
Contribution of Hypothyroidism to Cognitive Impairment and Hippocampal Synaptic Plasticity Regulation in an Animal Model of Depression

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Behavioral Study
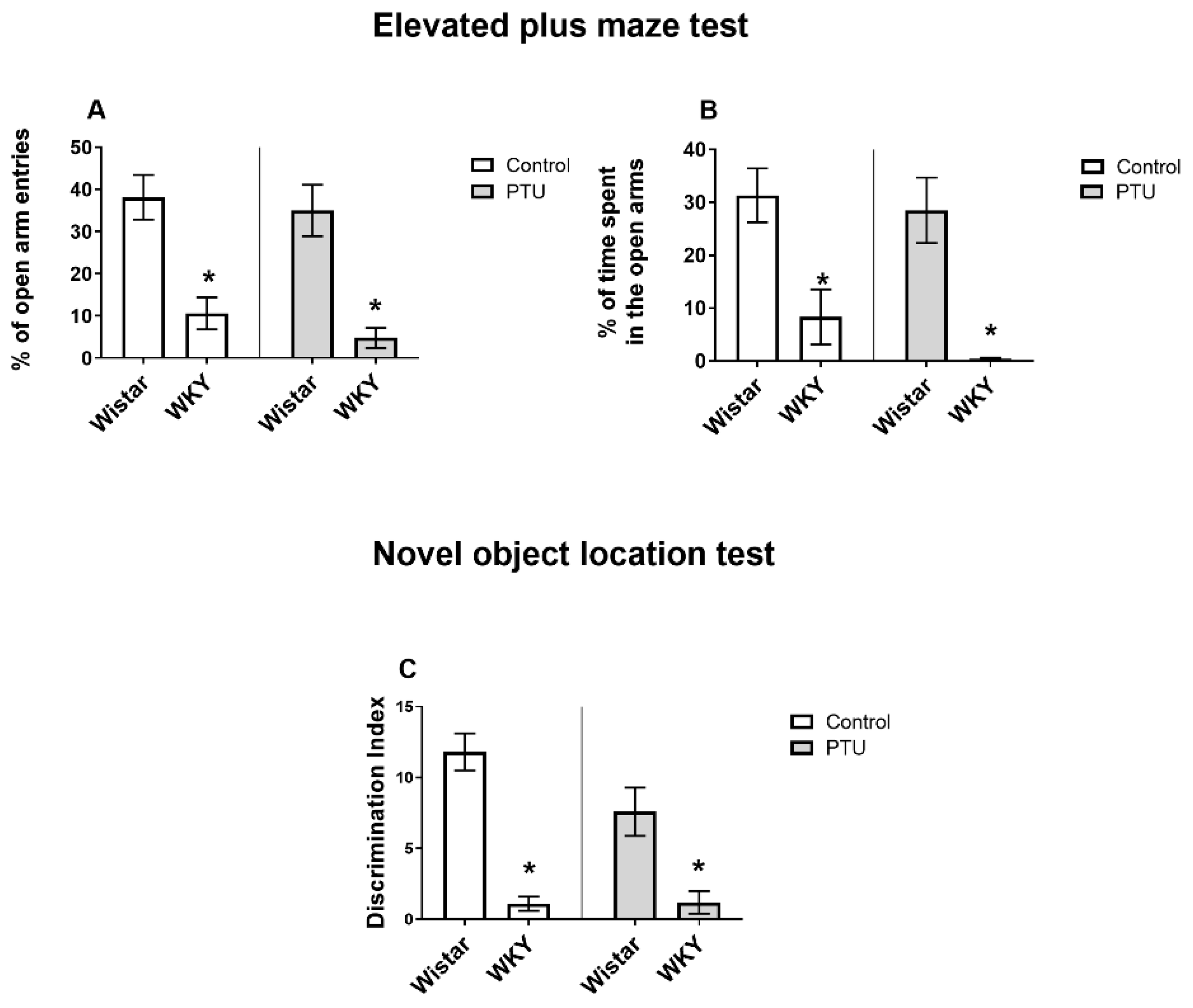
2.1.1. Anxiety Behavior Evaluated in the Elevated Plus Maze (EPM) Test
2.1.2. Spatial Memory Evaluated in the Novel Object Location (NOL) Test
2.2. Electrophysiological Study
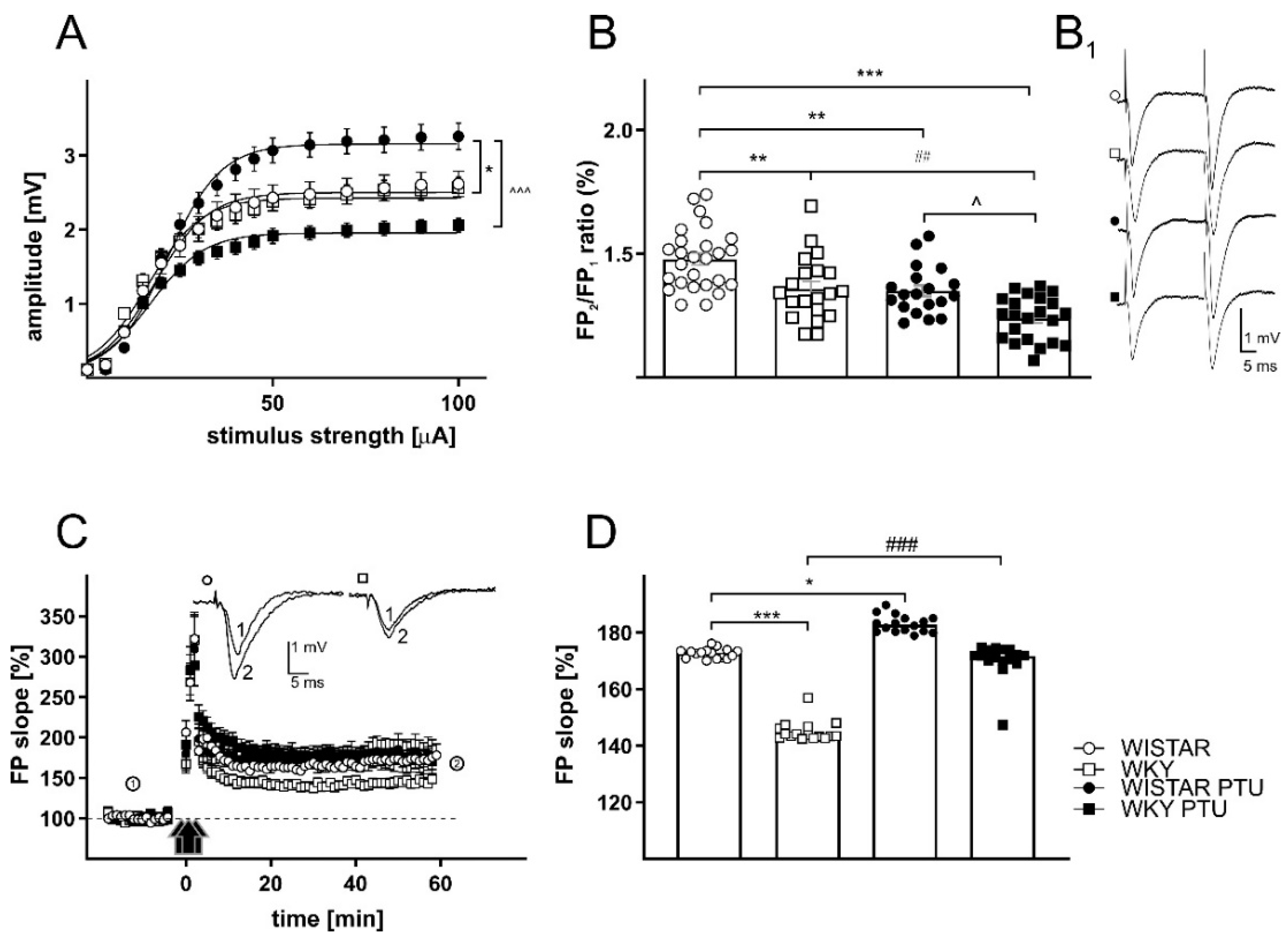
2.2.1. The Effects of PTU on Field Potentials (FPs) in the CA1 Region of the Hippocampus in Wistar and WKY Rats
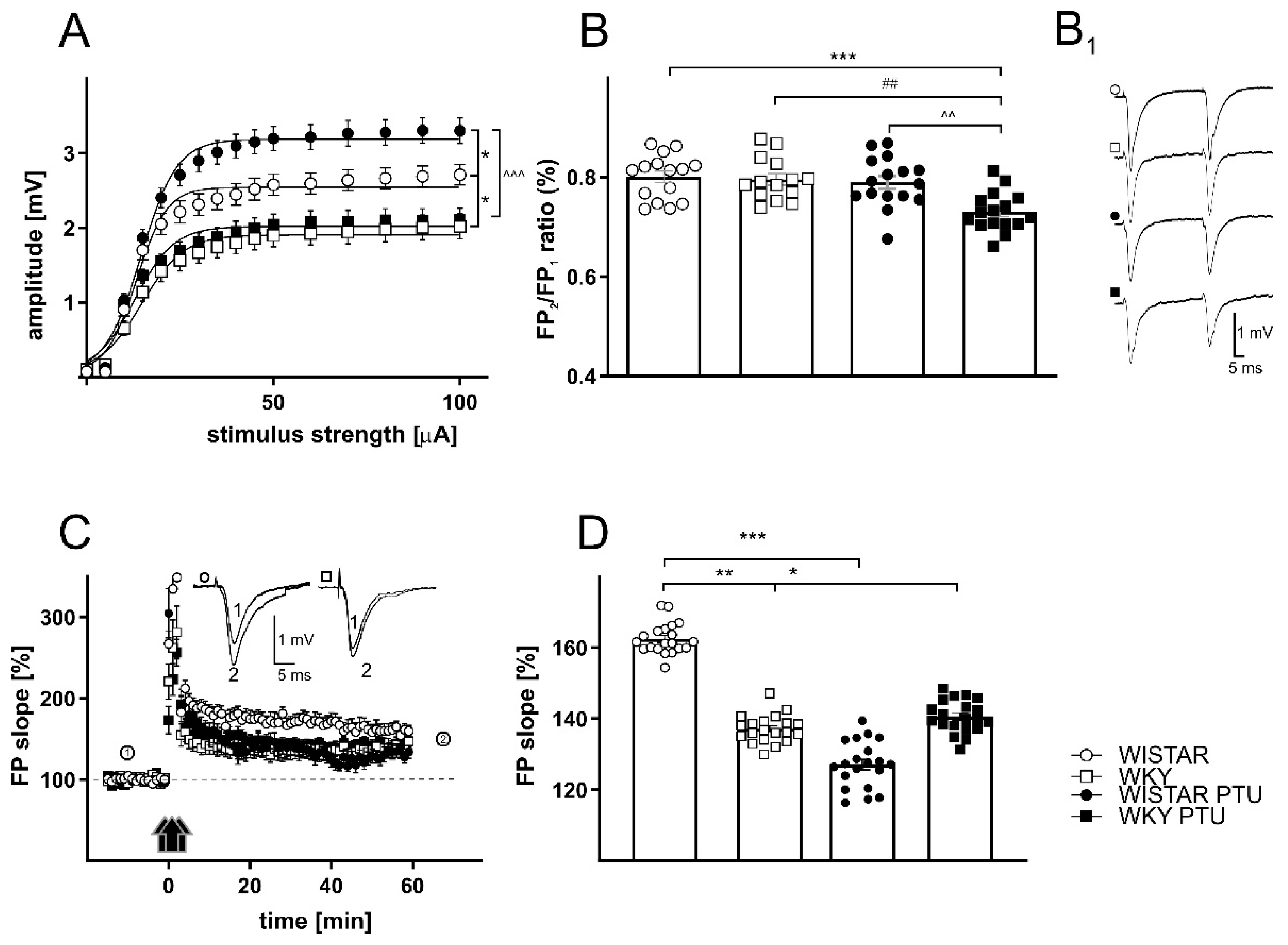
2.2.2. The Effects of PTU on Field Potentials (FPs) in the Dentate Gyrus (DG) Region of the Hippocampus in Wistar and WKY Rats
2.2.3. The Effects of PTU on Paired-Pulse Ratio (PPR)
2.2.4. The Effects of PTU on Long-Term Potentiation
2.3. Biochemical Study
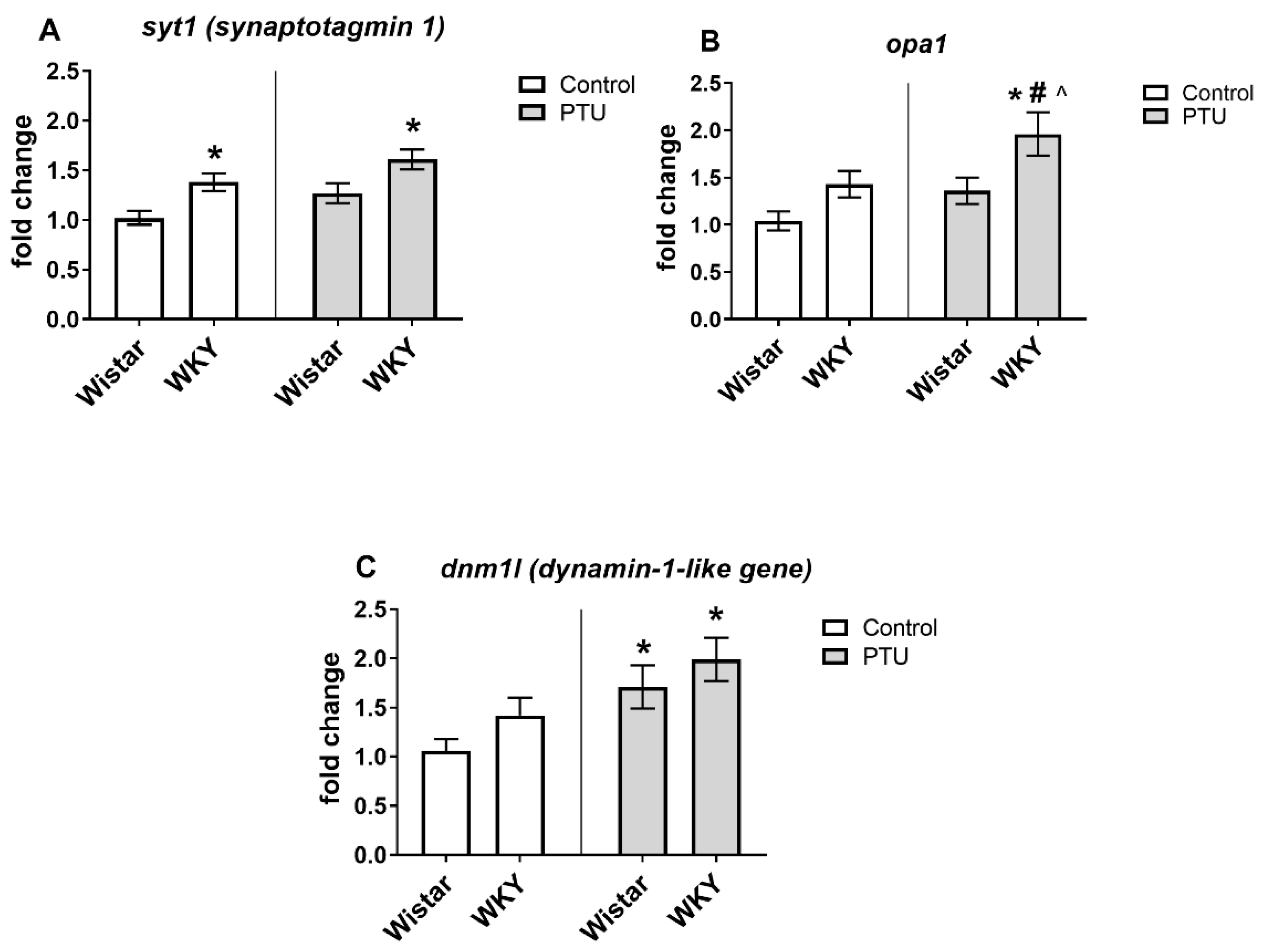
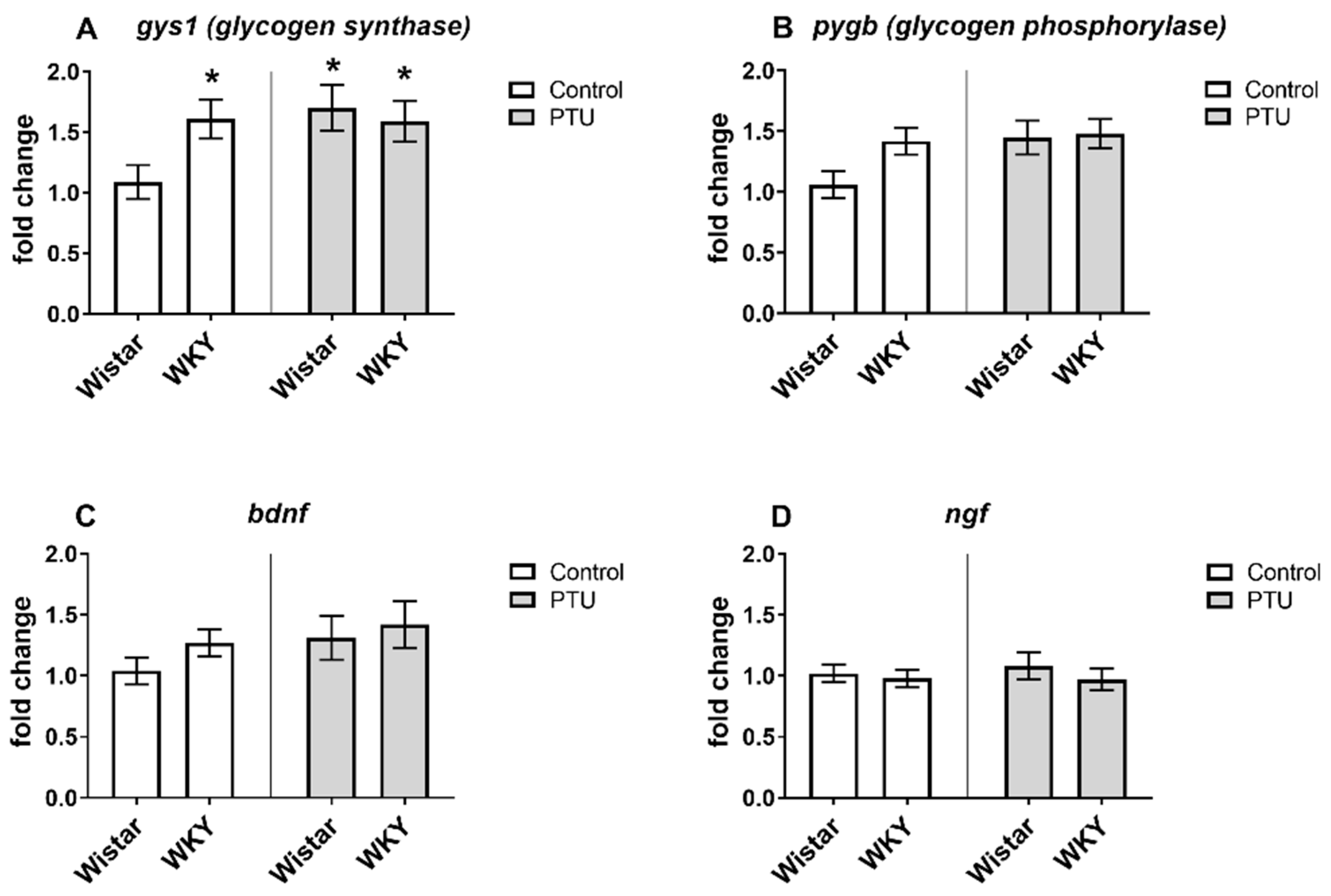
2.3.1. Gene Expression of Syt1, Opa1, Gys1, Dnm1l, Pygb, Bdnf, and Ngf
2.3.2. Protein Level of Subunits of NMDA Receptor (GluN1, GluN2A, GluN2B)
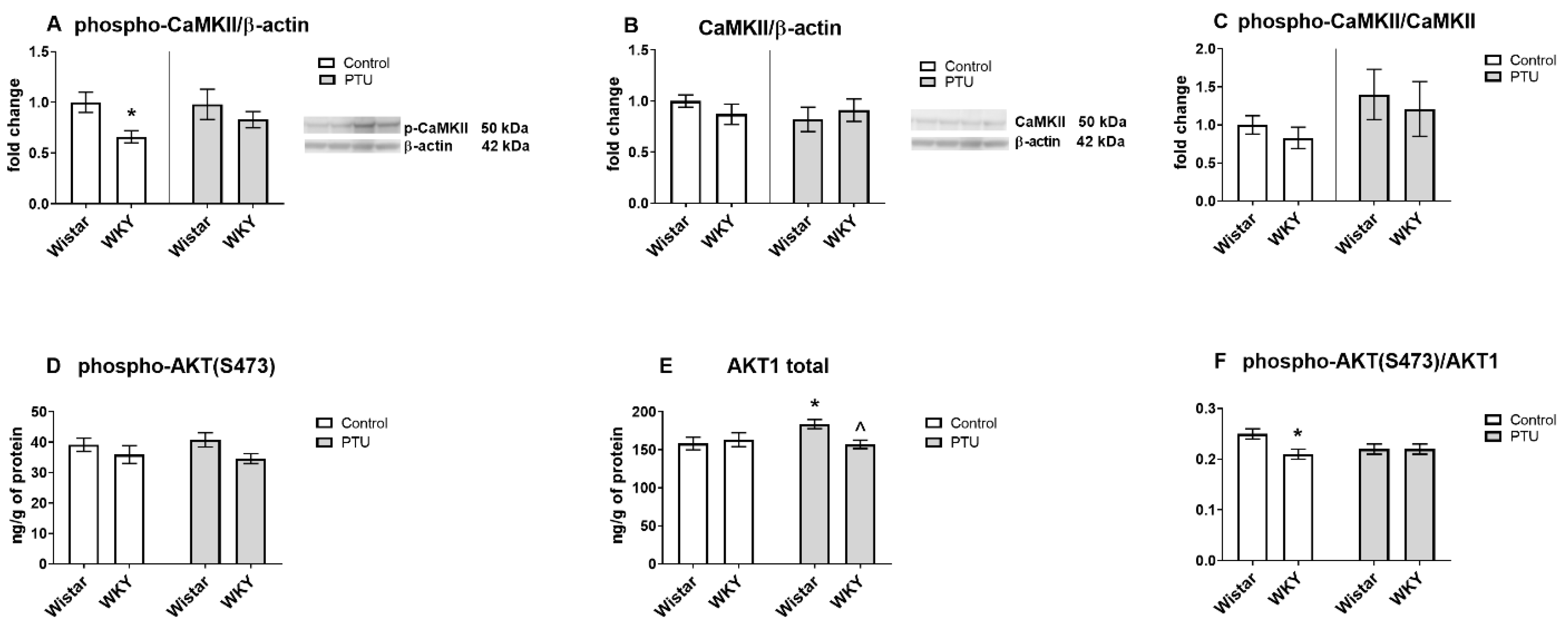
2.3.3. Protein Level of Calcium/Calmodulin-Dependent Protein Kinase II Isoform K II (CaMKII, Total and Phospho Form) and AKT1 (Total and Phospho (Ser473) Form)
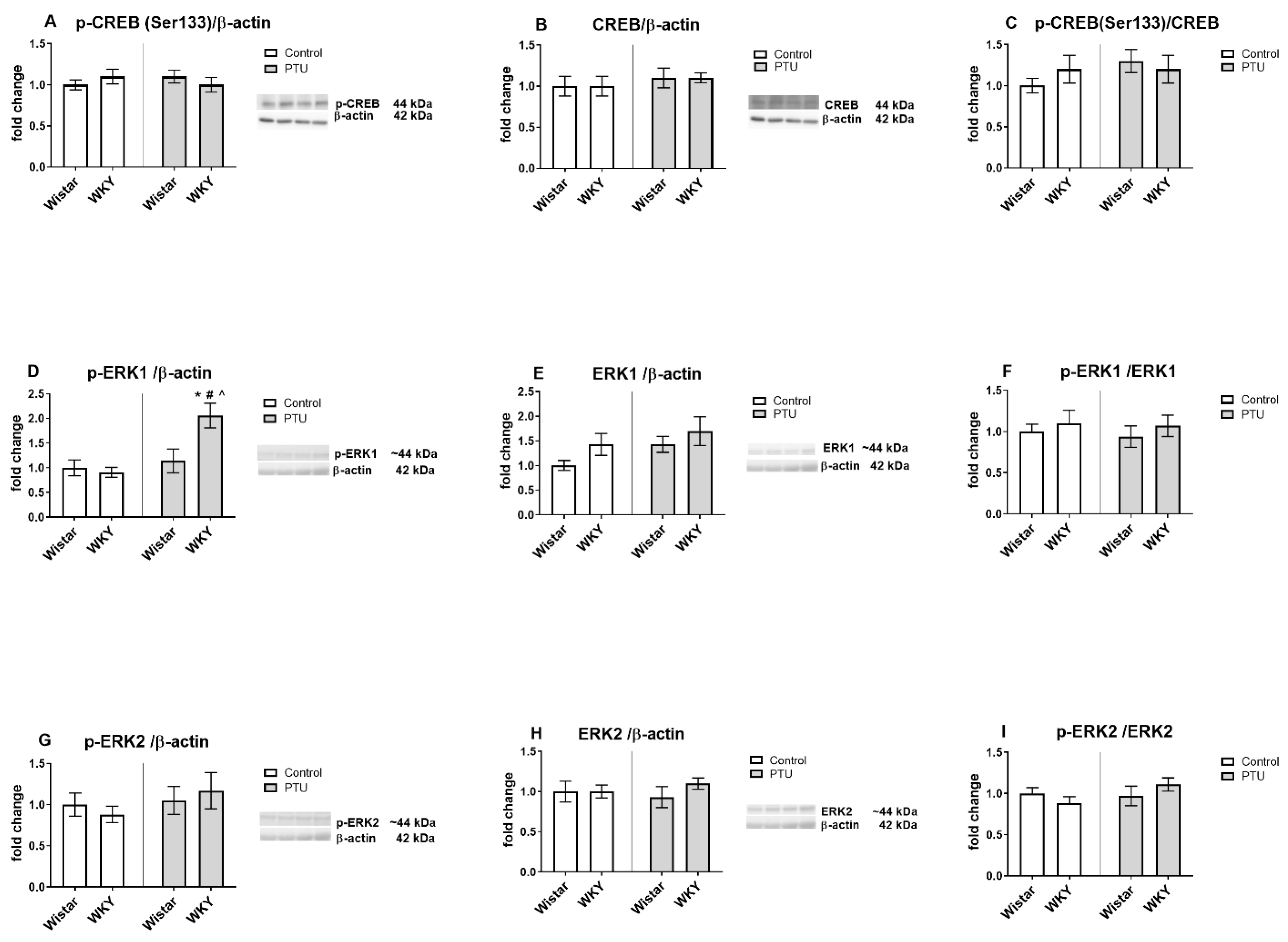
2.3.4. Protein Level of CREB (Total and Phospho (Ser133) Form), Extracellular Signal-Regulated Kinases 1/2 (ERK1/2, Phospho and Total Form)
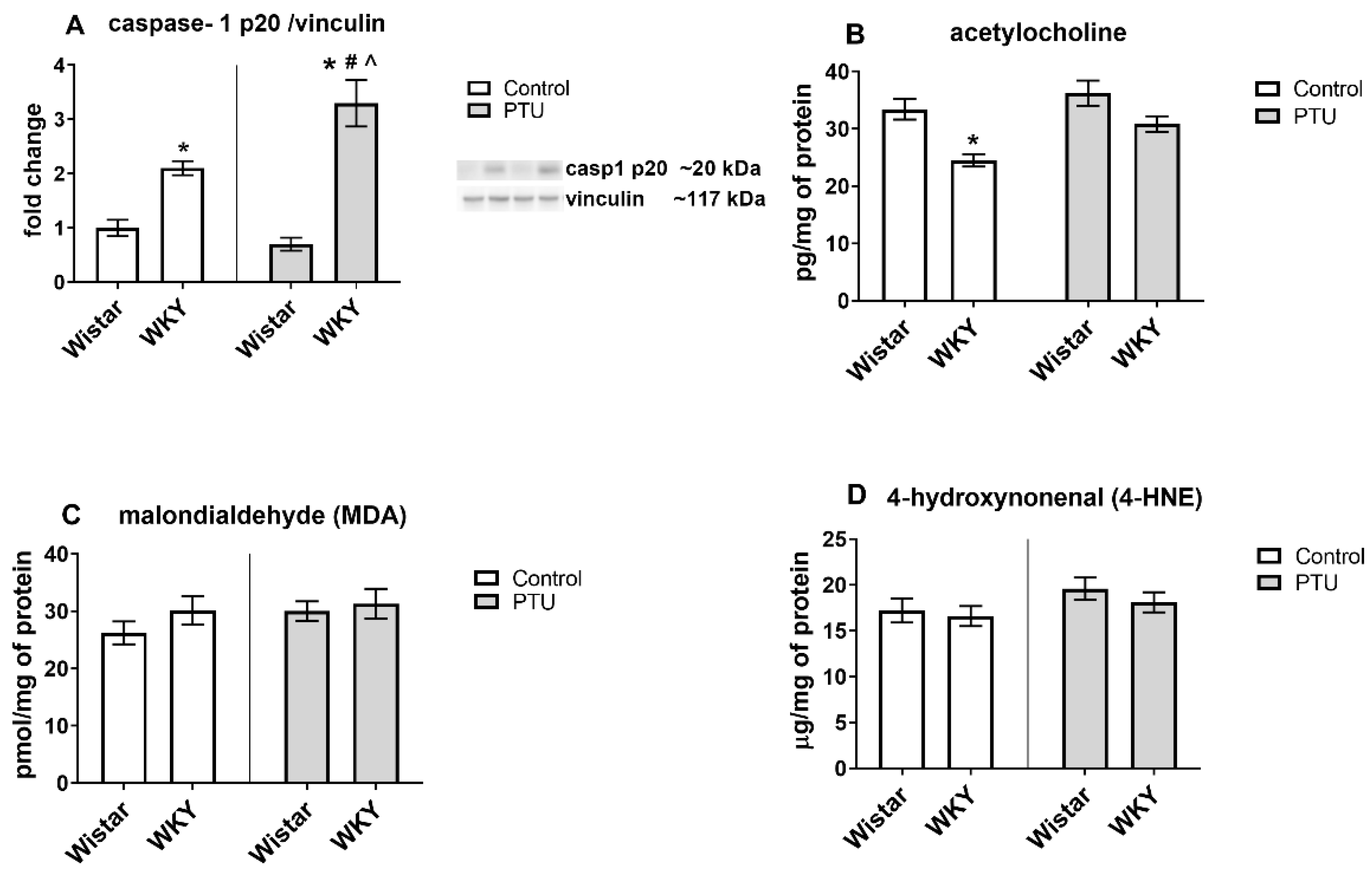
2.3.5. Protein Level of Caspase-1 p20
2.3.6. Acetylcholine Concentration
2.3.7. Protein Levels of Malondialdehyde (MDA) and 4-Hydroxynonenal (4-HNE)
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Behavioral Testing
