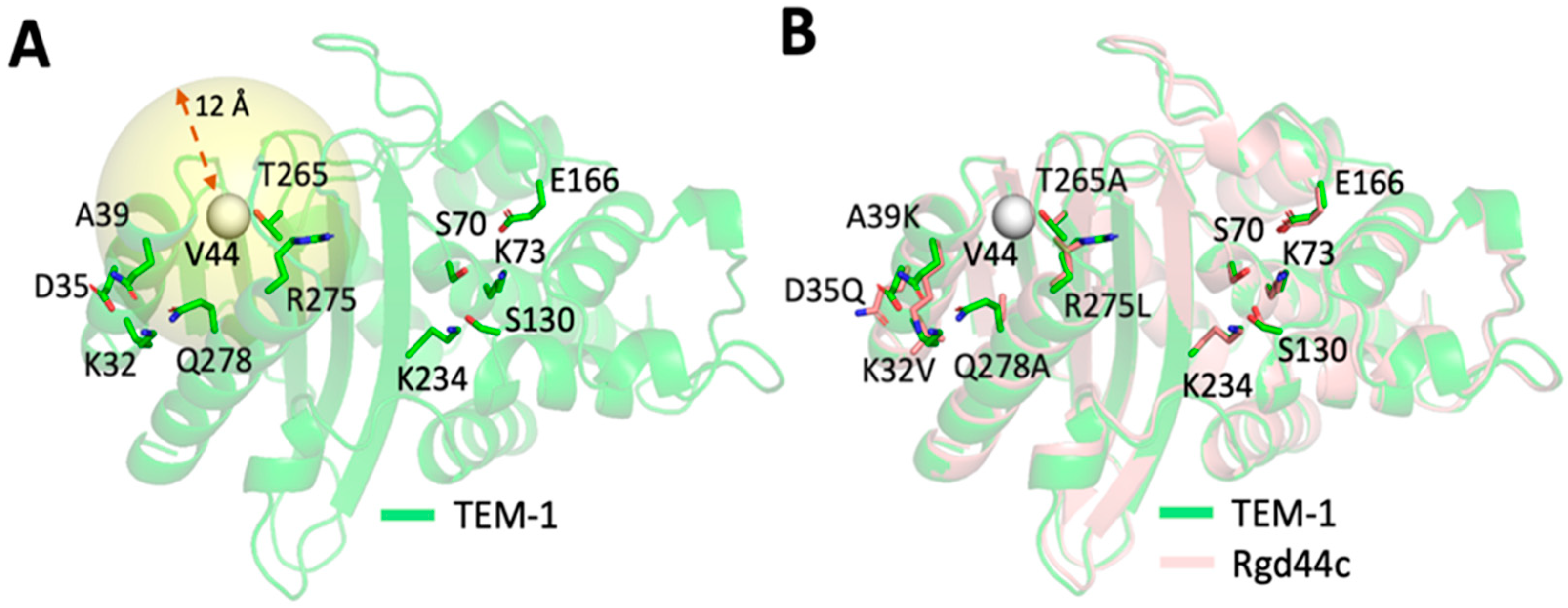
2.2. Computational Design of TEM-1 Variants
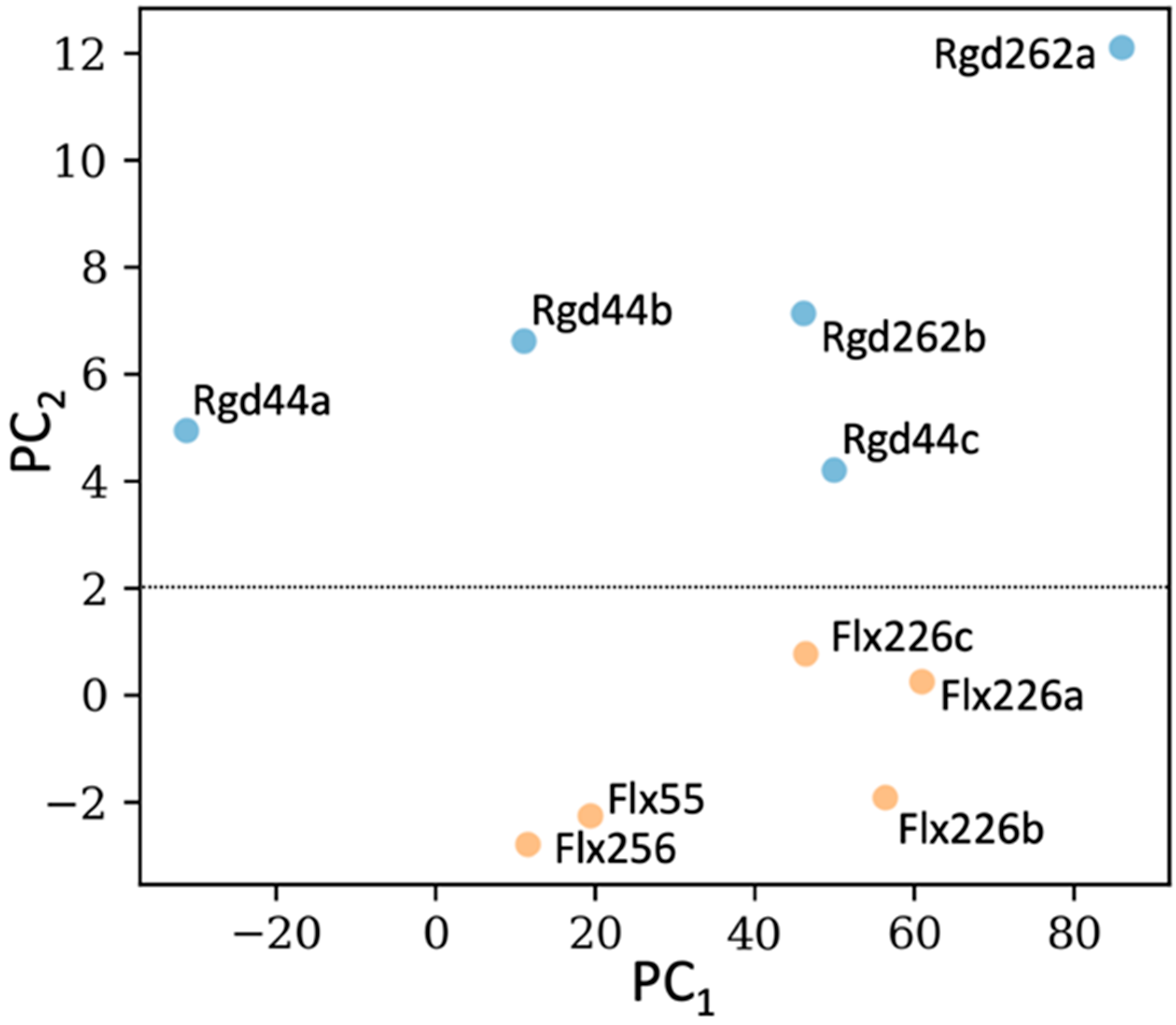
2.3. Selection of the Designed Proteins Using Flexibility Profiles
2.4. Experimental Analysis of the Designed Proteins
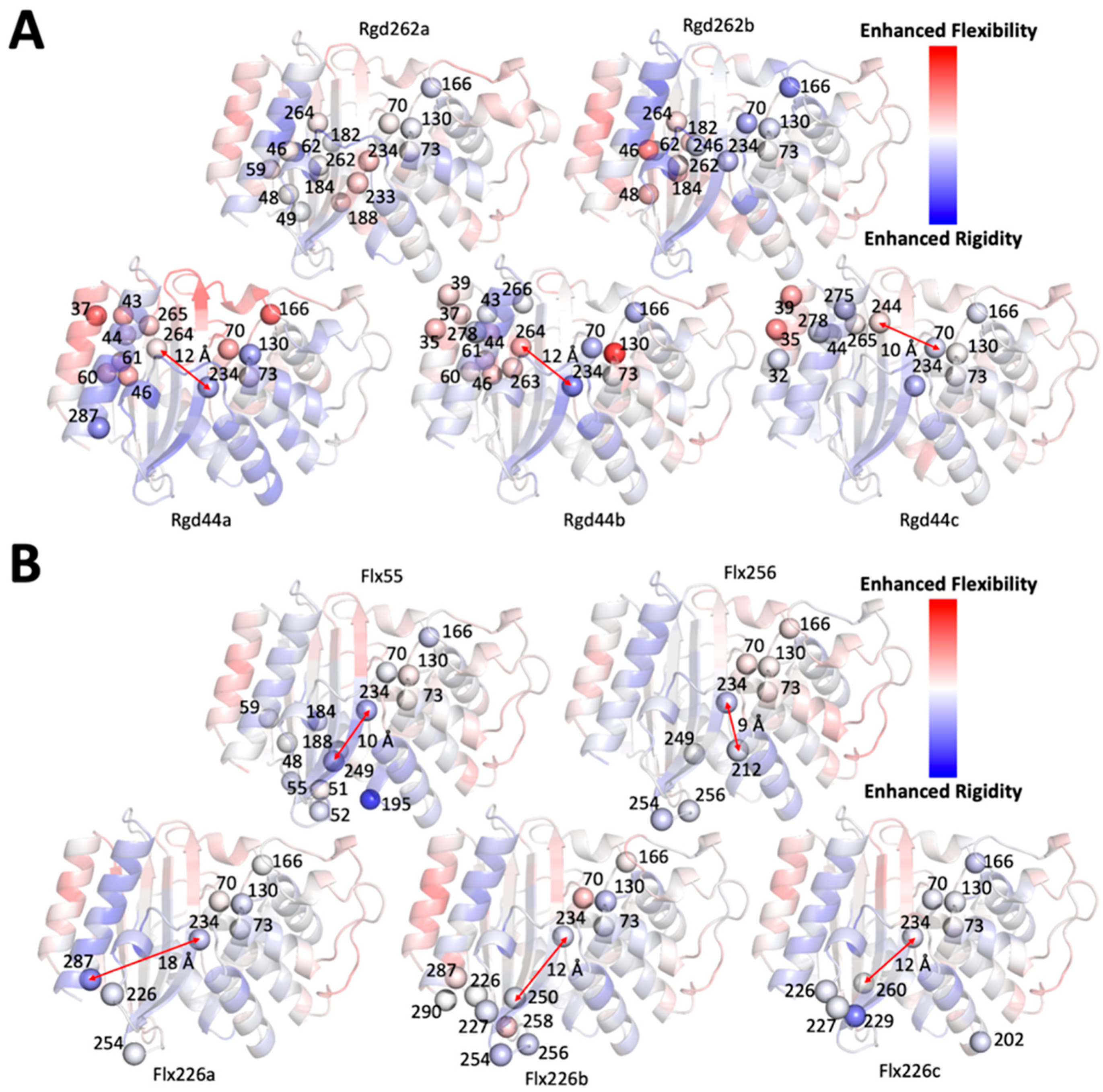
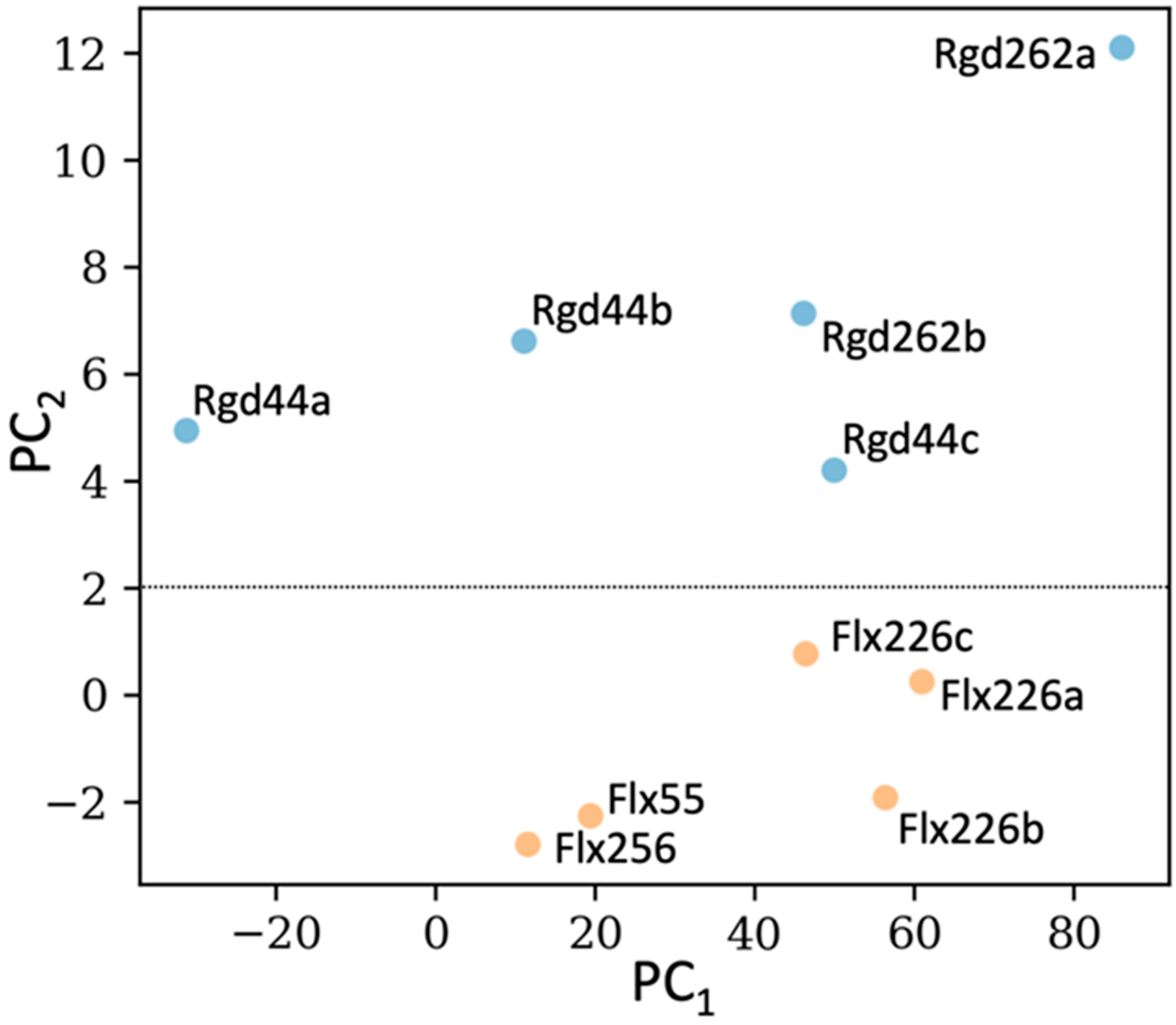
2.5. Dynamics Analysis of the Designed Proteins
3. Conclusions
4. Materials and Methods
4.1. Molecular Dynamics (MD)
4.2. Dynamic Flexibility Index (dfi)
4.3. Dynamic Coupling Index (dci)
4.4. Dynamic Distance Calculation
4.5. Rosetta Design Protocol
4.6. Protein Expression and Purification
4.7. Circular Dichroism Characterization of Protein Folding and Stability
4.8. MIC Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coulson, A. ß-Lactamases: Molecular Studies. Biotechnol. Genet. Eng. Rev. 1985, 3, 219–254. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother. 2018, 62, 01076-18. [Google Scholar] [CrossRef] [Green Version]
- Brandt, C.; Braun, S.D.; Stein, C.; Slickers, P.; Ehricht, R.; Pletz, M.W.; Makarewicz, O. In silico serine β-lactamases analysis reveals a huge potential resistome in environmental and pathogenic species. Sci. Rep. 2017, 7, srep43232. [Google Scholar] [CrossRef] [Green Version]
