
Age and Sex-Related Changes in Human First-Trimester Placenta Transcriptome and Insights into Adaptative Responses to Increased Oxygen

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification of Outliers in Samples
2.2. Identification of Key Genes and Term Enrichment Using DESeq2
2.3. Selection of Key Genes Based on WGCNA Method
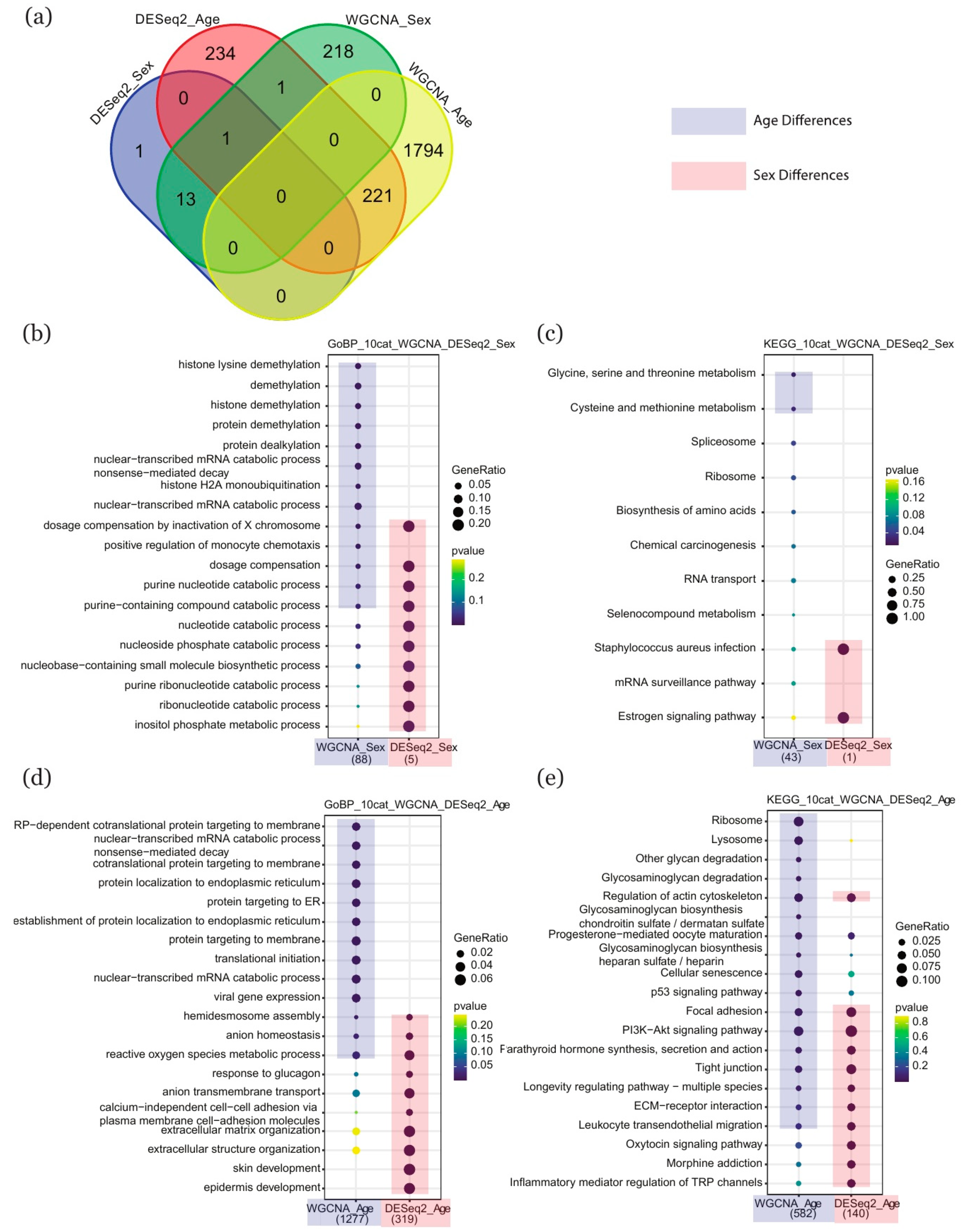
2.4. Comparison of Enriched Terms between DESeq2 and WGCNA
2.5. Enrichment in HIF Targets
2.6. Selection of GO Terms and Associated Genes Most Affected by HIF
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Ethics Statement
4.2. Cytotrophoblast Isolation and Culture
4.3. Fetal-Sex Determination by PCR
4.4. RNA-Seq Experiment and Data Processing
4.5. Clustering Analysis and Construction of Co-Expression Modules of Human Placental RNAseq Data
4.6. Analysis of Co-Expression Modules in Human Placental RNAseq Data
4.7. Enrichment and Differential Abundance of Key Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soncin, F.; Natale, D.; Parast, M.M. Signaling pathways in mouse and human trophoblast differentiation: A comparative review. Cell. Mol. Life Sci. 2015, 72, 1291–1302. [Google Scholar] [CrossRef]
- James, J.L.; Carter, A.M.; Chamley, L.W. Human placentation from nidation to 5 weeks of gestation. Part I: What do we know about formative placental development following implantation? Placenta 2012, 33, 327–334. [Google Scholar] [CrossRef]
- Patel, J.; Landers, K.; Mortimer, R.H.; Richard, K. Regulation of hypoxia inducible factors (HIF) in hypoxia and normoxia during placental development. Placenta 2010, 31, 951–957. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Goldman-Wohl, D.; Yagel, S. Regulation of trophoblast invasion: From normal implantation to pre-eclampsia. Mol. Cell. Endocrinol. 2002, 187, 233–238. [Google Scholar] [CrossRef]
- Ali, K.Z. Stereological study of the effect of altitude on the trophoblast cell populations of human term placental villi. Placenta 1997, 18, 447–450. [Google Scholar] [CrossRef]
- Todros, T.; Sciarrone, A.; Piccoli, E.; Guiot, C.; Kaufmann, P.; Kingdom, J. Umbilical Doppler waveforms and placental villous angiogenesis in pregnancies complicated by fetal growth restriction. Obstet. Gynecol. 1999, 93, 499–503. [Google Scholar] [PubMed]
- Schneider, H. Placental oxygen consumption. Part II: In vitro studies—A review. Placenta 2000, 21 (Suppl. A), S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Placental oxygen consumption. Part I: In vivo studies—A review. Placenta 2000, 21 (Suppl. A), S31–S37. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.N.; Dye, J.A.; Henriquez, A.R.; Stewart, E.J.; Lavrich, K.S.; Carswell, G.K.; Ren, H.; Freeborn, D.L.; Snow, S.J.; Schladweiler, M.C.; et al. Ozone-induced fetal growth restriction in rats is associated with sexually dimorphic placental and fetal metabolic adaptation. Mol. Metab. 2020, 42, 101094. [Google Scholar] [CrossRef]