4.2.1. Elevated Plus Maze (EPM) Test
4.2.2. Novel Object Location (NOL) Test
4.3. Electrophysiological Study
4.3.1. Slice Preparation
4.3.2. Field Potential (FP) Recording and LTP Induction
4.4. Biochemical Analysis
4.4.1. Gene Expression Measurement: RNA Extraction, cDNA Preparation and Real-Time PCR
4.4.2. Tissue Preparation and Determination of Protein Concentration
4.4.3. ELISA
4.4.4. Western Blot
4.4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| BDNF | brain-derived neurotrophic factor |
| CaMK II | calcium/calmodulin-dependent protein kinase II |
| CAPS | 3-clohexylaminopropanesulfonic acid |
| CREB | cAMP Response Element-Binding Protein |
| DG | dentate gyrus |
| Dnm1l | dynamin-1-like |
| ELISA | enzyme-linked immunosorbent assay |
| EPM | elevated plus maze |
| ERK | extracellular signal-regulated kinases |
| FP | field potential |
| Gys1 | glycogen sythase |
| HPT | Hypothalamic—pituitary—thyroid |
| HFS | high frequency stimulation |
| LTP | long-term potentiation |
| MDA | malondialdehyde |
| NGF | nerve growth factor |
| NMDA | N-methyl-d-aspartate receptor |
| NOL | novel object location |
| PCR | polymerase chain reaction |
| Pygb | glycogen phosphorylase |
| PPR | paired-pulse ratio |
| PTU | propylthiouracil |
| PVDF | polyvinylidene fluoride |
| SDS-PAGE | SDS poliacrylamide gel electrophoresis |
| Syt1 | synaptotagmin 1 |
| T3 | triiodothyronine |
| T4 | thyroxine |
| TBS | Tris-buffered saline |
| TMB | 3,3′,5,5′-tetramethylbenzidine |
| THs | thyroid hormones |
| TRE | thyroid response element |
| WKY | Wistar—Kyoto rats |
| 4-HNE | 4-hydroxynonenal |
References
