
Nuclear Receptor Nur77 Controls Cardiac Fibrosis through Distinct Actions on Fibroblasts and Cardiomyocytes

 and
and 
Abstract
1. Introduction
2. Results
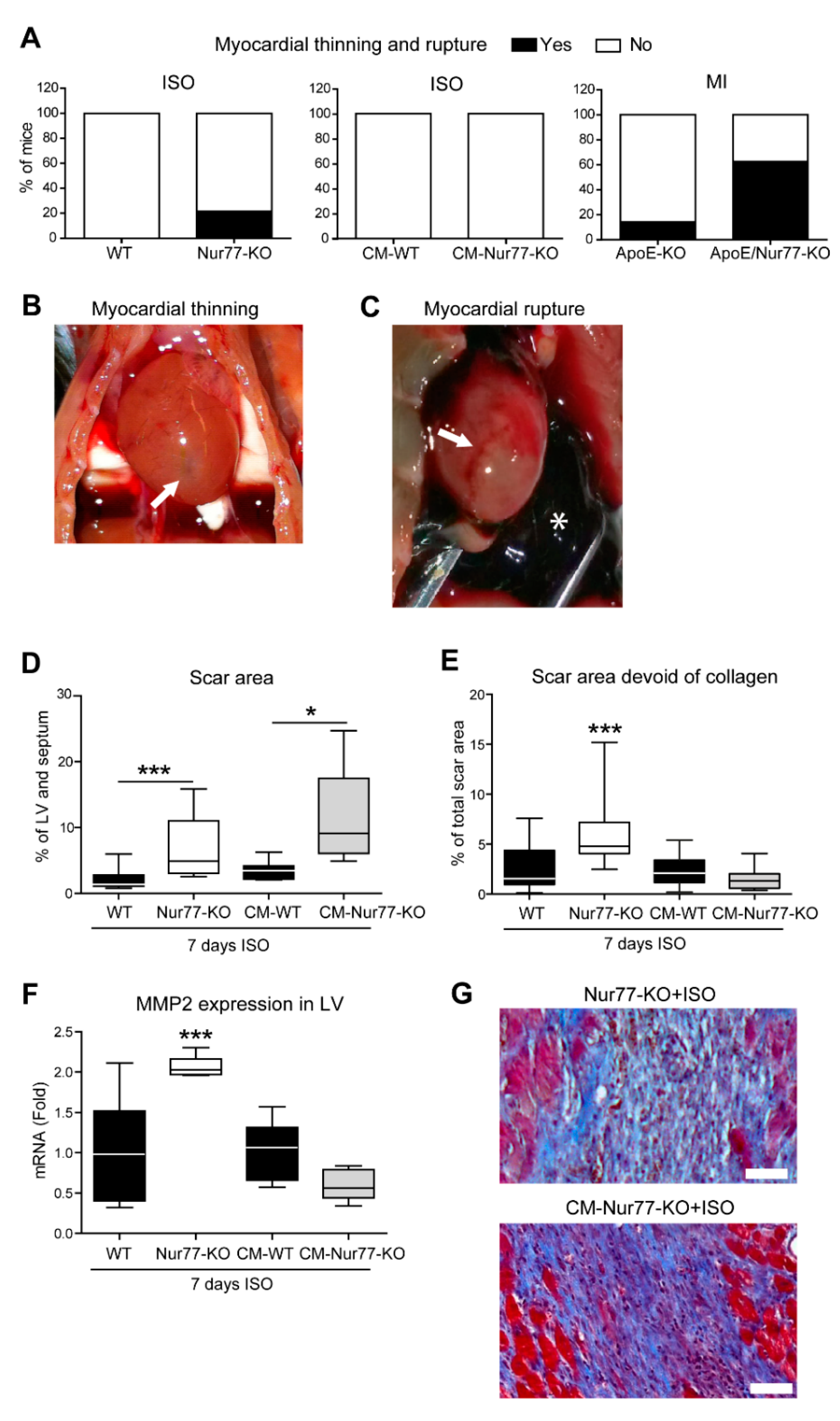
2.1. Cardiac Ruptures and Reduced Cardiac Scar Density Are Observed in Nur77-KO Mice
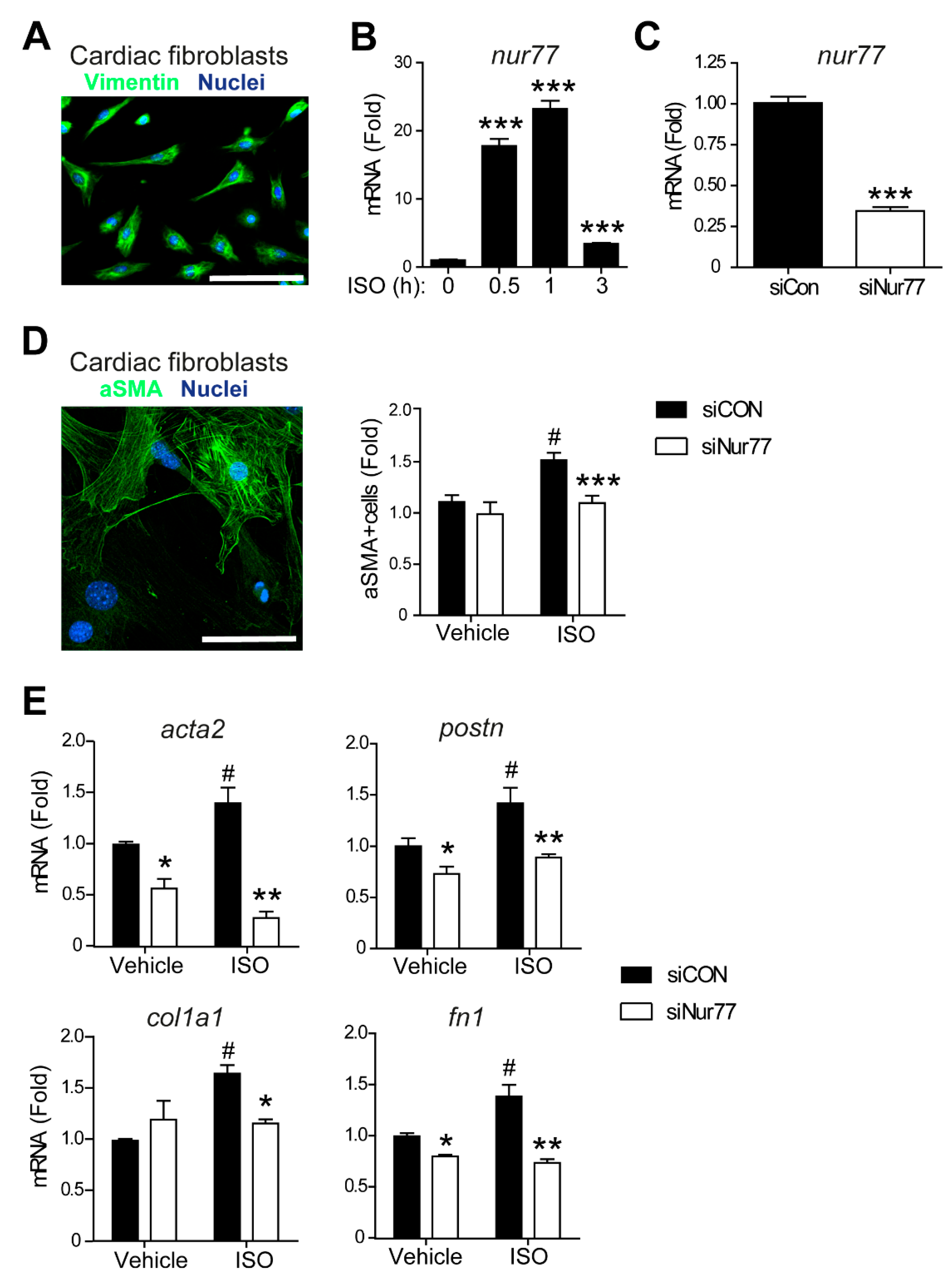
2.2. Nur77 Knockdown in CFs Represses MyoFB Marker Expression
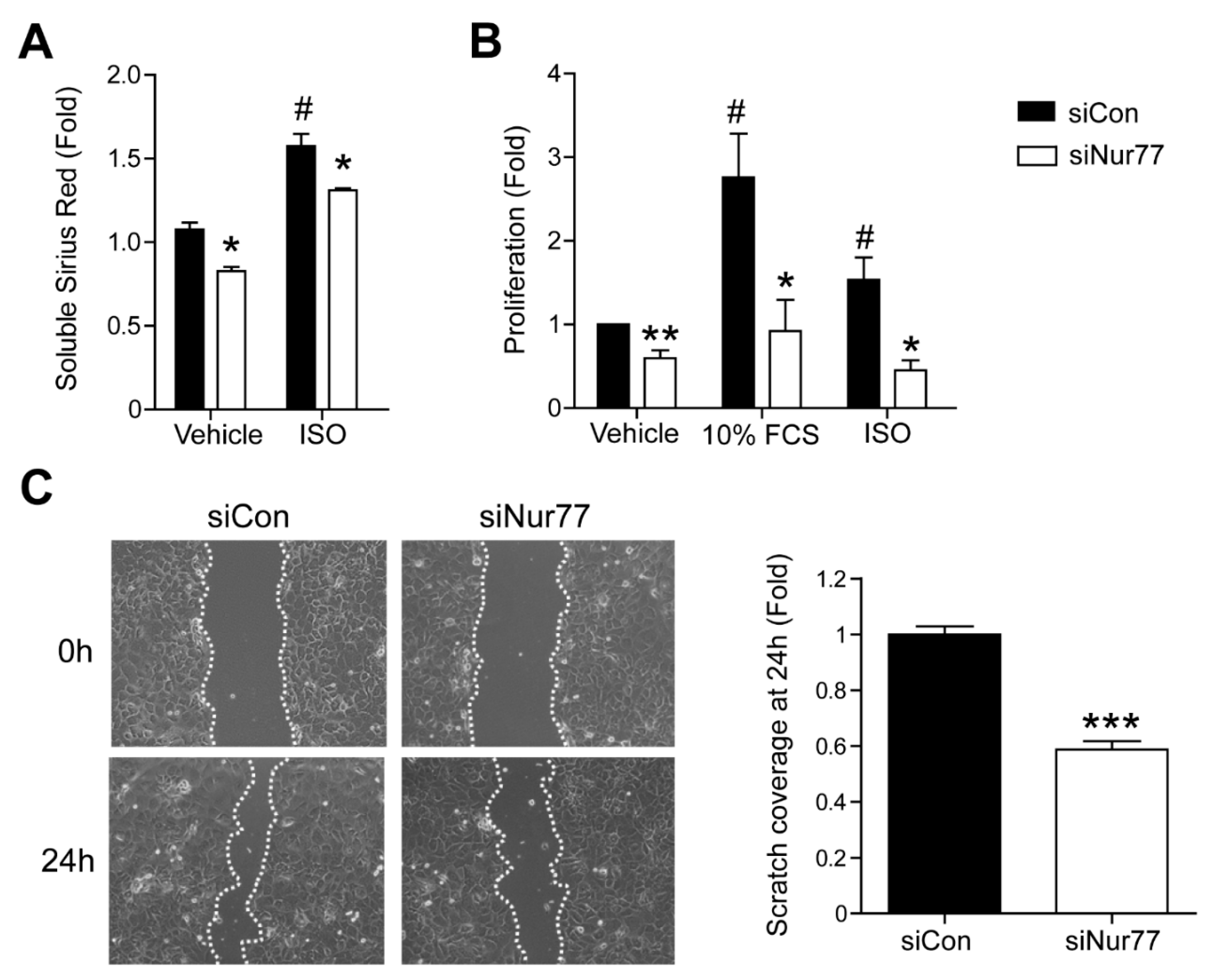
2.3. Nur77 Knockdown in CFs Represses MyoFB Functional Characteristics
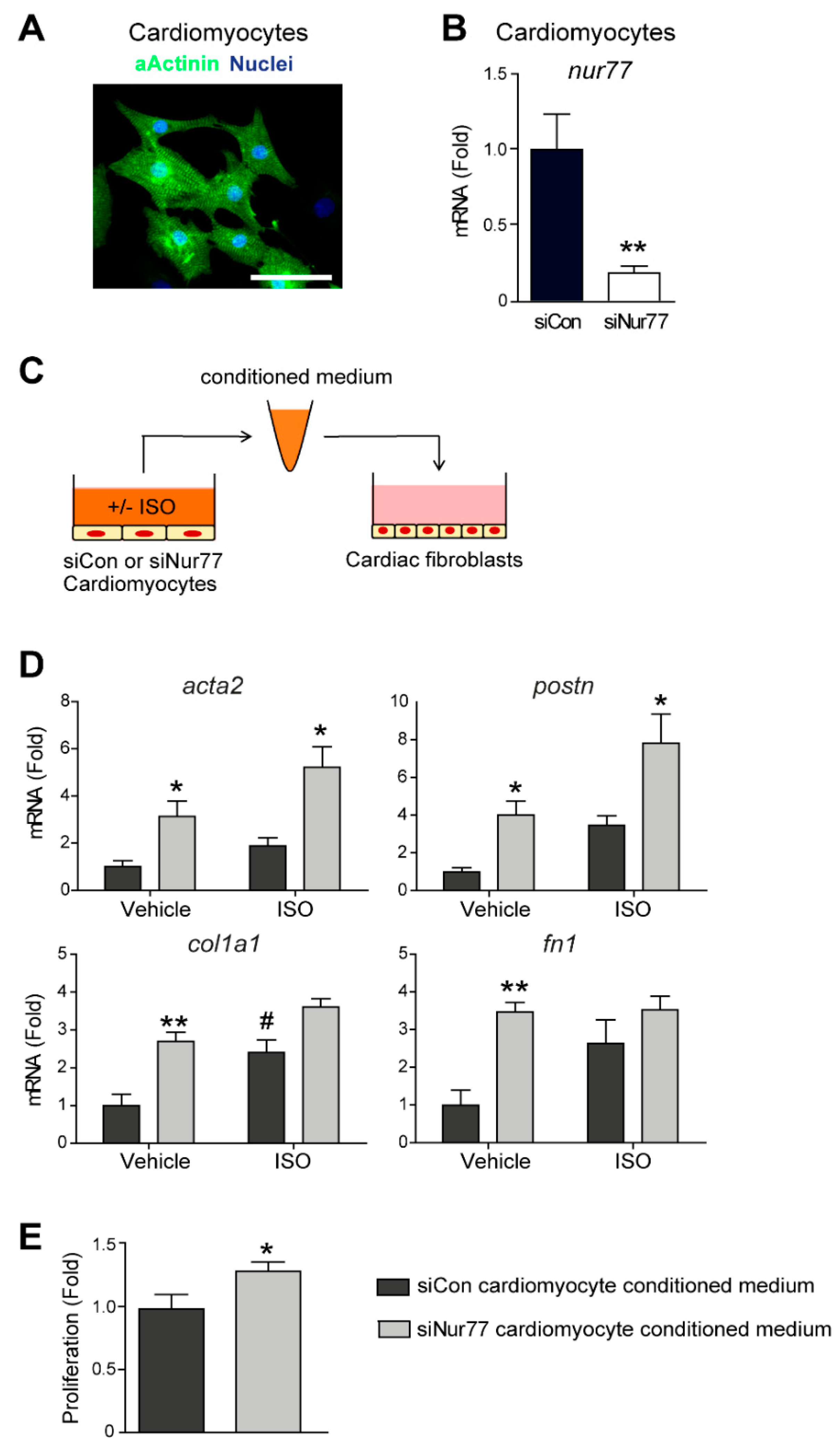
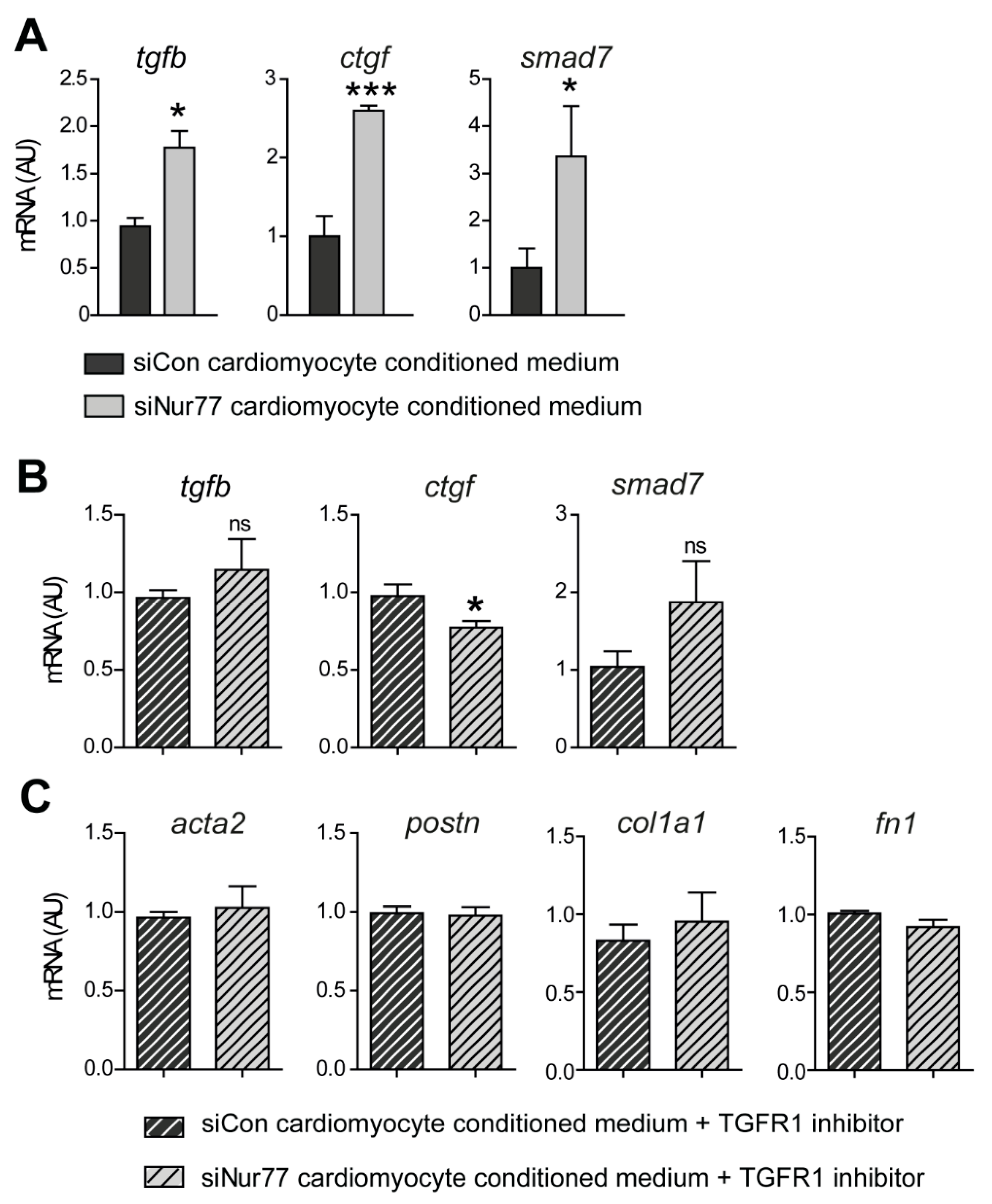
2.4. Paracrine Factors from Nur77-Silenced Cardiomyocytes Promote MyoFB Differentiation
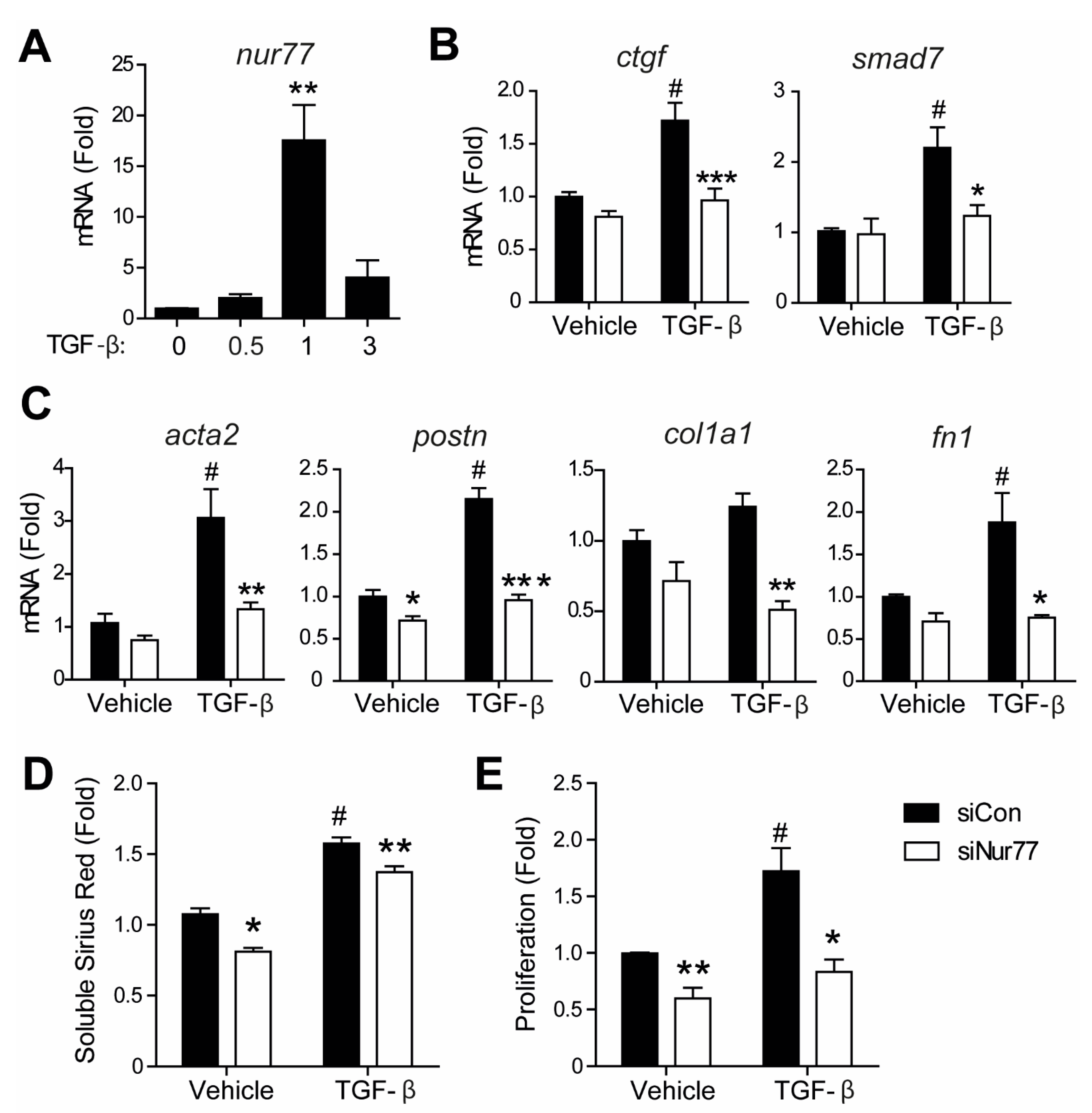
2.5. Nur77-Silenced Cardiomyocytes Promote a MyoFB Phenotype via TGF-β
2.6. Nur77 Silencing in CF Inhibits TGF-β-Induced Signaling and MyoFB Transition
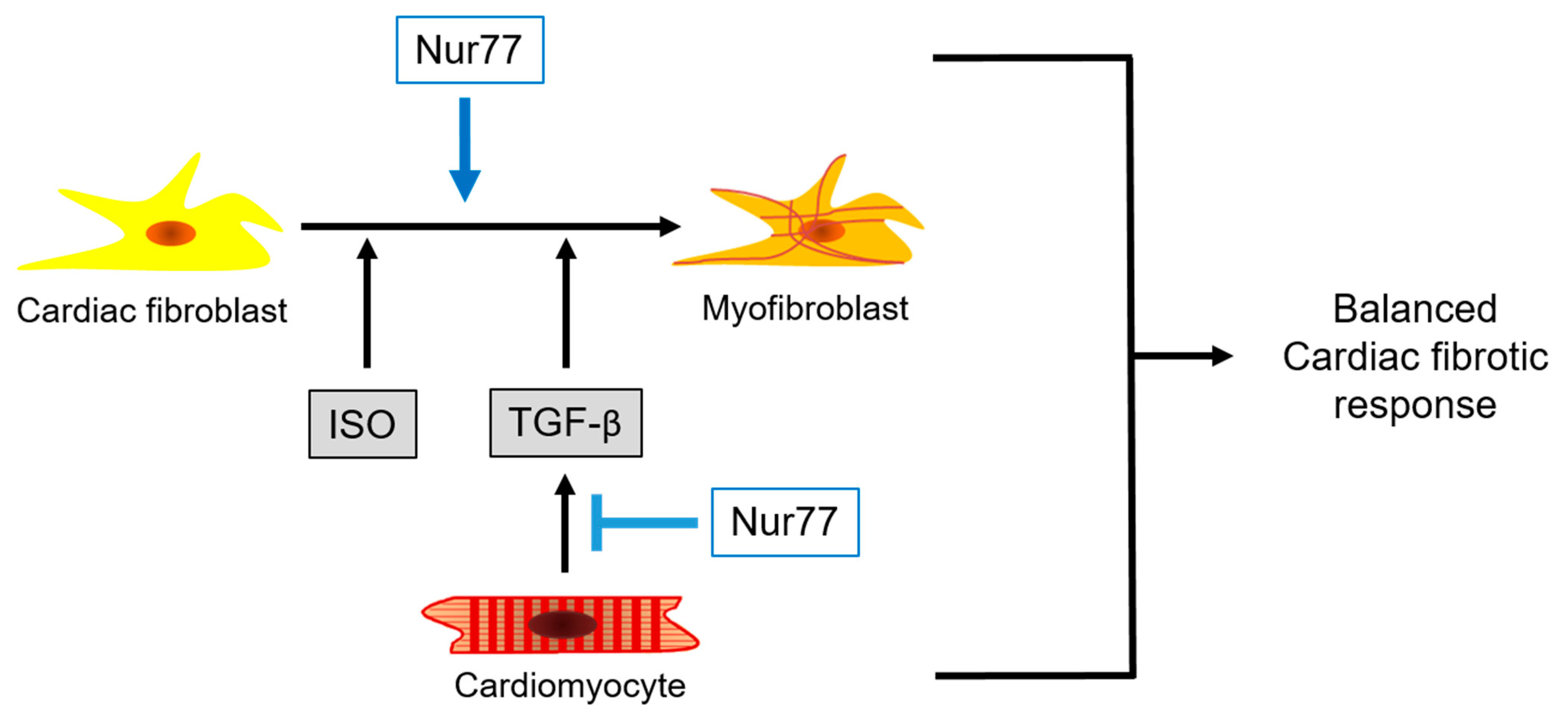
3. Discussion
4. Methods
4.1. Animal Experiments
4.2. LAD Ligation
4.3. In Vivo Isoproterenol-Induced Fibrosis
4.4. Autopsy
4.5. Collagen Density Measurements
4.6. Neonatal Rat Cardiomyocyte (NRCM) and Cardiac Fibroblast (CF) Isolation, Culture, and Stimulation
4.7. Nur77 Knockdown
4.8. RNA Isolation and qPCR
4.9. Immunofluorescence
4.10. Soluble Sirius Red Assay
4.11. Proliferation Assay
4.12. Scratch Wound Assay
4.13. Conditioned Medium Experiments
