The Odd Couple(s): An Overview of Beta-Lactam Antibiotics Bearing More Than One Pharmacophoric Group



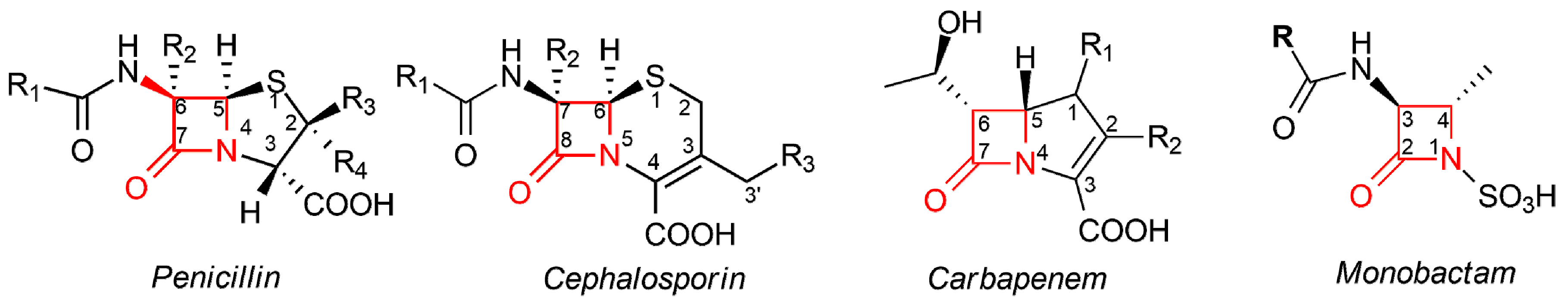{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. β-Lactam Antibiotics Bearing More Than One β-Lactam Ring
β-Lactam Nucleus on Calixarene Scaffolds
3. β-Lactam Antibiotics Bearing Additional Pharmacophores
3.1. Hybrid Antibiotics with a β-Lactam Moiety Associated with Another Antibiotic Moiety
3.2. Hybrid Antibiotics with a β-Lactam Moiety Associated with Other Non-Antibiotics Moieties
3.3. Conjugates with Other Non-Antibiotic Activities
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PBP | Penicillin-Binding Protein |
| 6-APA | 6-aminopenicillanic acid |
| 7-ACA | 7-aminocephalosporanic acid |
| MRSA | Methicillin-resistant strain of S. aureus |
| MSSA | Methicillin-sensitive strain of S. aureus |
| MIC | Minimum inhibitory concentration |
References
- Fleming, A. On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Bucci, R.; Galli, P. Vincenzo Tiberio: A misunderstood researcher. It. J. Public Health 2011, 8, 404–406. [Google Scholar]
- Hodgkin, D.C. The X-ray analysis of the structure of penicillin. Adv. Sci. 1949, 6, 85–89. [Google Scholar] [PubMed]
- Bo, G. Giuseppe Brotzu and the discovery of cephalosporins. Clin. Microbiol. Infect. 2000, 6 (Suppl. 3), 6–8. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Newton, G.G.F. The structure of cephalosporin C. Biochem. J. 1961, 79, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, D.C.; Maslen, E.N. The X-ray analysis of the structure of cephalosporin C. Biochem. J. 1961, 79, 393–402. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef]
- Sykes, R.B.; Cimarusti, C.M.; Bonner, D.P.; Bush, K.; Floyd, D.M.; Georgopapadakou, N.H.; Koster, W.M.; Liu, W.C.; Parker, W.L.; Principe, P.A.; et al. Monocyclic beta-lactam antibiotics produced by bacteria. Nature 1981, 291, 489–491. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P. β-lactams and β-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Marabotti, A. Cephalosporins. In Encyclopedia of Molecular Pharmacology, 3rd ed.; Offermanns, S., Rosenthal, W., Eds.; Springer Nature: Heidelberg, Germany, 2020; Chapter 5371–1. [Google Scholar]
- Nikolaou, K.C.; Montagnon, T. Molecules that Changed the World; Wiley VCH: Weinheim, Germany, 2008. [Google Scholar]
- Rodríguez-Tébar, A.; Arán, V.; Vázquez, D. Labelling and cross-linking of Escherichia coli penicillin-binding proteins with bis-β-lactam antibiotics. Eur. J. Biochem. 1984, 139, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Panunzio, M.; Malabarba, A.; Vicennati, P. Synthesis and antibacterial activity of new antibiotics arising from cephalosporin-monobactam coupling. ARKIVOC 2004, xiii, 36–47. [Google Scholar] [CrossRef]
- De Rosa, M.; Vigliotta, G.; Palma, G.; Saturnino, C.; Soriente, A. Novel penicillin-type analogues bearing a variable substituted 2-azetidinone ring at position 6: Synthesis and biological evaluation. Molecules 2015, 20, 22044–22057. [Google Scholar] [CrossRef]
- Verdino, A.; Vigliotta, G.; Giordano, D.; Caputo, I.; Soriente, A.; De Rosa, M.; Marabotti, A. Synthesis and biological evaluation of the progenitor of a new class of cephalosporin analogues, with a particular focus on structure-based computational analysis. PLoS ONE 2017, 12, e0181563. [Google Scholar] [CrossRef]
- Verdino, A.; Zollo, F.; De Rosa, M.; Soriente, A.; Hernández-Martínez, M.Á.; Marabotti, A. Computational analysis of the interactions of a novel cephalosporin derivative with β-lactamases. BMC Struct. Biol. 2018, 18, 13. [Google Scholar] [CrossRef]
- Vigliotta, G.; Giordano, D.; Verdino, A.; Caputo, I.; Martucciello, S.; Soriente, A.; Marabotti, A.; De Rosa, M. New compounds for a good old class: Synthesis of two β-lactam bearing cephalosporins and their evaluation with a multidisciplinary approach. Bioorg. Med. Chem. 2020, 28, 115302. [Google Scholar] [CrossRef]
- Pagadala, R.; Meshram, J.S.; Chopde, H.N.; Jetti, V. Synthesis of novel bis (β-lactam) from bis (ketene) and imines. Int. J. Chem. Tech. Res. 2010, 2, 1581–1585. [Google Scholar]
- Pagadala, R.; Kusampally, U.; Rajanna, K.C.; Meshram, J.S. Synthesis and antimicrobial studies of novel imidazole containing bisazetidinones and bisthiazolidinone derivatives. J. Heterocycl. Chem. 2015, 52, 403–410. [Google Scholar] [CrossRef]