- Parmentier, T.; Sienaert, P. The use of triiodothyronine (T3) in the treatment of bipolar depression: A review of the literature. J. Affect. Disord. 2018, 229, 410–414. [Google Scholar] [CrossRef]
- De Escobar, G.M.; Obregón, M.J.; Del Rey, F.E. Role of thyroid hormone during early brain development. Eur. J. Endocrinol. Suppl. 2004, 151, 25–37. [Google Scholar] [CrossRef]
- Giannocco, G.; Kizys, M.M.L.; Maciel, R.M.; de Souza, J.S. Thyroid hormone, gene expression, and Central Nervous System: Where we are. Semin. Cell Dev. Biol. 2020, 1–10. [Google Scholar] [CrossRef]
- Salazar, P.; Cisternas, P.; Martinez, M.; Inestrosa, N.C. Hypothyroidism and Cognitive Disorders during Development and Adulthood: Implications in the Central Nervous System. Mol. Neurobiol. 2019, 56, 2952–2963. [Google Scholar] [CrossRef]
- Bauer, M.; Goetz, T.; Glenn, T.; Whybrow, P.C. The thyroid-brain interaction in thyroid disorders and mood disorders. J. Neuroendocrinol. 2008, 20, 1101–1114. [Google Scholar] [CrossRef]
- Hage, M.P.; Azar, S.T. The Link between Thyroid Function and Depression. J. Thyroid Res. 2012, 2012, 590648. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Heinz, A.; Whybrow, P.C. Thyroid hormones, serotonin and mood: Of synergy and significance in the adult brain. Mol. Psychiatry 2002, 7, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.C.; Privalsky, M.L. Thyroid hormones, T3 and T4, in the brain. Front. Endocrinol. 2014, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.H.; Schuff, K.G.; Carlson, N.E.; Carello, P.; Janowsky, J.S. Health status, mood, and cognition in experimentally induced subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 2007, 92, 2545–2551. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Gerges, N.Z.; Aleisa, A.M.; Alkadhi, K.A. Levothyroxin restores hypothyroidism-induced impairment of hippocampus-dependent learning and memory: Behavioral, electrophysiological, and molecular studies. Hippocampus 2009, 19, 66–78. [Google Scholar] [CrossRef]