- Gobeil, S.M.C.; Ebert, M.C.C.J.C.; Park, J.; Gagné, D.; Doucet, N.; Berghuis, A.M.; Pleiss, J.; Pelletier, J.N. The Structural Dynamics of Engineered β-Lactamases Vary Broadly on Three Timescales yet Sustain Native Function. Sci. Rep. 2019, 9, 6656. [Google Scholar] [CrossRef]
- Brown, C.A.; Hu, L.; Sun, Z.; Patel, M.P.; Singh, S.; Porter, J.R.; Sankaran, B.; Prasad, B.V.V.; Bowman, G.R.; Palzkill, T. Antagonism between substitutions in β-lactamase explains a path not taken in the evolution of bacterial drug resistance. J. Biol. Chem. 2020, 295, 7376–7390. [Google Scholar] [CrossRef] [Green Version]
- Cortina, G.A.; Kasson, P.M. Predicting allostery and microbial drug resistance with molecular simulations. Curr. Opin. Struct. Biol. 2018, 52, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Cortina, G.A.; Hays, J.M.; Kasson, P.M. Conformational Intermediate That Controls KPC-2 Catalysis and Beta-Lactam Drug Resistance. ACS Catal. 2018, 8, 2741–2747. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Dominy, B.N. The Evolution of Cefotaximase Activity in the TEM β-Lactamase. J. Mol. Biol. 2012, 415, 205–220. [Google Scholar] [CrossRef]