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gabriel-Costa, D. The pathophysiology of myocardial infarction-induced heart failure. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2018. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic Nervous System in Heart Failure. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Burns, C. Cardiac Fibrosis: The Fibroblast Awakens. PubMed NCBI 2017, 118, 1021–1040. [Google Scholar] [CrossRef]
- Piek, A.; De Boer, R.A.; Silljé, H.H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Circ. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef]
- Forte, E.; Skelly, D.A.; Chen, M.; Daigle, S.; Morelli, K.A.; Hon, O.; Philip, V.M.; Costa, M.W.; Rosenthal, N.A.; Furtado, M.B. Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Rep. 2020, 30, 3149–3163.e6. [Google Scholar] [CrossRef]
- Leicht, M.; Greipel, N.; Zimmer, H. Comitogenic effect of catecholamines on rat cardiac fibroblasts in culture. Circ. Res. 2000, 48, 274–284. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E.; Smith, W.H.T.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Circ. Res. 2003, 57, 784–792. [Google Scholar]
- Cartledge, J.E.; Kane, C.; Dias, P.; Tesfom, M.; Clarke, L.; McKee, B.; Al Ayoubi, S.; Chester, A.; Yacoub, M.H.; Camelliti, P.; et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Circ. Res. 2015, 105, 260–270. [Google Scholar] [CrossRef]