- Sui, L.; Anderson, W.L.; Gilbert, M.E. Impairment in short-term but enhanced long-term synaptic potentiation and ERK activation in adult hippocampal area CA1 following developmental thyroid hormone insufficiency. Toxicol. Sci. 2005, 85, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Głombik, K.; Detka, J.; Kurek, A.; Budziszewska, B. Impaired Brain Energy Metabolism: Involvement in Depression and Hypothyroidism. Front. Neurosci. 2020, 14, 1320. [Google Scholar] [CrossRef]
- Armario, A.; Gavaldà, A.; Martí, J. Comparison of the behavioural and endocrine response to forced swimming stress in five inbred strains of rats. Psychoneuroendocrinology 1995, 20, 879–890. [Google Scholar] [CrossRef]
- Nam, H.; Clinton, S.M.; Jackson, N.L.; Kerman, I.A. Learned helplessness and social avoidance in the Wistar-Kyoto rat. Front. Behav. Neurosci. 2014, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Paré, W.P.; Redei, E. Depressive behavior and stress ulcer in Wistar Kyoto rats. J. Physiol. Paris 1993, 87, 229–238. [Google Scholar] [CrossRef]
- Will, C.C.; Aird, F.; Redei, E.E. Selectively bred Wistar-Kyoto rats: An animal model of depression and hyper-responsiveness to antidepressants. Mol. Psychiatry 2003, 8, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Bhatti, B.H.; Manaye, K.F.; Das, J.R.; Akinfiresoye, L. Antidepressant-like effects of low ketamine dose is associated with increased hippocampal AMPA/NMDA receptor density ratio in