- Ramsinghani, R.G.; Filmwala, Z.A. Synthesis of novel 1,1′-(ethane-1,2-diylbis(4,1-phenylene))bis(4-substituted-phenyl-3-chloroazetidin-2-ones)form1,2-dian-ilino ethane, their characterization and evaluation of their antibacterial activity. J. Emerg. Technol. Innov. Res. 2018, 5, 590–600. [Google Scholar]
- Meenakshisundaram, S.; Manickam, M.; Vinayagam, V. Synthesis, antibacterial and anticancer activity of novel bis-azetidinones. J. Chem. Pharm. Res. 2016, 8, 733–742. [Google Scholar]
- Kovács-Kulyassa, A.; Herczegh, P.; Sztaricskai, F.J.; Szabó, P. Cephalosporin podand derivatives. J. Antibiot. 2000, 53, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Agócs, A.; Herczegh, P.; Sztaricskai, F. A trimer of phenoxymethyl penicillin sulphone: Synthesis of a new β-lactam podand. J. Antibiot. 2002, 55, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Dive, G.; Bouillon, C.; Sliwa, A.; Valet, B.; Verlaine, O.; Sauvage, E.; Marchand-Brynaert, J. Macrocycle-embedded β-lactams as novel inhibitors of the penicilling binding protein PBP2a from MRSA. Eur. J. Med. Chem. 2013, 64, 365–376. [Google Scholar] [CrossRef]
- Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Rajesh, R.; Periyasami, G.; Raghunathan, R. Synthesis and antimicrobial evaluation of novel bis-β-lactam grafted macrocycles. Med. Chem. 2014, 10, 730–737. [Google Scholar] [CrossRef]
- Vatmurge, N.S.; Hazra, B.G.; Pore, V.S.; Shirazi, F.; Deshpande, M.V.; Kadreppa, S.; Chattopadhyay, S.; Gonnade, R.G. Synthesis and biological evaluation of bile acid dimers linked with 1,2,3-triazole and bis-β-lactam. Org. Biomol. Chem. 2008, 6, 3823–3830. [Google Scholar] [CrossRef]
- Böhmer, V. Calixarenes, macrocycles with (almost) unlimited possibilities. Angew. Chem. Int. Ed. Engl. 1995, 34, 713–745. [Google Scholar] [CrossRef]
- Neri, P.; Sessler, J.L.; Wang, M.-X. Calixarenes and Beyond; Springer International Publisher: Cham, Switzerland, 2016. [Google Scholar]
- De Rosa, M.; La Manna, P.; Soriente, A.; Gaeta, C.; Talotta, C.; Hickey, N.; Geremia, S.; Neri, P. A simple tetraminocalix[4]arene as a highly efficient catalyst under “on water“ conditions through hydrophobic amplification of weak hydrogen bonds. Chem. Eur. J. 2017, 23, 7142–7151. [Google Scholar] [CrossRef]
- De Rosa, M.; La Manna, P.; Talotta, C.; Soriente, A.; Gaeta, C.; Neri, P. Supramolecular organocatalysis in water mediated by macrocyclic compounds. Front. Chem. 2018, 6, 84. [Google Scholar] [CrossRef]
- Naseer, M.M.; Ahmed, M.; Hameed, S. Functionalized calix[4]arenes as potential therapeutics agents. Chem. Biol. Drug Des. 2017, 89, 243–256. [Google Scholar] [CrossRef]
- Español, E.S.; Villamil, M.M. Calixarenes: Generalities and their role in improving the solubility, biocompatibility, stability, bioavailability, detection, and transport of biomolecules. Biomolecules 2019, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Shurpik, D.N.; Padnya, P.L.; Stoikov, I.I.; Cragg, P.J. Antimicrobial activity of calixarenes and related macrocycles. Molecules 2020, 25, 5145–5177. [Google Scholar] [CrossRef] [PubMed]
- Pur, F.N.; Dilmaghani, K.A. Calixpenams: Synthesis, characterization, and biological evaluation of penicillins V and X clustered scaffold. Turk. J. Chem. 2014, 38, 288–296. [Google Scholar]
- Pur, F.N.; Dilmaghani, K.A. Calixcephems: Clustered cephalosporins analogues to calixpenams as novel potential anti-MRSA agents. Turk. J. Chem. 2014, 38, 850–858. [Google Scholar]
- Bushby, S.R.; Hitchings, G.H. Trimethoprim, a sulphonamide potentiator. Br. J. Pharmacol. Chemother. 1968, 33, 72–90. [Google Scholar] [CrossRef]
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef]
- Parkes, A.; Yune, I.A. Hybrid antibiotics—Clinical progress and novel designs. Expert. Opin. Drug Discov. 2016, 11, 665–680. [Google Scholar] [CrossRef]
- Long, D.D.; Marquess, D.G. Novel heterodimer antibiotics: A review of recent patent literature. Future Med. Chem. 2009, 1, 1037–1050. [Google Scholar] [CrossRef]
- Domalaon, R.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Antibiotic hybrids: The next generation of agents and adjuvants against Gram-negative pathogens? Clin. Microbiol. Rev. 2018, 31, e00077-17. [Google Scholar] [CrossRef]
- Greenwood, D.; O’Grady, F. Dual-action cephalosporin utilizing a novel therapeutic principle. Antimicrob. Agents Chemother. 1976, 10, 249–252. [Google Scholar] [CrossRef]