- Nuamnaichati, N.; Sato, V.H.; Moongkarndi, P.; Parichatikanond, W.; Mangmool, S. Sustained β-AR stimulation induces synthesis and secretion of growth factors in cardiac myocytes that affect on cardiac fibroblast activation. Life Sci. 2018, 193, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Circ. Res. 2007, 74, 207–212. [Google Scholar] [CrossRef]
- Chao, L.C.; Wroblewski, K.; Ilkayeva, O.R.; Stevens, R.D.; Bain, J.; Meyer, G.A.; Schenk, S.; Martinez, L.; Vergnes, L.; Narkar, V.A.; et al. Skeletal muscle Nur77 expression enhances oxidative metabolism and substrate utilization. J. Lipid Res. 2012, 53, 2610–2619. [Google Scholar] [CrossRef]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef]
- Fernandez, P.M.; Brunel, F.; Saez, J.M.; Cereghini, S.; Zakin, M.M. Albeit Distinct, Roles in the Hypothalamo-Pituitary-Adrenal Axis *. Society 2000, 141, 2392–2400. [Google Scholar]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef]
- Hiwatashi, N.; Bing, R.; Kraja, I.; Branski, R.C. NR4A1 is an endogenous inhibitor of vocal fold fibrosis. Laryngoscope 2017, 127, E317–E323. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yue, Z.; Gao, Y.; Jiang, G.; Zeng, F.; Shao, Y.; Huang, J.; Yin, M.; Li, Y. NR4A1 is Involved in Fibrogenesis in Ovarian Endometriosis. Cell. Physiol. Biochem. 2018, 46, 1078–1090. [Google Scholar] [CrossRef]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.A.; De Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.A.C.; Sheppard, K.-A.; Goumans, M.-J.; Luwor, R.B.; De Vries, C.J.; et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014, 5, 3388. [Google Scholar] [CrossRef]