- Holland, O.; Dekker Nitert, M.; Gallo, L.A.; Vejzovic, M.; Fisher, J.J.; Perkins, A.V. Review: Placental mitochondrial function and structure in gestational disorders. Placenta 2017, 54, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Telugu, B.P.; Thompson, L.P. Sexual dimorphism of mitochondrial function in the hypoxic guinea pig placenta. Biol. Reprod. 2019, 100, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Sferruzzi-Perri, A.N.; Camm, E.J. The Programming Power of the Placenta. Front. Physiol. 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L. The placenta: A multifaceted, transient organ. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef]
- Chang, C.W.; Wakeland, A.K.; Parast, M.M. Trophoblast lineage specification, differentiation and their regulation by oxygen tension. J. Endocrinol. 2018, 236, R43–R56. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.; Depping, R. Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1beta): Is it a rare exception? Mol. Med. 2014, 20, 215–220. [Google Scholar] [CrossRef]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Adelman, D.M.; Gertsenstein, M.; Nagy, A.; Simon, M.C.; Maltepe, E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000, 14, 3191–3203. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Myatt, L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta 2013, 34, 1183–1189. [Google Scholar] [CrossRef]
- Huppertz, B. Placental origins of preeclampsia: Challenging the current hypothesis. Hypertension 2008, 51, 970–975. [Google Scholar] [CrossRef]
- Roberts, J.M.; Cooper, D.W. Pathogenesis and genetics of pre-eclampsia. Lancet 2001, 357, 53–56. [Google Scholar] [CrossRef]
- Braun, A.E.; Muench, K.L.; Robinson, B.G.; Wang, A.; Palmer, T.D.; Winn, V.D. Examining Sex Differences in the Human Placental Transcriptome During the First Fetal Androgen Peak. Reprod. Sci. 2021, 28, 801–818. [Google Scholar] [CrossRef]
- Gonzalez, T.L.; Sun, T.; Koeppel, A.F.; Lee, B.; Wang, E.T.; Farber, C.R.; Rich, S.S.; Sundheimer, L.W.; Buttle, R.A.; Chen, Y.I.; et al. Sex differences in the late first trimester human placenta transcriptome. Biol. Sex Differ. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Sitras, V.; Fenton, C.; Paulssen, R.; Vartun, A.; Acharya, G. Differences in gene expression between first and third trimester human placenta: A microarray study. PLoS ONE 2012, 7, e33294. [Google Scholar] [CrossRef]