- O’Callaghan, C.H.; Sykes, R.B.; Staniforth, S.E. A new cephalosporin with a dual mode of action. Antimicrob. Agents Chemother. 1976, 10, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Lopez Quezada, L.; Li, K.; McDonald, S.L.; Nguyen, Q.; Perkowski, A.J.; Pharr, C.W.; Gold, B.; Roberts, J.; McAulay, K.; Saito, K.; et al. Dual-pharmacophore pyrithione-containing cephalosporins kill both replicating and nonreplicating Mycobacterium tuberculosis. ACS Infect. Dis. 2019, 5, 1433–1445, Erratum in: ACS Infect. Dis. 2019, 5, 2175. [Google Scholar] [CrossRef] [PubMed]
- Mobashery, S.; Lerner, S.A.; Johnston, M. Conscripting β-lactamase for use in drug delivery. Synthesis and biological activity of a cephalosporin C10-ester of an antibiotic dipeptide. J. Am. Chem. Soc. 1986, 108, 1685–1686. [Google Scholar] [CrossRef]
- Grapsas, I.; Lerner, S.A.; Mobashery, S. Conjoint molecules of cephalosporins and aminoglycosides. Arch. Pharm. 2001, 334, 295–301. [Google Scholar] [CrossRef]
- Perrone, E.; Jabés, D.; Alpegiani, M.; Andreini, B.P.; Della Bruna, C.; Del Nero, S.; Rossi, R.; Visentin, G.; Zarini, F.; Franceschi, G. Dual-action penems. J. Antibiot. (Tokyo) 1992, 45, 589–594. [Google Scholar] [CrossRef]
- Rao, V.S.; Fung-Tome, J.C.; Desiderio, J.V. Use of amino acid N-carboxy anhydride in the synthesis of peptide prodrug derivatives (including β-chloroalanyl) of C4-β-aminoalkylcarbapenems. In vitro and in vivo activities. J. Antib. 1993, 46, 167–176. [Google Scholar] [CrossRef][Green Version]
- Hamilton-Miller, J.M.T. Dual action antibiotic hybrids. J. Antimicrob. Chemother. 1994, 33, 197–202. [Google Scholar] [CrossRef]
- Hwu, J.R.; Moshfegh, A.A.; Tsay, S.-C.; Lin, C.C.; Tseng, W.N.; Azaripour, A.; Mottaghian, H.; Hakimelahi, G.H. Cephalosporin 3’-phloroglucide esters and 7-(phloroglucidamido) cephalosporins as novel antibacterial agents. J. Med. Chem. 1997, 40, 3434–3441. [Google Scholar] [CrossRef]
- Li, Q.; Lee, J.Y.; Castillo, R.; Hixon, M.S.; Pujol, C.; Doppalapudi, V.R.; Shepard, H.M.; Wahl, G.M.; Lobl, T.J.; Chan, M.F. NB2001, a novel antibacterial agent with broad-spectrum activity and enhanced potency against beta-lactamase-producing strains. Antimicrob. Agents Chemother. 2002, 46, 1262–1268. [Google Scholar] [CrossRef]
- Stone, G.W.; Zhang, Q.; Castillo, R.; Doppalapudi, V.R.; Bueno, A.R.; Lee, J.Y.; Li, Q.; Sergeeva, M.; Khambatta, G.; Georgopapadakou, N.H. Mechanism of action of NB2001 and NB2030, novel antibacterial agents activated by beta-lactamases. Antimicrob. Agents Chemother. 2004, 48, 477–483. [Google Scholar] [CrossRef]
- Tevyashova, A.N.; Olsufyeva, E.N.; Preobrazhenskaya, M.N. Design of dual action antibiotics as an approach to search for new promising drugs. Russ. Chem. Rev. 2015, 84, 61–97. [Google Scholar] [CrossRef]
- Bremner, J.B.; Ambrus, J.I.; Samosorn, S. Dual action-based approaches to antibacterial agents. Curr. Med. Chem. 2007, 14, 1459–1477. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, H.A.; Beskid, G.; Chan, K.-K.; Christenson, J.G.; Cleeland, R.; Deitcher, K.H.; Georgopapadakou, N.H.; Keith, D.D.; Pruess, D.L.; Sepinwall, J.; et al. Cephalosporin 3’-quinolone esters with a dual mode of action. J. Med. Chem. 1990, 33, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, H.A.; Beskid, G.; Christenson, J.G.; Durkin, J.W.; Fallat, V.; Georgopapadakou, N.H.; Keith, D.D.; Konzelmann, F.M.; Lipschitz, E.R.; McGarry, D.H.; et al. Dual action cephalosporins: Cephalosporin 3’-quaternary ammonium quinolones. J. Med. Chem. 1991, 34, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, H.A.; Beskid, G.; Christenson, J.G.; Georgopapadakou, N.H.; Keith, D.D.; Konzelmann, F.M.; Pruess, D.I.; Rossman, P.I.; Wei, C.C. Dual action cephalosporins: Cephalosporin 3’-quinolone carbamates. J. Med. Chem. 1991, 34, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Georgopapadakou, N.H.; Bertasso, A. Mechanisms of action of cephalosporin 3’-quinolone esters, carbamates, and tertiary amines in Escherichia coli. Antimicrob. Agents Chemother. 1993, 37, 559–565. [Google Scholar] [CrossRef]
- Bryskier, A. Dual β-lactam-fluoroquinolone compounds: A novel approach to antibacterial treatment. Exp. Opin. Investig. Drugs 2005, 6, 1479–1499. [Google Scholar] [CrossRef]
- Dax, S.L.; Pruess, D.L.; Rossman, P.L.; Wei, C.-C. Synthesis and mechanistic studies of a “tetrazole-tethered” cephalosporin-quinolone hybrid. Bioorg. Med. Chem. Lett. 1993, 3, 209–214. [Google Scholar] [CrossRef]