- Hedrick, E.; Lee, S.-O.; Doddapaneni, R.; Singh, M.; Safe, S. NR4A1 Antagonists Inhibit β1-Integrin-Dependent Breast Cancer Cell Migration. Mol. Cell. Biol. 2016, 36, 1383–1394. [Google Scholar] [CrossRef]
- Medzikovic, L.; Schumacher, C.A.; Verkerk, A.O.; Van Deel, E.D.; Wolswinkel, R.; Van Der Made, I.; Bleeker, N.; Cakici, D.; Hoogenhof, M.M.G.V.D.; Meggouh, F.; et al. Orphan nuclear receptor Nur77 affects cardiomyocyte calcium homeostasis and adverse cardiac remodelling. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.H.; Jebessa, Z.H.; Kreusser, M.M.; Horsch, A.; Hermann-Josef, G.; Kronlage, M.; Dewenter, M.; Sramek, V.; Oehl, U.; Krebs-Haupenthal, J.; et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 2018, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Völkers, M.; Din, S.; Avitabile, D.; Khan, M.; Gude, N.; Mohsin, S.; Bo, T.; Truffa, S.; Alvarez, R.; et al. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur. Heart J. 2011, 32, 2179–2188. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.S.; Tan, T.C.; Winter, C.; Holderried, T.A.W.