female Wistar-Kyoto rats. Neuroscience 2012, 213, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, L.R.; Wang, Y.T.; Phillips, A.G. Evaluation of the Wistar-Kyoto rat model of depression and the role of synaptic plasticity in depression and antidepressant response. Neurosci. Biobehav. Rev. 2019, 105, 1–23. [Google Scholar] [CrossRef]
- Luo, J.; Min, S.; Wei, K.; Cao, J.; Wang, B.; Li, P.; Dong, J.; Liu, Y. Behavioral and molecular responses to electroconvulsive shock differ between genetic and environmental rat models of depression. Psychiatry Res. 2015, 226, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Taşkin, E.; Artis, A.S.; Bitiktas, S.; Dolu, N.; Liman, N.; Süer, C. Experimentally induced hyperthyroidism disrupts hippocampal long-term potentiation in adult rats. Neuroendocrinology 2011, 94, 218–227. [Google Scholar] [CrossRef]
- Fernández-Lamo, I.; Montero-Pedrazuela, A.; Delgado-García, J.M.; Guadaño-Ferraz, A.; Gruart, A. Effects of thyroid hormone replacement on associative learning and hippocampal synaptic plasticity in adult hypothyroid rats. Eur. J. Neurosci. 2009, 30, 679–692. [Google Scholar] [CrossRef]
- Yousef, M.; Babür, E.; Delibaş, S.; Tan, B.; Çimen, A.; Dursun, N.; Süer, C. Adult-Onset Hypothyroidism Alters the Metaplastic Properties of Dentate Granule Cells by Decreasing Akt Phosphorylation. J. Mol. Neurosci. 2019, 68, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Alkadhi, K.A. Levothyroxin replacement therapy restores hypothyroidism induced impairment of L-LTP induction: Critical role of CREB. Brain Res. Bull. 2014, 100, 29–37. [Google Scholar] [CrossRef]