- Jones, R.N.; Barry, A.L.; Thornsberry, C. Antimicrobial activity of Ro 23–9424, a novel ester-linked codrug of fleroxacin and desacetylcefotaxime. Antimicrob. Agents Chemother. 1989, 33, 944–950. [Google Scholar] [CrossRef]
- Jones, R.N.; Barry, A.L. In vitro activity of Ro 23–9424, ceftazidime, and eight other newer beta-lactams against 100 gram-positive blood culture isolates. Diagn. Microbiol. Infect. Dis. 1989, 12, 143–147. [Google Scholar] [CrossRef]
- Beskid, G.; Fallat, V.; Lipschitz, E.R.; McGarry, D.H.; Cleeland, R.; Chan, K.; Keith, D.D.; Unowsky, J. In vitro activities of a dual-action antibacterial agent, Ro 23–9424, and comparative agents. Antimicrob. Agents Chemother. 1989, 33, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Georgopapadakou, N.H.; Bertasso, A.; Chan, K.K.; Chapman, J.S.; Cleeland, R.; Cummings, L.M.; Dix, B.A.; Keith, D.D. Mode of action of the dual-action cephalosporin Ro 23–9424. Antimicrob. Agents Chemother. 1989, 33, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.W.; Neu, H.C. In vitro activity of Ro 23–9424, a dual-action cephalosporin, compared with activities of other antibiotics. Antimicrob. Agents Chemother. 1990, 34, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.; Bertasso, A.; Georgopapadakou, N.H. Escherichia coli resistant to cephalosporins and quinolones is still susceptible to the cephalosporin-quinolone ester Ro 23–9424. Antimicrob. Agents Chemother. 1991, 35, 910–915. [Google Scholar] [CrossRef]
- Evans, L.E.; Krishna, A.; Ma, Y.; Webb, T.E.; Marshall, D.C.; Tooke, C.L.; Spencer, J.; Clarke, T.B.; Armstrong, A.; Edwards, A.M. Exploitation of antibiotic resistance as a novel drug target: Development of a β-lactamase-activated antibacterial prodrug. J. Med. Chem. 2019, 62, 4411–4425. [Google Scholar] [CrossRef]
- Corraz, A.J.; Dax, S.L.; Dunlap, N.K.; Georgopapadakou, N.H.; Keith, D.D.; Pruess, D.L.; Rossman, P.L.; Then, R.; Unowsky, J.; Wei, C.C. Dual-action penems and carbapenems. J. Med. Chem. 1992, 35, 1828–1839. [Google Scholar] [CrossRef]
- Alex, R.R.; Kulkarni, V.M. Design and synthesis of penicilloyl oxymethyl quinolone carbamates as a new class of dual-acting antibacterials. Eur. J. Med. Chem. 1995, 20, 815–818. [Google Scholar] [CrossRef]
- Smyth, T.P.; O’Donnell, M.E.; O’Connor, M.J.; St. Ledger, J.O. S-Aminosulfeniminopenicillins: Multimode β-lactamase inhibitors and template structures for penicillin-based β-lactamase substrates as prodrugs. J. Org. Chem. 1998, 63, 7600–7618. [Google Scholar] [CrossRef]
- Long, D.D.; Aggen, J.B.; Christensen, B.G.; Judice, J.K.; Hegde, S.S.; Kaniga, K.; Krause, K.M.; Linsell, M.S.; Moran, E.J.; Pace, J.L. A multivalent approach to drug discovery for novel antibiotics. J. Antibiot. (Tokyo) 2008, 61, 595–602. [Google Scholar] [CrossRef]
- Long, D.D.; Aggen, J.B.; Chinn, J.; Choi, S.K.; Christensen, B.G.; Fatheree, P.R.; Green, D.; Hegde, S.S.; Judice, J.K.; Kaniga, K.; et al. Exploring the positional attachment of glycopeptide/beta-lactam heterodimers. J. Antibiot. (Tokyo) 2008, 61, 603–614. [Google Scholar] [CrossRef]
- Leuthner, K.D.; Vidaillac, C.; Cheung, C.M.; Rybak, M.J. In vitro activity of the new multivalent glycopeptide-cephalosporin antibiotic TD-1792 against vancomycin-nonsusceptible Staphylococcus isolates. Antimicrob. Agents Chemother. 2010, 54, 3799–3803. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blais, J.; Lewis, S.R.; Krause, K.M.; Benton, B.M. Antistaphylococcal activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic. Antimicrob. Agents Chemother. 2012, 56, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, K.L.; Citron, D.M.; Warren, Y.A.; Goldstein, E.J.C. In vitro activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic, against 377 strains of anaerobic bacteria and 34 strains of Corynebacterium species. Antimicrob. Agents Chemother. 2012, 56, 2194–2197. [Google Scholar] [CrossRef] [PubMed]
- Hedge, S.S.; Okusanya, O.O.; Skinner, R.; Shaw, J.-P.; Obedenclo, G.; Ambrose, P.G.; Blais, J.; Bhavnani, S.M. Pharmacodynamics of TD-1792, a novel glycopeptide-cephalosporin heterodimer antibiotic used against Gram-positive bacteria, in a neutropenic murine thigh model. Antimicrob. Agents Chemother. 2012, 56, 1578–1583. [Google Scholar]
- Stryjewski, M.E.; Potgieter, P.D.; Li, Y.-P.; Barriere, S.L.; Churukian, A.; Kingsley, J.; Corey, G.R. TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections. Antimicrob. Agents Chemother. 2012, 56, 5476–5483. [Google Scholar] [CrossRef] [PubMed]