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6C high Monocytes Depend on Nr4a1 to Balance Both Inflammatory and Reparative Phases in the Infarcted Myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Medzikovic, L.; van Roomen, C.; Baartscheer, A.; van Loenen, P.B.; de Vos, J.; Bakker, E.N.T.P.; Koenis, D.S.; Damanafshan, A.; Creemers, E.E.; Arkenbout, E.K.; et al. Nur77 protects against adverse cardiac remodelling by limiting Neuropeptide Y signalling in the sympathoadrenal-cardiac axis. Circ. Res. 2018, 114, 1617–1628. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Y-Hassan, S.; Tornvall, P. Epidemiology, pathogenesis, and management of takotsubo syndrome. Clin. Auton. Res. Off. J. Clin. Auton. Res. Soc. 2018, 28, 53–65. [Google Scholar] [CrossRef]
- Kumar, S.; Kaushik, S.; Nautiyal, A.; Choudhary, S.K.; Kayastha, B.L.; Mostow, N.; Lazar, J.M. Cardiac Rupture in Takotsubo Cardiomyopathy: A Systematic Review. Clin. Cardiol. 2011, 34, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Van Craeyveld, E.; Jacobs, F.; Gordts, S.C.; De Geest, B. Low-density lipoprotein receptor gene transfer in hypercholesterolemic mice improves cardiac function after myocardial infarction. Gene Ther. 2012, 19, 860–871. [Google Scholar] [CrossRef]
- Pellman, J.; Zhang, J.; Sheikh, F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J. Mol. Cell. Cardiol. 2016, 94, 22–31. [Google Scholar] [CrossRef]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (New York, NY) 2013, 339, 161–166. [Google Scholar] [CrossRef]