- Soncin, F.; Khater, M.; To, C.; Pizzo, D.; Farah, O.; Wakeland, A.; Arul Nambi Rajan, K.; Nelson, K.K.; Chang, C.W.; Moretto-Zita, M.; et al. Comparative analysis of mouse and human placentae across gestation reveals species-specific regulators of placental development. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Gonzalez, T.L.; Deng, N.; DiPentino, R.; Clark, E.L.; Lee, B.; Tang, J.; Wang, Y.; Stripp, B.R.; Yao, C.; et al. Sexually Dimorphic Crosstalk at the Maternal-Fetal Interface. J. Clin. Endocrinol. Metab. 2020, 105, e4831–e4847. [Google Scholar] [CrossRef] [PubMed]
- Brew, O.; Sullivan, M.H.F. Oxygen and tissue culture affect placental gene expression. Placenta 2017, 55, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cross, C.E.; Tolba, M.F.; Rondelli, C.M.; Xu, M.; Abdel-Rahman, S.Z. Oxidative Stress Alters miRNA and Gene Expression Profiles in Villous First Trimester Trophoblasts. BioMed Res. Int. 2015, 2015, 257090. [Google Scholar] [CrossRef]
- Treissman, J.; Yuan, V.; Baltayeva, J.; Le, H.T.; Castellana, B.; Robinson, W.P.; Beristain, A.G. Low oxygen enhances trophoblast column growth by potentiating differentiation of the extravillous lineage and promoting LOX activity. Development 2020, 147. [Google Scholar] [CrossRef]
- Liu, F.; Zhu, W.; Shoaito, H.; Chissey, A.; Degrelle, S.A.; Fournier, T. Mining of combined human placental gene expression data across pregnancy, applied to PPAR signaling pathway. Placenta 2020, 99, 157–165. [Google Scholar] [CrossRef]
- Qi, L.; Liu, B.; Chen, X.; Liu, Q.; Li, W.; Lv, B.; Xu, X.; Wang, L.; Zeng, Q.; Xue, J.; et al. Single-Cell Transcriptomic Analysis Reveals Mitochondrial Dynamics in Oocytes of Patients With Polycystic Ovary Syndrome. Front. Genet. 2020, 11, 396. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, W.; Yuan, J.; Qiang, B.; Han, W.; Peng, X. Integrated Analysis of RNA-Binding Proteins in Glioma. Cancers 2020, 12, 892. [Google Scholar] [CrossRef]
- Koh, M.J.; Shin, D.H.; Lee, S.J.; Hwang, C.S.; Lee, H.J.; Kim, A.; Park, W.Y.; Lee, J.H.; Choi, K.U.; Kim, J.Y.; et al. Gastric-type gene expression and phenotype in non-terminal respiratory unit type adenocarcinoma of the lung with invasive mucinous adenocarcinoma morphology. Histopathology 2020, 76, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Broderick, S.R.; Wijeratne, S.; Wijeratn, A.J.; Chapin, L.J.; Meulia, T.; Jones, M.L. RNA-sequencing reveals early, dynamic transcriptome changes in the corollas of pollinated petunias. BMC Plant Biol. 2014, 14, 307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, L.J.; Liu, X.Q.; Hu, Z.Y.; Jiang, Y.; Gao, R.F.; Feng, Q.; Lan, X.; Geng, Y.Q.; Chen, X.M.; et al. nm23 regulates decidualization through the PI3K-Akt-mTOR signaling pathways in mice and humans. Hum. Reprod. 