- Guang, X.; McLeod, X. Strategies for enzyme/prodrug cancer therapy. Clin. Cancer Res. 2001, 7, 3314–3324. [Google Scholar]
- Wang, Y.; Lambert, P.; Zhao, L.; Wang, D. Synthesis and antibacterial activity of dual-action agents of a β-lactam antibiotic with cytotoxic agent mitozolomide or temozolomide. Eur. J. Med. Chem. 2002, 37, 323–332. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Jalde, S.S.; Choi, H.K. Recent advances in the development of β-lactamase inhibitors. J. Microbiol. 2020, 58, 633–647. [Google Scholar] [CrossRef]
- Graef, F.; Gordon, S.; Lehr, C.M. Anti-infectives in drug delivery—Overcoming the Gram-negative bacterial cell envelope. Curr. Top. Microbiol. Immunol. 2016, 398, 475–496. [Google Scholar]
- Hider, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Pramanik, A.; Gwinner, T.; Köberle, M.; Bohn, E. Sideromycins: Tools and antibiotics. Biometals 2009, 22, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Page, M.G.P. The role of iron and siderophores in infection, and the development of siderophore antibiotics. Clin. Infect. Dis. 2019, 69 (Suppl. 7), S529–S537. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Ghosh, M.; Miller, P.A.; Möllmann, U.; Miller, M.J. Synthetic sideromycins (skepticism and optimism): Selective generation of either broad or narrow spectrum Gram-negative antibiotics. Biometals 2019, 32, 425–451. [Google Scholar] [CrossRef] [PubMed]
- Basker, M.J.; Branch, C.L.; Finch, S.C.; Guest, A.W.; Milner, P.H.; Pearson, M.J.; Ponsford, R.J.; Smale, T.C. Studies on semi-synthetic 7 alpha-formamidocephalosporins. I. Structure-activity relationships in some semi-synthetic 7 alpha-formamidocephalosporins. J. Antibiot. (Tokyo) 1986, 39, 1788–1791. [Google Scholar] [CrossRef][Green Version]
- Burton, G.; Best, D.J.; Dixon, R.A.; Kenyon, R.F.; Lashford, A.G. Studies on 6 alpha-substituted penicillins. II. Synthesis and structure-activity relationships of 6 beta-(2-aryl-2-sulfoacetamido)-6 alpha-methoxy penicillanic acids. J. Antibiot. (Tokyo) 1986, 39, 1419–1429. [Google Scholar] [CrossRef]
- Ohi, N.; Aoki, B.; Shinozaki, T.; Moro, K.; Noto, T.; Nehashi, T.; Okazaki, H.; Matsunaga, I. Semisynthetic beta-lactam antibiotics. I. Synthesis and antibacterial activity of new ureidopenicillin derivatives having catechol moieties. J. Antibiot. (Tokyo) 1986, 39, 230–241. [Google Scholar] [CrossRef]
- Ohi, N.; Aoki, B.; Moro, K.; Kuroki, T.; Sugimura, N.; Noto, T.; Nehashi, T.; Matsumoto, M.; Okazaki, H.; Matsunaga, I. Semisynthetic beta-lactam antibiotics. II. Effect on antibacterial activity of ureido N-substituents in the 6-[(R)-2-[3-(3,4-dihydroxybenzoyl)-1- ureido]-2-phenylacetamido]penicillanic acids. J. Antibiot. (Tokyo) 1986, 39, 242–250. [Google Scholar] [CrossRef]
- Ohi, N.; Aoki, B.; Kuroki, T.; Matsumoto, M.; Kojima, K.; Nehashi, T. Semisynthetic beta-lactam antibiotics. III. Effect on antibacterial activity and comt-susceptibility of chlorine-introduction into the catechol nucleus of 6-[(R)-2-[3-(3,4-dihydroxybenzoyl)-3-(3-hydroxypropyl)-1-ureido]-2-phenylacetamido]penicillanic acid. J. Antibiot. (Tokyo) 1987, 40, 22–28. [Google Scholar]
- Ohi, N.; Aoki, B.; Shinozaki, T.; Moro, K.; Kuroki, T.; Noto, T.; Nehashi, T.; Matsumoto, M.; Okazaki, H.; Matsunaga, I. Semisynthetic beta-lactam antibiotics. IV. Synthesis and antibacterial activity of new ureidocephalosporin and ureidocephamycin derivatives containing a catechol moiety or its acetate. Chem. Pharm. Bull. (Tokyo) 1987, 35, 1903–1909. [Google Scholar] [CrossRef]
- Mochida, K.; Shiraki, C.; Yamasaki, M.; Hirata, T.; Sato, K.; Okachi, R. Aminothiazolylglycyl derivatives of carbacephems. I. Synthesis and antibacterial activity of novel carbacephems with substituted aminothiazolyl groups. J. Antibiot. (Tokyo) 1987, 40, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Ono, Y.; Yamasaki, M.; Shiraki, C.; Hirata, T.; Sato, K.; Okachi, R. Aminothiazolylglycyl derivatives of carbacephem antibiotics. II. Synthesis and antibacterial activity of novel aminothiazolyl cephem compounds with hydroxypyridone moiety. J. Antibiot. (Tokyo) 1987, 40, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Sanada, M.; Matsuda, K.; Hazumi, N.; Tanaka, N. Biological activity of BO-1236, a new antipseudomonal cephalosporin. Antimicrob. Agents Chemother. 1987, 31, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.A.; Nagasu, T.; Katsu, K.; Kitoh, K. E-0702, a new cephalosporin, is incorporated into Escherichia coli cells via the tonB-dependent iron transport system. Antimicrob. Agents Chemother. 1987, 31, 497–504. [Google Scholar] [CrossRef]