- Marinković, G.; Koenis, D.S.; De Camp, L.; Jablonowski, R.; Graber, N.; De Waard, V.; De Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Chen, J.; Jia, J.; Ma, L.; Li, B.; Qin, Q.; Ge, J.; Ge, J. Nur77 deficiency exacerbates cardiac fibrosis after myocardial infarction by promoting endothelial-to-mesenchymal transition. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.M.; Dronkers, E.; Goumans, M.-J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Koenis, D.S.; Medzikovic, L.; Van Loenen, P.B.; Van Weeghel, M.; Huveneers, S.; Vos, M.; Gogh, I.J.E.-V.; Bossche, J.V.D.; Speijer, D.; Kim, Y.; et al. Nuclear Receptor Nur77 Limits the Macrophage Inflammatory Response through Transcriptional Reprogramming of Mitochondrial Metabolism. Cell Rep. 2018, 24, 2127–2140.e7. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Hamers, A.A.J.; Argmann, C.; Moerland, P.D.; Koenis, D.S.; Marinković, G.; Sokolović, M.; De Vos, A.F.; De Vries, C.J.M.; Van Tiel, C.M. Nur77-deficiency in bone marrow-derived macrophages modulates inflammatory responses, extracellular matrix homeostasis, phagocytosis and tolerance. BMC Genom. 2016, 17, 162. [Google Scholar] [CrossRef] [PubMed]
- Bonta, P.I.; Matlung, H.L.; Vos, M.; Peters, S.L.; Pannekoek, H.; Bakker, E.N.T.P.; De Vries, C.J.M. Nuclear receptor Nur77 inhibits vascular outward remodelling and reduces macrophage accumulation and matrix metalloproteinase levels. Cardiovasc. Res. 2010, 87, 561–568. [Google Scholar] [CrossRef]