- Orencia, M.C.; Yoon, J.S.; Ness, J.E.; Stemmer, W.P.; Stevens, R.C. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Genet. 2001, 8, 238–242. [Google Scholar] [CrossRef]
- Wang, X.; Minasov, G.; Shoichet, B.K. Evolution of an Antibiotic Resistance Enzyme Constrained by Stability and Activity Trade-offs. J. Mol. Biol. 2002, 320, 85–95. [Google Scholar] [CrossRef]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Med. Chem. 2014, 6, PMC.S14459-64. [Google Scholar] [CrossRef] [Green Version]
- Doucet, N.; Savard, P.-Y.; Pelletier, J.N.; Gagné, S.M. NMR Investigation of Tyr105 Mutants in TEM-1 β-Lactamase. J. Biol. Chem. 2007, 282, 21448–21459. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Zou, T.; Modi, C.; Dörner, K.; Grunkemeyer, T.J.; Chen, L.; Fromme, R.; Matz, M.V.; Ozkan, S.B.; Wachter, R.M. A Hinge Migration Mechanism Unlocks the Evolution of Green-to-Red Photoconversion in GFP-like Proteins. Structure 2015, 23, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Modi, T.; Huihui, J.; Ghosh, K.; Ozkan, S.B. Ancient thioredoxins evolved to modern-day stability–function requirement by altering native state ensemble. Philos. Trans. R. Soc. B: Biol. Sci. 2018, 373, 