- Curtis, N.A.; Eisenstadt, R.L.; East, S.J.; Cornford, R.J.; Walker, L.A.; White, A.J. Iron-regulated outer membrane proteins of Escherichia coli K-12 and mechanism of action of catechol-substituted cephalosporins. Antimicrob. Agents Chemother. 1988, 32, 1879–1886. [Google Scholar]
- Negash, K.H.; Norris, J.K.S.; Hodgkinson, J.T. Siderophore-antibiotic conjugate design: New drugs for bad bugs? Molecules 2019, 24, 3314. [Google Scholar] [CrossRef]
- Kong, H.; Cheng, W.; Wei, H.; Yuan, Y.; Yang, Z.; Zhang, X. An overview of recent progress in siderophore-antibiotic conjugates. Eur. J. Med. Chem. 2019, 182, 111615. [Google Scholar] [CrossRef]
- Page, M.G.P. Siderophore conjugates. Ann. N. Y. Acad. Sci. 2013, 1277, 115–126. [Google Scholar]
- Sykes, R.B.; Koster, W.H.; Bonner, D.P. The new monobactams: Chemistry and biology. J. Clin. Pharmacol. 1988, 28, 113–119. [Google Scholar] [CrossRef]
- Barbachyn, M.R.; Tuominen, T.C. Synthesis and structure-activity relationships of monocarbams leading to U-78608. J. Antibiot. (Tokyo) 1990, 43, 1199–1202. [Google Scholar] [CrossRef]
- Zurenko, G.E.; Truesdell, S.E.; Yagi, B.H.; Mourey, R.J.; Laborde, A.L. In vitro antibacterial activity and interactions with beta-lactamases and penicillin-binding proteins of the new monocarbam antibiotic U-78608. Antimicrob. Agents Chemother. 1990, 34, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 21, 22002–22007. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; Crandon, J.L.; McPherson, C.J.; Nicolau, P. Potentiation of antibacterial activity of the MB-1 siderophore-monobactam conjugate using an efflux pump inhibitor. Antimicrob. Agents Chemother. 2015, 59, 2439–2442. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Miller, P.A.; Vakulenko, S.B.; Stewart, N.K.; Boggess, W.C.; Miller, M.J. A synthetic dual drug sideromycin induces Gram-negative bacteria to commit suicide with a Gram-positive antibiotic. J. Med. Chem. 2018, 61, 3845–3854. [Google Scholar] [CrossRef]
- Brochu, A.; Brochu, N.; Nicas, T.; Parr, T.R., Jr.; Minnick, A.A.J.; Dolence, E.K.; McKee, J.A.; Miller, M.J.; Lavoie, M.C.; Malouin, F. Modes of action and inhibitory activities of new siderophore-beta-lactam conjugates that use specific iron uptake pathways for entry into bacteria. Antimicrob. Agents Chemother. 1992, 36, 2166–2175. [Google Scholar] [CrossRef]
- Wencewicz, T.A.; Möllmann, U.; Long, T.E.; Miller, M.J. Is drug release necessary for antimicrobial activity of siderophore-drug conjugates? Syntheses and biological studies of the naturally occurring salmycin “Trojan Horse” antibiotics and synthetic desferridanoxamine-antibiotic conjugates. Biometals 2009, 22, 633–648. [Google Scholar] [CrossRef]
- Zheng, N.T.; Nolan, E.M. Enterobactin-mediated delivery of β-lactam antibiotics enhances antibacterial activity against pathogenic Escherichia coli. J. Am. Chem. Soc. 2014, 136, 9677–9691. [Google Scholar] [CrossRef]
- McKee, J.A.; Sharma, S.K.; Miller, M.J. Iron transport mediated drug delivery systems: Synthesis and antibacterial activity of spemidine- and lysine-based siderophore-beta-lactam conjugates. Bioconjugate Chem. 1991, 2, 281–291. [Google Scholar] [CrossRef]
- Möllmann, U.; Ghosh, A.; Dolence, E.K.; Dolence, J.A.; Ghosh, M.; Miller, M.J.; Reissbrodt, R. Selective growth promotion and growth inhibition of gram-negative and gram-positive bacteria by synthetic siderophore-beta-lactam conjugates. Biometals 1998, 11, 1–12. [Google Scholar]
- Nikaido, H.; Rosenberg, E.Y. Cir and Fiu proteins in the outer membrane of Escherichia coli catalyze transport of monomeric catechols: Study with beta-lactam antibiotics containing catechol and analogous groups. J. Bacteriol. 1990, 172, 1361–1367. [Google Scholar] [CrossRef]
- Minnick, A.A.; McKee, J.A.; Dolence, E.K.; Miller, M.J. Iron transport-mediated antibacterial activity of and development of resistance to hydroxamate and catechol siderophore-carbacephalosporin conjugates. Antimicrob. Agents Chemother. 1992, 36, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; Crandon, J.L.; McPherson, C.J.; Banevicius, M.A.; Finegan, S.M.; Irvine, R.L.; Brown, M.F.; O’Donnell, J.P.; Nicolau, D.P. Adaptation-based resistance to siderophore-conjugated antibacterial agents by Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4197–4207. [Google Scholar] [CrossRef]