- Koenis, D.S.; Medzikovic, L.; Vos, M.; Beldman, T.J.; Van Loenen, P.; Van Tiel, C.M.; Hamers, A.A.; Rubio, I.O.; De Waard, V.; De Vries, C.J. Nur77 variants solely comprising the amino-terminal domain activate hypoxia-inducible factor-1α and affect bone marrow homeostasis in mice and humans. J. Biol. Chem. 2018, 293, 15070–15083. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat Primer | Forward | Reverse |
| acta2 | TTCAATGTCCCTGCCATGTA | GAAGGAATAGCCACGCTCAG |
| col1a1 | TGCTGCCTTTTCTGTTCCTT | AAGGTGCTGGGTAGGGAAGT |
| ctgf | TAGCAAGAGCTGGGTGTGTG | TTCACTTGCCACAAGCTGTC |
| fn1 | GAAAGGCAACCAGCAGAGTC | CTGGAGTCAAGCCAGACACA |
| nur77 | TGTTGCTAGAGTCCGCCTTT | CAGTGATGAGGACCAGAGCA |
| postn | TCCTGAATACCCTCCAGTGC | AGGTCCGTGAAAGTGGTTTG |
| rpl13 | AAAAAGGAGAAGGCCAGAGC | CCGCGCATTATTTCTTCTTC |
| rplp0 | CTCAGTGCCTCACTCCATCA | CTTCCTTTGCTTCGACCTTG |
| smad7 | TCCTGCTGTGCAAAGTGTTC | TCTGGACAGTCTGCAGTTGG |
| tgfb | ATACGCCTGAGTGGCTGTCT | TGGGACTGATCCCATTGATT |
| Mouse primer | Forward | Reverse |
| mmp2 | GACCTTGACCAGAACACCATC | CATCCACGGTTTCAGGGTCC |
| Rpl13 | GGGCAGGTTCTGGTATTGGAT | GGCTCGGAAGTGGTAGGGG |