2016, 31, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Ganguly, A.; Home, P.; Bhattacharya, B.; Ray, S.; Ghosh, A.; Rumi, M.A.K.; Marsh, C.; French, V.A.; Gunewardena, S.; et al. TEAD4 ensures postimplantation development by promoting trophoblast self-renewal: An implication in early human pregnancy loss. Proc. Natl. Acad. Sci. USA 2020, 117, 17864–17875. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Cheong, M.L.; Chang, G.D.; Tsai, M.S.; Chen, H. Involvement of Epac1/Rap1/CaMKI/HDAC5 signaling cascade in the regulation of placental cell fusion. Mol. Hum. Reprod. 2013, 19, 745–755. [Google Scholar] [CrossRef]
- Gerbaud, P.; Tasken, K.; Pidoux, G. Spatiotemporal regulation of cAMP signaling controls the human trophoblast fusion. Front. Pharmacol. 2015, 6, 202. [Google Scholar] [CrossRef]
- Haider, S.; Meinhardt, G.; Velicky, P.; Otti, G.R.; Whitley, G.; Fiala, C.; Pollheimer, J.; Knofler, M. Notch signaling plays a critical role in motility and differentiation of human first-trimester cytotrophoblasts. Endocrinology 2014, 155, 263–274. [Google Scholar] [CrossRef]
- Ietta, F.; Bechi, N.; Romagnoli, R.; Bhattacharjee, J.; Realacci, M.; Di Vito, M.; Ferretti, C.; Paulesu, L. 17β-Estradiol modulates the macrophage migration inhibitory factor secretory pathway by regulating ABCA1 expression in human first-trimester placenta. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E411–E418. [Google Scholar] [CrossRef]
- Kho, A.T.; Chhabra, D.; Sharma, S.; Qiu, W.; Carey, V.J.; Gaedigk, R.; Vyhlidal, C.A.; Leeder, J.S.; Tantisira, K.G.; Weiss, S.T. Age, Sexual Dimorphism, and Disease Associations in the Developing Human Fetal Lung Transcriptome. Am. J. Respir. Cell. Mol. Biol. 2016, 54, 814–821. [Google Scholar] [CrossRef]
- Baczyk, D.; Audette, M.C.; Coyaud, E.; Raught, B.; Kingdom, J.C. Spatiotemporal distribution of small ubiquitin-like modifiers during human placental development and in response to oxidative and inflammatory stress. J. Physiol. 2018, 596, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Bebington, C.; Doherty, F.J.; Fleming, S.D. Ubiquitin cross-reactive protein gene expression is increased in decidualized endometrial stromal cells at the initiation of pregnancy. Mol. Hum. Reprod. 1999, 5, 966–972. [Google Scholar] [CrossRef]
- Anton, L.; Brown, A.G.; Bartolomei, M.S.; Elovitz, M.A. Differential methylation of genes associated with cell adhesion in preeclamptic placentas. PLoS ONE 2014, 9, e100148. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, N.; Yaqinuddin, A.; Kakal, F.; Sheikh, L.; Qureshi, R.; Somani, M. Frequent hypomethylation of PTGS2 gene promoter in human term placenta. Ital. J. Anat. Embryol. 2013, 118, 211–216. [Google Scholar]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka-Tatematsu, M.; Nakashima, A.; Fujita, N.; Shima, T.; Yoshimori, T.; Saito, S. Autophagy induced by HIF1alpha overexpression supports trophoblast invasion by supplying cellular energy. PLoS ONE 2013, 8, e76605. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Maninger, S.; Siwetz, M.; Deutsch, A.; El-Heliebi, A.; Kolb-Lenz, D.; Hiden, U.; Desoye, G.; Herse, F.; Prokesch, A. Downregulation of p53 drives autophagy during human trophoblast differentiation. Cell. Mol. Life Sci. 2018, 75, 1839–1855. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.; St John, J.; Ahmed, A. Modulation of trophoblast cell death by oxygen and EGF. Mol. Med. 2002, 8, 847–856. [Google Scholar] [CrossRef]
- Cvitic, S.; Longtine, M.S.; Hackl, H.; Wagner, K.; Nelson, M.D.; Desoye, G.; Hiden, U. The human placental sexome differs between trophoblast epithelium and villous vessel endothelium. PLoS ONE 2013, 8, e79233. [Google Scholar] [CrossRef] [PubMed]
- Degrelle, S.A.; Fournier, T. Fetal-sex determination of human placental tissues. Placenta 2018, 61, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Hoch, D.; Novakovic, B.; Cvitic, S.; Saffery, R.; Desoye, G.; Majali-Martinez, A. Sex matters: XIST and DDX3Y gene expression as a tool to determine fetal sex in human first trimester placenta. Placenta 2020, 97, 68–70. [Google Scholar] [CrossRef]
- Rachdi, L.; Maugein, A.; Pechberty, S.; Armanet, M.; Hamroune, J.; Ravassard, P.; Marullo, S.; Albagli, O.; Scharfmann, R. Regulated expression and function of the GABAB receptor in human pancreatic beta cell line and islets. Sci. Rep. 2020, 10, 13469. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Simasotchi, C.; Vibert, F.; Zhu, W.; Gil, S.; Degrelle, S.A.; Fournier, T. Age and Sex-Related Changes in Human First-Trimester Placenta Transcriptome and Insights into Adaptative Responses to Increased Oxygen. Int. J. Mol. Sci. 2021, 22, 2901. https://doi.org/10.3390/ijms22062901
Liu F, Simasotchi C, Vibert F, Zhu W, Gil S, Degrelle SA, Fournier T. Age and Sex-Related Changes in Human First-Trimester Placenta Transcriptome and Insights into Adaptative Responses to Increased Oxygen. International Journal of Molecular Sciences. 2021; 22(6):2901. https://doi.org/10.3390/ijms22062901
Chicago/Turabian StyleLiu, Fulin, Christelle Simasotchi, Françoise Vibert, Wencan Zhu, Sophie Gil, Séverine A. Degrelle, and Thierry Fournier. 2021. "Age and Sex-Related Changes in Human First-Trimester Placenta Transcriptome and Insights into Adaptative Responses to Increased Oxygen" International Journal of Molecular Sciences 22, no. 6: 2901. https://doi.org/10.3390/ijms22062901
APA StyleLiu, F., Simasotchi, C., Vibert, F., Zhu, W., Gil, S., Degrelle, S. A., & Fournier, T. (2021). Age and Sex-Related Changes in Human First-Trimester Placenta Transcriptome and Insights into Adaptative Responses to Increased Oxygen. International Journal of Molecular Sciences, 22(6), 2901. https://doi.org/10.3390/ijms22062901