- Page, M.G.P.; Dantier, C.; Desarbre, E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant Gram-negative bacteria. Antimicrob. Agents Chemother. 2010, 54, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S.; Warner, M.; Livermore, D. Activity of the siderophore monobactam BAL30072 against multiresistant non-fermenters. J. Antimicrob. Chemother. 2010, 65, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Mima, T.; Kvitko, B.H.; Rholl, D.A.; Page, M.G.; Desarbre, E.; Schweizer, H.P. In vitro activity of BAL30072 against Burkholderia pseudomallei. Int. J. Antimicrob. Agents 2011, 38, 157–159. [Google Scholar] [CrossRef]
- Russo, T.A.; Page, M.G.P.; Beanan, J.M.; Olson, R.; Hujer, A.M.; Hujer, K.M.; Jacobs, M.; Bajaksouzian, S.; Endimiani, A.; Bonomo, R.A. In vivo and in vitro activity of the siderophore monosulfactam BAL30072 against Acinetobacter baumannii. J. Antimicrob. Chemother. 2011, 66, 867–873. [Google Scholar] [CrossRef]
- Higgins, P.G.; Stefanik, D.; Page, M.G.P.; Hackel, M.; Seifert, H. In vitro activity of the siderophore monosulfactam BAL30072 against meropenem-non-suceptible Acinetobacter baumannii. J. Antimicrob. Chemother. 2012, 67, 1167–1169. [Google Scholar] [CrossRef]
- Bird, T.G.; Arnould, J.C.; Bertrandie, A.; Jung, F.H. Pharmacokinetics of catechol cephalosporins. The effect of incorporating substituents into the catechol moiety on pharmacokinetics in a marmoset model. J. Med. Chem. 1992, 35, 2643–2651. [Google Scholar]
- Abdul-Mutakabbir, J.C.; Alosaimy, S.; Morrisette, T.; Kebriaei, R.; Rybak, M.J. Cefiderocol: A novel siderophore cephalosporin against multidrug-resistant Gram-negative pathogens. Pharmacotherapy 2020, 40, 1228–1247. [Google Scholar] [CrossRef]
- Dobias, J.; Denervaud-Tendon, V.; Poirel, L.; Nordmann, P. Activity of the novel siderophore cephalosporin cefiderocol against multidrug-resistant Gram-negative pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2319–2327. [Google Scholar] [CrossRef]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In vitro antibacterial properties of Cefiderocol, a novel siderophore cephalosporin, against Gram-negative bacteria. Antimicrob. Agents Chemother. 2017, 62, e0145-17. [Google Scholar] [CrossRef] [PubMed]
- Hackel, M.A.; Tsuji, M.; Yamano, Y.; Echols, R.; Karlowsky, J.A.; Sahm, D.F. In vitro activity of the siderophore cephalosporin, Cefiderocol, against a recent collection of clinically relevant Gram-negative bacilli from North America and Europe, including carbapenem-nonsusceptible isolates (SIDERO-WT-2014 Study). Antimicrob. Agents Chemother. 2017, 61, e00093-17. [Google Scholar] [CrossRef] [PubMed]
- Hackel, M.A.; Tsuji, M.; Yamano, Y.; Echols, R.; Karlowsky, J.A.; Sahm, D.F. In vitro activity of the siderophore cephalosporin, Cefiderocol, against carbapenem-nonsusceptible and multidrug-resistant isolates of Gram-negative bacilli collected worldwide in 2014 to 2016. Antimicrob. Agents Chemother. 2018, 62, e01968-17. [Google Scholar] [CrossRef] [PubMed]
- Karlowsky, J.A.; Hackel, M.A.; Tsuji, M.; Yamano, Y.; Echols, R.; Sahm, D.F. In vitro activity of Cefiderocol, a siderophore cephalosporin, against Gram-negative bacilli isolated by clinical laboratories in North America and Europe in 2015–2016: SIDERO-WT-2015. Int. J. Antimicrob. Agents 2019, 53, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, K.M.; Tsuji, M.; Wise, M.G.; Hackel, M.; Yamano, Y.; Echols, R.; Sahm, D.F. In vitro activity of cefiderocol, a siderophore cephalosporin, against a recent collection of clinically relevant carbapenem-non-susceptible Gram-negative bacilli, including serine carbapenemase- and metallo-beta-lactamase-producing isolates (SIDERO-WT-2014 Study). Int. J. Antimicrob. Agents 2019, 53, 177–184. [Google Scholar] [PubMed]
- Yamano, Y. In vitro activity of Cefiderocol against a broad range of clinically important Gram-negative bacteria. Clin. Infect. Dis. 2019, 69 (Suppl. 7), S544–S551. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.K.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A.; et al. Cefiderocol: A siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-negative bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef]