- Salazar, P.; Cisternas, P.; Codocedo, J.F.; Inestrosa, N.C. Induction of hypothyroidism during early postnatal stages triggers a decrease in cognitive performance by decreasing hippocampal synaptic plasticity. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 870–883. [Google Scholar] [CrossRef]
- Solberg, L.C.; Olson, S.L.; Turek, F.W.; Redei, E. Altered hormone levels and circadian rhythm of activity in the WKY rat, a putative animal model of depression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, 786–794. [Google Scholar] [CrossRef]
- Freund, T.F.; Buzsáki, G. Interneurons of the Hippocampus. Hippocampus 1996, 6, 347–470. [Google Scholar] [CrossRef]
- Vara, H.; Martínez, B.; Santos, A.; Colino, A. Thyroid hormone regulates neurotransmitter release in neonatal rat hippocampus. Neuroscience 2002, 110, 19–28. [Google Scholar] [CrossRef]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef]
- Lai, Y.; Lin, P.; Chen, M.; Zhang, Y.; Chen, J.; Zheng, M.; Liu, J.; Du, H.; Chen, R.; Pan, X.; et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020, 34, 101503. [Google Scholar] [CrossRef]
- Memarpour, S.; Beheshti, F.; Baghcheghi, Y.; Vafaei, A.A.; Hosseini, M.; Rashidy-Pour, A. Neuronal Nitric Oxide Inhibitor 7-Nitroindazole Improved Brain-Derived Neurotrophic Factor and Attenuated Brain Tissues Oxidative Damage and Learning and Memory Impairments of Hypothyroid Juvenile Rats. Neurochem. Res. 2020, 45, 2775–2785. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CAMKII actio in LTP. Nat. Rev. Neurosci. 2014, 13, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Calloni, G.W.; Penno, C.A.; Cordova, F.M.; Trentin, A.G.; Neto, V.M.; Leal, R.B. Congenital hypothyroidism alters the phosphorylation of ERK1/2 and p38 MAPK in the hippocampus of neonatal