20170184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, T.; Risso, V.A.; Gavira, J.A.; Sanchez-Ruiz, J.M.; Ozkan, S.B. Evolution of Conformational Dynamics Determines the Conversion of a Promiscuous Generalist into a Specialist Enzyme. Mol. Biol. Evol. 2014, 32, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerek, Z.N.; Ozkan, S.B. Change in Allosteric Network Affects Binding Affinities of PDZ Domains: Analysis through Perturbation Response Scanning. PLoS Comput. Biol. 2011, 7, e1002154. [Google Scholar] [CrossRef]
- Larrimore, K.E.; Kazan, I.C.; Kannan, L.; Kendle, R.P.; Jamal, T.; Barcus, M.; Bolia, A.; Brimijoin, S.; Zhan, C.-G.; Ozkan, S.B.; et al. Plant-expressed cocaine hydrolase variants of butyrylcholinesterase exhibit altered allosteric effects of cholinesterase activity and increased inhibitor sensitivity. Sci. Rep. 2017, 7, 10419. [Google Scholar] [CrossRef] [Green Version]
- Gerek, Z.N.; Keskin, O.; Ozkan, S.B. Identification of specificity and promiscuity of PDZ domain interactions through their dynamic behavior. Proteins: Struct. Funct. Bioinform. 