- Golden, A.R.; Adam, H.J.; Baxter, M.; Walkty, A.; Lagacé-Wiens, P.; Karlowsky, J.A.; Zhanel, G.G. In vitro activity of Cefiderocol, a novel siderophore cephalosporin, against Gram-negative bacilli isolated from patients in Canadian intensive care units. Diagn. Microbiol. Infect. Dis. 2020, 97, 115012. [Google Scholar] [CrossRef]
- Ito-Horiyama, T.; Ishii, Y.; Ito, A.; Sato, T.; Nakamura, R.; Fukuhara, N.; Tsuji, M.; Yamano, Y.; Yamaguchi, K.; Tateda, K. Stability of novel siderophore cephalosporin S-649266 against clinically relevant carbapenemases. Antimicrob. Agents Chemother. 2016, 60, 4384–4386. [Google Scholar] [CrossRef]
- Ito, A.; Nishikawa, T.; Ota, M.; Ito-Horiyama, T.; Ishibashi, N.; Sato, T.; Tsuji, M.; Yamano, Y. Stability and low induction propensity of cefiderocol against chromosomal AmpC beta-lactamases of Pseudomonas aeruginosa and Enterobacter cloacae. J. Antimicrob. Chemother. 2018, 73, 3049–3052. [Google Scholar] [CrossRef]
- Poirel, L.; Kieffer, N.; Nordmann, P. Stability of cefiderocol against clinically significant broad-spectrum oxacillinases. Int. J. Antimicrob. Agents 2018, 52, 866–867. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Li, Q.; Ju, J. Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 2014, 4, 50566. [Google Scholar] [CrossRef]
- Rosett, T.; Sankhala, R.H.; Stickings, C.E.; Taylor, M.E.; Thomas, R. Studies in the biochemistry of micro-organisms. 103. Metabolites of Alternaria tenuis auct; culture filtrate products. Biochem. J. 1957, 67, 390–400. [Google Scholar] [CrossRef]
- Cherian, P.T.; Deshpande, A.; Cheramie, M.N.; Bruhn, D.F.; Hurdle, J.G.; Lee, R.E. Design, synthesis and microbiological evaluation of ampicillin tetramic acid hybrid antibiotics. J. Antibiot. (Tokyo) 2017, 70, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Cui, Z.; Gu, Y.; Liu, Y.; Wang, Q. The phytotoxicity of natural tetramic acid derivatives. Pest. Manag. Sci. 2011, 67, 1059–1061. [Google Scholar] [CrossRef]
- Steyn, P.S.; Rabie, C.J. Characterization of magnesium and calcium tenuazonate from Phoma sorghina. Phytochemistry 1976, 15, 1977–1979. [Google Scholar] [CrossRef]
- Lebrun, M.-H.; Duvert, P.; Gaudemer, F.; Gaudemer, A.; Deballon, C.; Boucly, P. Complexation of the fungal metabolite tenuazonic acid with copper (II), iron (III), nickel (II), and magnesium (II) ions. J. Inorg. Biochem. 1985, 24, 167–181. [Google Scholar] [CrossRef]
- Vinale, F.; Nigro, M.; Sivasithamparam, K.; Flematti, G.; Ghisalberti, E.L.; Ruocco, M.; Varlese, R.; Marra, R.; Lanzuise, S.; Eid, A.; et al. Harzianic acid: A novel siderophore from Trichoderma harzianum. FEMS Microbiol. Lett. 2013, 347, 123–129. [Google Scholar] [CrossRef]
- Ferres, H.; Basker, M.J.; Best, D.J.; Harrington, F.P.; O’Hanlon, P.J. Beta-lactam antibiotics. II Structure-activity relationships of 6-[alpha-(alpha-ureidoacylamino)acylamino] penicillanic acids. J. Antibiot. (Tokio) 1978, 31, 1013–1022. [Google Scholar] [CrossRef]
- Cherian, P.T.; Cheramie, M.N.; Marreddy, R.K.R.; Fernando, D.M.; Hurdle, J.G.; Lee, R.E. New β-lactam-tetramic acid hybrids show promising antibacterial activities. Bioorg. Med. Chem. Lett. 2018, 28, 3105–3112. [Google Scholar] [CrossRef]
- Projan, S. Why is big Pharma getting out of antibacterial drug discovery? Curr. Opin. Microbiol. 2003, 6, 427–430. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rosa, M.; Verdino, A.; Soriente, A.; Marabotti, A. The Odd Couple(s): An Overview of Beta-Lactam Antibiotics Bearing More Than One Pharmacophoric Group. Int. J. Mol. Sci. 2021, 22, 617. https://doi.org/10.3390/ijms22020617
De Rosa M, Verdino A, Soriente A, Marabotti A. The Odd Couple(s): An Overview of Beta-Lactam Antibiotics Bearing More Than One Pharmacophoric Group. International Journal of Molecular Sciences. 2021; 22(2):617. https://doi.org/10.3390/ijms22020617
Chicago/Turabian StyleDe Rosa, Margherita, Anna Verdino, Annunziata Soriente, and Anna Marabotti. 2021. "The Odd Couple(s): An Overview of Beta-Lactam Antibiotics Bearing More Than One Pharmacophoric Group" International Journal of Molecular Sciences 22, no. 2: 617. https://doi.org/10.3390/ijms22020617
APA StyleDe Rosa, M., Verdino, A., Soriente, A., & Marabotti, A. (2021). The Odd Couple(s): An Overview of Beta-Lactam Antibiotics Bearing More Than One Pharmacophoric Group. International Journal of Molecular Sciences, 22(2), 617. https://doi.org/10.3390/ijms22020617