| Rplp0 | GGACCCGAGAAGACCTCCTT | GCACATCACTCAGAATTTCAATGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medzikovic, L.; Heese, H.; van Loenen, P.B.; van Roomen, C.P.A.A.; Hooijkaas, I.B.; Christoffels, V.M.; Creemers, E.E.; de Vries, C.J.M.; de Waard, V. Nuclear Receptor Nur77 Controls Cardiac Fibrosis through Distinct Actions on Fibroblasts and Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 1600. https://doi.org/10.3390/ijms22041600
Medzikovic L, Heese H, van Loenen PB, van Roomen CPAA, Hooijkaas IB, Christoffels VM, Creemers EE, de Vries CJM, de Waard V. Nuclear Receptor Nur77 Controls Cardiac Fibrosis through Distinct Actions on Fibroblasts and Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(4):1600. https://doi.org/10.3390/ijms22041600
Chicago/Turabian StyleMedzikovic, Lejla, Hylja Heese, Pieter B. van Loenen, Cindy P. A. A. van Roomen, Ingeborg B. Hooijkaas, Vincent M. Christoffels, Esther E. Creemers, Carlie J. M. de Vries, and Vivian de Waard. 2021. "Nuclear Receptor Nur77 Controls Cardiac Fibrosis through Distinct Actions on Fibroblasts and Cardiomyocytes" International Journal of Molecular Sciences 22, no. 4: 1600. https://doi.org/10.3390/ijms22041600
APA StyleMedzikovic, L., Heese, H., van Loenen, P. B., van Roomen, C. P. A. A., Hooijkaas, I. B., Christoffels, V. M., Creemers, E. E., de Vries, C. J. M., & de Waard, V. (2021). Nuclear Receptor Nur77 Controls Cardiac Fibrosis through Distinct Actions on Fibroblasts and Cardiomyocytes. International Journal of Molecular Sciences, 22(4), 1600. https://doi.org/10.3390/ijms22041600