2009, 77, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Modi, T.; Ozkan, S.B. Mutations Utilize Dynamic Allostery to Confer Resistance in TEM-1 β-lactamase. Int. J. Mol. Sci. 2018, 19, 3808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campitelli, P.; Modi, T.; Kumar, S.; Ozkan, S.B. The Role of Conformational Dynamics and Allostery in Modulating Protein Evolution. Annu. Rev. Biophys. 2020, 49, 267–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Butler, B.M.; Kumar, S.; Ozkan, S.B. Integration of structural dynamics and molecular evolution via protein interaction networks: A new era in genomic medicine. Curr. Opin. Struct. Biol. 2015, 35, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campitelli, P.; Guo, J.; Zhou, H.-X.; Ozkan, S.B. Hinge-Shift Mechanism Modulates Allosteric Regulations in Human Pin1. J. Phys. Chem. B 2018, 122, 5623–5629. [Google Scholar] [CrossRef] [PubMed]
- Risso, V.A.; Gavira, J.A.; Mejia-Carmona, D.F.; Gaucher, E.A.; Sanchez-Ruiz, J.M. Hyperstability and Substrate Promiscuity in Laboratory Resurrections of Precambrian β-Lactamases. J. Am. Chem. Soc. 2013, 135, 2899–2902. [Google Scholar] [CrossRef]