rats. Dev. Brain Res. 2005, 154, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Pellow, S.; Chopin, P.; File, S.E.; Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 1985, 14, 149–167. [Google Scholar] [CrossRef]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, 1–20. [Google Scholar] [CrossRef]
- Bobula, B.; Sowa, J.; Hess, G. Anti-interleukin-1β antibody prevents the occurrence of repeated restraint stress-induced alterations in synaptic transmission and long-term potentiation in the rat frontal cortex. Pharmacol. Rep. 2015, 67, 123–128. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Amplitude Vmax (mV) | U50 (µA) | Slope | Number of Slices |
|---|---|---|---|---|
| Wistar | 2.50 ± 0.17 | 18.54 ± 1.10 | 7.02 ± 0.55 | 26 |
| WKY | 2.42 ± 0.13 | 16.54 ± 1.30 | 7.03 ± 0.66 | 24 |
| Wistar + PTU | 3.15 ± 0.17 * | 21.79 ± 1.20 | 7.05 ± 0.50 | 22 |
| WKY + PTU | 1.95 ± 0.09 ^^^ | 17.45 ± 1.02 | 7.05 ± 0.61 | 18 |
| Group | Amplitude Vmax (mV) | U50 (µA) | Slope | Number of Slices |
|---|---|---|---|---|
| Wistar | 2.54 ± 0.13 | 13.51 ± 0.96 | 3.96 ± 0.53 | 15 |
| WKY | 1.90 ± 0.15 * | 15.01 ± 1.00 | 5.42 ± 0.51 | 20 |
| Wistar + PTU | 3.18 ± 0.16 * | 14.71 ± 0.65 | 4.69 ± 0.37 | 24 |
| WKY + PTU | 2.02 ± 0.14 ^^^ | 13.14 ± 0.78 | 5.12 ± 0.59 | 16 |
| Group | CA1 | DG |
|---|---|---|
| Wistar | 1.478 ± 0.024 | 0.8007 ± 0.011 |
| WKY | 1.359 ± 0.029 ** | 0.7955 ± 0.011 |
| Wistar + PTU | 1.347 ± 0.022 ** | 0.7898 ± 0.012 |
| WKY + PTU | 1.239 ± 0.019 *** ## ^ | 0.7304 ± 0.01 *** ## ^^ |
| Group | CA1—fEPSP Slope | DG—fEPSP Slope |
|---|---|---|
| Wistar | 172.7 ± 1.93 | 162.5 ± 0.965 |
| WKY | 145.4 ± 1.91 *** | 137.1 ± 0.87 ** |
| Wistar + PTU | 181.2 ± 2.16 * | 127.1 ± 1.46 *** |