- Salverda, M.L.; De Visser, J.A.G.; Barlow, M. Natural evolution of TEM-1 β-lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010, 34, 1015–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiffler, M.A.; Hekstra, D.R.; Ranganathan, R. Evolvability as a Function of Purifying Selection in TEM-1 β-Lactamase. Cell 2015, 160, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bolia, A.; Maxwell, J.D.; Bobkov, A.A.; Ghirlanda, G.; Ozkan, S.B.; Margulis, C.J. A Rigid Hinge Region Is Necessary for High-Affinity Binding of Dimannose to Cyanovirin and Associated Constructs. Biochemistry 2015, 54, 6951–6960. [Google Scholar] [CrossRef]
- Modi, T.; Campitelli, P.; Kazan, I.C.; Ozkan, S.B. Protein folding stability and binding interactions through the lens of evolution: A dynamical perspective. Curr. Opin. Struct. Biol. 2021, 66, 207–215. [Google Scholar] [CrossRef]
- Gerek, Z.N.; Kumar, S.; Ozkan, S.B. Structural dynamics flexibility informs function and evolution at a proteome scale. Evol. Appl. 2013, 6, 423–433. [Google Scholar] [CrossRef]
- Kumar, A.; Glembo, T.J.; Ozkan, S.B. The Role of Conformational Dynamics and Allostery in the Disease Development of Human Ferritin. Biophys. J. 2015, 109, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- NCBI BioProject Database. Available online: https://www.ncbi.nlm.nih.gov/bioproject/ (accessed on 4 May 2019).
- Ambler, R.P.; Coulson, A.F.W.; Frère, J.M.; Ghuysen, J.M.; Joris, B.; Forsman, M.; Levesque, R.C.; Tiraby, G.; Waley, S.G. A standard numbering scheme for the class A β-lactamases. Biochem. J. 1991, 276, 269–270. [Google Scholar] [CrossRef]
- Jelsch, C.; Mourey, L.; Masson, J.M.; Samama, J.P. Crystal Structure of Escherichia Coli TEM1 Beta-Lactamase at 1.8 A Resolution. Proteins 1993, 16, 364–383. [Google Scholar] [CrossRef]
- Risso, V.A.; Gavira, J.A.; Gaucher, E.A.; Sanchez-Ruiz, J.M. Phenotypic comparisons of consensus variants versus laboratory resurrections of Precambrian proteins. Proteins Struct. Funct. Bioinform. 2014, 82, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Butler, B.M.; Gerek, Z.N.; Kumar, S.; Ozkan, S.B. Conformational dynamics of nonsynonymous variants at protein interfaces reveals disease association. Proteins Struct. Funct. Bioinform. 2014, 83, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Atilgan, C.; Gerek, Z.; Ozkan, S.; Atilgan, A. Manipulation of Conformational Change in Proteins by Single-Residue Perturbations. Biophys. J. 2010, 99, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Leaver-Fay, A.; Tyka, M.D.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Rosetta3. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Knies, J.L.; Cai, F.; Weinreich, D.M. Enzyme Efficiency but Not Thermostability Drives Cefotaxime Resistance Evolution in TEM-1 β-Lactamase. Mol. Biol. Evol. 2017, 34, 1040–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Doruker, P.; Kaynak, B.; Zhang, S.; Krieger, J.; Li, H.; Bahar, I. Intrinsic dynamics is evolutionarily optimized to enable allosteric behavior. Curr. Opin. Struct. Biol. 2020, 62, 14–21. [Google Scholar] [CrossRef]
- Ma, B.; Tsai, C.-J.; Haliloğlu, T.; Nussinov, R. Dynamic Allostery: Linkers Are Not Merely Flexible. Structure 2011, 19, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Campitelli, P.; Ozkan, S.B.; Swint-Kruse, L. Asymmetry in Dynamic Allosteric Residue Coupling (DARC) Interactions Captures Evolutionary Landscape. Biophys. J. 2020, 118, 52a. [Google Scholar] [CrossRef]
- Bishop, C. Pattern Recognition and Machine Learning, 1st ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomic Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Minimal Inhibitory Concentration of Ampicillin MICamp (µg/mL) | Melting Temperature Tm (°C) |
|---|---|---|
| GNCA | 43 | 90.3 |
| TEM-1 | 1500 | 56.4 |
| Rdg44a | <2 ** | NM |
| Rdg44b | <2 | 63.1 |
| Rdg44c | 26 | 66.4 |
| Rdg262a | <2 ** | NM |
| Rdg262b | 26 | 56.4 |
| Flx226a | 1500 | 57.4 |
| Flx226b | 375 | 53.2 |
| Flx226c | 1500 | 55.6 |
| Flx256 | 750 | 58.1 |
| Flx55 | 750 | 58.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolbaba-Kartchner, B.; Kazan, I.C.; Mills, J.H.; Ozkan, S.B. The Role of Rigid Residues in Modulating TEM-1 β-Lactamase Function and Thermostability. Int. J. Mol. Sci. 2021, 22, 2895. https://doi.org/10.3390/ijms22062895
Kolbaba-Kartchner B, Kazan IC, Mills JH, Ozkan SB. The Role of Rigid Residues in Modulating TEM-1 β-Lactamase Function and Thermostability. International Journal of Molecular Sciences. 2021; 22(6):2895. https://doi.org/10.3390/ijms22062895
Chicago/Turabian StyleKolbaba-Kartchner, Bethany, I. Can Kazan, Jeremy H. Mills, and S. Banu Ozkan. 2021. "The Role of Rigid Residues in Modulating TEM-1 β-Lactamase Function and Thermostability" International Journal of Molecular Sciences 22, no. 6: 2895. https://doi.org/10.3390/ijms22062895
APA StyleKolbaba-Kartchner, B., Kazan, I. C., Mills, J. H., & Ozkan, S. B. (2021). The Role of Rigid Residues in Modulating TEM-1 β-Lactamase Function and Thermostability. International Journal of Molecular Sciences, 22(6), 2895. https://doi.org/10.3390/ijms22062895