| WKY + PTU | 175.3 ± 1.62 ### | 140.7 ± 1.018 * |
| Group | GluN1 | GluN2A | GluN2B |
|---|---|---|---|
| Wistar | 1.00 ± 0.09 | 1.00 ± 0.07 | 1.00 ± 0.05 |
| WKY | 1.02 ± 0.07 | 1.03 ± 0.06 | 0.87 ± 0.05 |
| Wistar + PTU | 0.91 ± 0.09 | 0. 99 ± 0.08 | 0.93 ± 0.09 |
| WKY + PTU | 1.02 ± 0.11 | 1.07 ± 0.08 | 0.86 ± 0.06 |
 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Głombik, K.; Detka, J.; Bobula, B.; Bąk, J.; Kusek, M.; Tokarski, K.; Budziszewska, B. Contribution of Hypothyroidism to Cognitive Impairment and Hippocampal Synaptic Plasticity Regulation in an Animal Model of Depression. Int. J. Mol. Sci. 2021, 22, 1599. https://doi.org/10.3390/ijms22041599
Głombik K, Detka J, Bobula B, Bąk J, Kusek M, Tokarski K, Budziszewska B. Contribution of Hypothyroidism to Cognitive Impairment and Hippocampal Synaptic Plasticity Regulation in an Animal Model of Depression. International Journal of Molecular Sciences. 2021; 22(4):1599. https://doi.org/10.3390/ijms22041599
Chicago/Turabian StyleGłombik, Katarzyna, Jan Detka, Bartosz Bobula, Joanna Bąk, Magdalena Kusek, Krzysztof Tokarski, and Bogusława Budziszewska. 2021. "Contribution of Hypothyroidism to Cognitive Impairment and Hippocampal Synaptic Plasticity Regulation in an Animal Model of Depression" International Journal of Molecular Sciences 22, no. 4: 1599. https://doi.org/10.3390/ijms22041599
APA StyleGłombik, K., Detka, J., Bobula, B., Bąk, J., Kusek, M., Tokarski, K., & Budziszewska, B. (2021). Contribution of Hypothyroidism to Cognitive Impairment and Hippocampal Synaptic Plasticity Regulation in an Animal Model of Depression. International Journal of Molecular Sciences, 22(4), 1599. https://doi.org/10.3390/ijms22041599